

Abstract
The growth factor independence-1 (Gfi1) transcription factor is required for proper development of neuroendocrine cells, sensory neurons, and blood. Patients with mutations in Gfi1 exhibit severe congenital neutropenia (SCN) or non-immune chronic idiopathic neutropenia of adults. Gfi1 was initially described as an oncoprotein that mediates tumor progression in a mouse model of leukemia; however, recent data suggest that Gfi1 may act as either an oncogene or an anti-proliferative tumor suppressor gene depending on the cell type. Here we review the latest literature on Gfi1, and emphasize its role in the hematopoietic, sensory and neuroendocrine systems.
Keywords: Growth factor independence-1, Notch, Ash1, T lymphopoiesis, Hematopoiesis, Peripheral nervous system, Pulmonary neuroendocrine cells, Oncogene, Tumor suppressor
1. Clinical relevance
Growth factor independence-1 (Gfi1) is a transcriptional repressor protein that plays important biological roles in hematopoietic and neuronal cell development [1–4]. Specifically, mice deleted for Gfi1 not only lack neutrophils [2,3], but have defective hematopoietic stem cell maintenance [5,6], and developmental deficits in sensory neurons [7,8] and neuroendocrine cells [9]. Gfi1 mutations in humans were recently shown to correlate to severe congenital neutropenia (SCN) and non-immune chronic idiopathic neutropenia of adults (NI-CINA) [10]. Interestingly, Gfi1 is also expressed and is oncogenic in human small cell lung cancer (SCLC), the deadliest neuroendocrine tumor [9]. Thus, understanding the biological and biochemical activities of Gfi1 may provide a unique opportunity to understand these human diseases.
2. Gfi1 mediates tumor progression as a frequent oncogenic target of the Moloney murine leukemia virus
The Moloney murine leukemia virus (MoMLV) mediates oncogenic function through insertion mutagenesis of cellular proto-oncogenes and tumor suppressors. MoMLV infection leads to aggressive leukemias which accumulate proviral insertions and progress from interleukin-2 (IL2)-dependent to IL2-independent growth [11]. Growth factor independence-1 (Gfi1) was first identified as the gene activated by a MoMLV proviral insertion site (gfi-1) in a screen for MoMLV-target genes capable of inducing progression of rat T cell leukemias to IL2-independent growth [12]. Activating MoMLV insertions in the gfi-1 locus mapped to the promoter of the affected Gfi1 gene, and result in long terminal repeat (LTR)-driven overexpression of an unaltered Gfi1 open reading frame [12]. Gfi1 overexpression does not induce IL2 expression. Instead, IL2-dependent cell lines transduced with Gfi1-expressing retroviral vectors either frequently gave rise to IL2-independent cell lines [12], or showed an impaired ability to maintain cell cycle checkpoints upon IL2 withdrawal [13,14]. Thus, in the context of the in vitro screen, Gfi1 overexpression is the effector of MoMLV insertion at this locus.
Naturally occurring IL2-independent MoMLV tumors also display insertions at gfi-1 [12]. In fact, murine retroviruses that cause T and B cell leukemias upregulate Gfi1 by occupying multiple common proviral insertion loci in rodent genomes [12,15–17]. Leukemias in mice transgenic for Myc or Pim oncoprotein overexpression and infected with MoMLV display frequent MoMLV proviral integrations in the pal-1 common integration locus [15,18], the extra integration site-1 (eis-1) locus, and the T cell Moloney integration site (tmi-1) locus [15]. The common mink-cell focus-forming (MCF) virus insertion locus, evi-5, in AKXD mouse T cell leukemias, maps 18 kilobases upstream of Gfi1 [19]. Notably, tmi-1 and evi-5 loci are different names for the same locus [16], and the gene disrupted by MoMLV insertion encodes a ubiquitously expressed gene (Evi5) which is homologous to the Tre 2 oncogene. However, it is not known if Evi5 is itself involved in leukemogenesis or if proviral insertions within the Evi5 locus simply serve to enhance Gfi1 expression [17]. Each of the MoMLV insertion loci gfi-1, eis-1, evi-5/tmi-1, and pal-1 are distinct chromosomal locations surrounding the Gfi1 gene, and proviral occupancy in any of these loci results in the induction of Gfi1 overexpression in leukemias [14,15]. A majority of MoMLV-induced T cell leukemias have integrations in one of the gfi-1, eis-1, evi-5/tmi-1, and pal-1 loci (mouse retroviral tagged cancer gene database, http://rtcgd.ncifcrf.gov/ [20]). Interestingly, retroviral insertions in these loci are also associated with MoMLV-induced marginal zone B cell lymphomas [21], and murine acquired immunodeficiency syndrome (MAIDS) virus-induced lymphomas [17]. In sharp contrast, proviral occupancy of these loci is extremely rare in myeloid tumors [20]. Thus, the oncogenic function of Gfi1 overexpression may be restricted to lymphocytes.
Gfi1 is a tumor progression oncoprotein. MoMLV insertion mediated overexpression of Gfi1 suggests that simply deregulating expression of Gfi1 is oncogenic: however, overexpression of Gfi1 alone is not a potent initiating oncogenic event. Mice transgenic for Gfi1 expression do not show an unusual incidence of leukemia [22,23]. Moreover, while chromosomal irregularities in a variety of human cancers map to 1p22, the location of Gfi1 [24,25], no mutations that directly affect Gfi1 have been identified. To confirm a role for Gfi1 in MoMLV leukemogenesis, mice transgenic for Gfi1 overexpression were infected with MoMLV. While Moloney virus infection of wild type mice leads to leukemia in about 6 months, Gfi1-transgenic mice were leukemic in one-third the time [14]. Thus, while Gfi1 overexpression is a significant mechanism utilized during MoMLV-induced leukemogenesis, Gfi1 oncogenic function is only unleashed upon transformation that is initiated by other oncogenes. These data are in agreement with the identification of Gfi1 as a tumor progression oncoprotein.
Clues to the oncogenic action of Gfi1 lie in the pattern of insertion sites within MoMLV leukemias. At least three groups of MoMLV-target genes can be specified from the common proviral insertion sites in transgenic mice: the Pim family of serine-threonine kinases, the Myc family transcription factors, and a group consisting of Bmi1, Gfi1, and Gfi1b [26]. Gfi1 and Gfi1b are encoded by different genes, but are both transcription factors with nearly identical transcription-repression domains and zinc-finger DNA-binding motifs [27] (Fig. 1). However, insertions that activate Gfi1b expression are exceedingly rare [27]. It is currently unclear whether Gfi1b is a poor oncogene. Given that Gfi1b transgenic mice show a mild acceleration of naturally occurring lymphomagenesis [22], it is more likely that local chromatin and/or potentially tumor suppressive genes surrounding Gfi1b makeitan infrequent MoMLV target. In contrast to either Gfi1 or Gfi1b, overexpression of Bmi1 alone induces leukemia in mice [28]. Unlike members of the Myc or Pim oncogenic groups, Bmi1 and Gfi1/Gfi1b are not structurally related. Bmi1 (B-cell-specific Moloney murine leukemia virus insertion site-1) is a nuclear protein with an amino terminal Cys/His metal binding RING-zinc-finger motif and a helix-turn-helix-turn-helix-turn DNA binding motif. Bmi1 is a member of the polycomb family of negative transcriptional regulators of both homeotic genes [29–31], and the p16ink4a/p19arf tumor suppressor locus [32]. Proviral insertions in the gfi-1, eis-1, evi-5/tmi-1, and pal-1 loci show very little overlap with integrations activating Bmi1 overexpression, suggesting that the activation of the Bmi1 and Gfi1 genes are mutually exclusive. Thus, Bmi1 and Gfi1 may act on identical or complementary targets to perform the same cellular function [15].
Fig. 1.
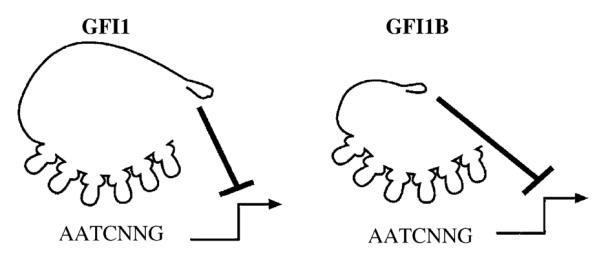
Schematic representation of the transcriptional repressors Gfi1 and Gfi1B. Both Gfi1 and Gfi1B bind to the consensus-DNA binding site 5′-AATCNNG-3′ with C2H2 type zinc-fingers on target genes. Once bound to DNA, Gfi1 and Gfi1B repress transcription through the N-terminal SNAG repressor domain.
Both Gfi1 and Bmi1 cooperate with Myc oncogenesis [16,33]. Overexpression of the Myc proteins induces cell cycle progression and apoptosis [34]. Activation of Myc overexpression is a central mechanism of MoMLV leukemogenesis [20]. Notably, both Gfi1 and Bmi1 can induce both cell cycle progression and cell survival [33,35,36], which may explain why they cooperate with Myc-induced oncogenesis. While Bmi1 cooperation with Myc may rely on transcriptional repression of the p16ink4a/p19arf tumor suppressor locus [33], there is no evidence for Gfi1 regulation of these genes.
3. Gfi1 is a transcriptional repressor
Gfi1 is a DNA-binding protein with six C2H2-type, C-terminal zinc-finger motifs that bind the DNA sequence TAAATCAC(A/T)GCA [12,37]. Position- and orientation-independent active transcriptional repression activity by Gfi1 depends on a 20 amino acid N-terminal repressor domain that is coincident with a nuclear localization motif [13] (Fig. 1). The amino acid sequence of this domain is nearly identical in Gfi1B, and similar in the insulinoma-associated protein IA-1, the homeobox protein Gsh-1, the Ovo-like proteins, and all the vertebrate members of the Snail/Slug family of mesoderm-determining transcription factors (Snail + Gfi1 = SNAG domain) [13] (Table 1). Deletion or mutation of the first seven amino acids in the Gfi1 or Gfi1B SNAG domain destroys the repression activity of either protein [13,27]. Notably, mutation of the proline at position 2 to alanine (P2A) in Gfi1 significantly impaired repression function without disturbing nuclear localization [13]. In contrast to wild-type Gfi1, the P2A mutant fails to mediate IL-2 independent growth of MoMLV leukemias [13]. Similarly, the transcriptional repressor activity of the SNAG domain in Snail has been confirmed [38]. While Snail represses the promoter of the E-cadherin gene, deletion of the first seven amino acids of Snail, or a P2A-mutant Snail, failed to affect E-cadherin transcription [38]. Thus, for both Gfi1 and Snail, SNAG transcription repression function and oncogenic biology appear to be linked. Moreover, the Gfi1 SNAG domain is the prototype of a novel family of evolutionarily conserved repressor domains that operate in multiple cell lineages and are critical mediators of oncogenic function in both lymphoid and solid tumors (Table 1).
Table I.
SNAG domain containing family of proteins from representative species
| SPECIES | PROTEIN | 1 5 10 15 20 |
|---|---|---|
| Homo sapiens* | Gfil | MPRSFLVKSKKAHSYHQPRSP |
| Homo sapiens | GfilB | MPRSFLVKSKKAHTYHQPRVQ |
| Gallus gallus | ZFP | MPRSFLVKSKKAHTYHQHRFV |
| Xenopus laevis | Znf544-prov P | MPRSFLVKSKKTHTYNQHRYV |
| Homo sapiens | SNAIL 1 | MPRSFLVRKPSDPNRKPNYSE |
| Homo sapiens | SNAIL 2 | MPRSFLVKKHFNASKKPNYSE |
| Gallus gallus | Snail like P | MPRSFLVKKHFSASKKPNYSE |
| Danio rerio | Snail 1 | MPRSFLVKKYFTSKRPNYSEL |
| Danio rerio | Snail 2 | MPRSFLVKKYFTNKKPNYSEL |
| Xenopus laevis | xSna1 | MPRSFLVKKHFSASKKPNYSE |
| Xenopus laevis | xSna2 | MPRSFLVKKHFNSAKKPNYGE |
| Nematostella vectensis | Snail ZFP-A | MPRSFLVKKTCDKKALLLRNG |
| Nematostella vectensis | Snail ZFP-B | MPRSFLVKTKTERCSHFDSPI |
| Branchiostoma Floridae | Snail | MPRSFLIKKKLHHAAKKPRFD |
| Homo sapiens** | Slug | MPRSFLVKKHFNASKKPNYSE |
| Gallus gallus | Slug P | MPRSFLVKKHFNSSKKPNYSE |
| Danio rerio | Zgc:92564 | MPRSFLVKKHFNAAKKPNYSE |
| Xenopus laevis | ZFTF Slug β | MPRSFLVKKHFNTTKKPNYGE |
| Homo sapiens | Scratch 1 | MPRSFLVKKVKLDAFSSADLE |
| Homo sapiens | Scratch 2 | MPRSFLVKKIKGDGFQCSGVP |
| Danio rerio | Scratch-a | MPRSFLVKKIKLDDFSSSPVS |
| Mus Musclus | Smuc | MPRSFLVKTHSSHRVPNYGKL |
| Tetraodon nigroviridis | (unnamed) | MPRSFLVKKKRGSSGGWQWKE |
| Lytechinus variegatus | ZFTF | MPRSFLVKKNAKQAASGLKKL |
| Mus Musclus | Gsh1 | MPRSFLVDSLVLREASDKKAP |
| Danio rerio | P for MGC: 110817 |
MPRSFLVDSLILRENSEKGTE |
| Homo sapiens | INSM1 | MPRGFLVKRSKKSTPVSYRVR |
| Tetraodon nigroviridis | (unnamed) | MPRGFLVKRNKKANAVSYRVR |
| Homo sapiens# | OVO-like 1 Binding P |
MPRAFLVKKPCVSTCKRNWSE |
| Homo sapiens | ZFP 339 | MPKVFLVKRRSLGVSVRSWDE |
| Tetraodon nigroviridis | (unnamed) | MPRAFLVKKANVSPGKRNWSE |
| Xenopus tropicalis | MGC89597 P | MPRAFLVKRRSPPPVIRNWDE |
ZFTF, zinc finger transcription factor; ZFP, zinc finger protein; P, protein.
Mus musclus, Rattus norvegicus, Canis familiaris, and Danio rerio have identical N-terminal 20 amino acids to Homo sapiens Gfi1.
Mus Musclus and Rattus norvegicus have identical N-terminal 20 amino acids to Homo sapiens Slug.
Mus musclus has identical N-terminal 20 amino acids to Homo sapiens OVO-like 1 binding P.
The SNAG domain mediates repression function, but is constrained by positional effects. While structure-function analyses of Gfi1, Gfi1b and Snail confirm SNAG activity [13,27,38], fusion proteins containing the SNAG domain of either the Scratch or IA-1 transcription factors did not validate SNAG domain activity [39,40]. A fusion protein consisting of the Gfi1 SNAG domain, the DNA binding domain of the PAX3 transcription factor and the hormone-binding domain of the estrogen receptor (SNAG/PAX3/ER) displayed hormone-inducible transcriptional repression of PAX3 target genes, as well as inhibition of growth and survival of PAX3-FKHR oncoprotein-expressing rhabdomyosarcomas [41]. Similarly, fusion of the Gfi1 amino terminus to the amino terminus of the LexA DNA-binding domain transformed LexA into a transcriptional repressor [13]. However, fusion of the same Gfi1 amino terminus to the carboxy terminus of LexA did not yield a repressor protein (Grimes, unpublished data). Fusion of immuno-epitope-tags to the amino-terminal SNAG domain of Gfi1 also significantly impairs repressor function (Grimes, unpublished data). Given the sensitivity of the proline at position 2 in the SNAG domain, it may not be surprising that the SNAG domain often fails to function in contexts other than the amino terminus. Since the Scratch and IA-1 SNAG domains were fused to the carboxy-terminus of a DNA binding protein [39,40], the function of these SNAG domains remains to be determined.
Gfi1 mediates reversible transcriptional repression through recruitment of the eight 21 corepressor (ETO), histone deacetylase (HDAC) enzymes and the G9a histone lysine methyltransferase. The SNAG/PAX3/ER fusion protein described above represses transcription of PAX3 target genes in a hormone-inducible manner, but these genes were quickly reactivated upon withdrawal of the inducing hormone [41]. SNAG-mediated repression is therefore, mediated by reversible chromatin changes, such as local histone deacetylation, and is unlikely to induce long-term silencing of target genes. The ETO family of proteins bind to the zinc fingers of Gfi1 and Gfi1b, and a subset of both ETO and Gfi1 protein are attached to the nuclear matrix [42]. Both ETO and Gfi1 coimmunoprecipitate with HDAC1, HDAC2 and HDAC3 [43,42], and Gfi1 transcriptional repression is significantly impaired by the HDAC inhibitor Trichostatin A [42]. It is possible that the SNAG domain tethers a transcriptional corepressor that mediates the chromatin changes that precede HDAC recruitment, or stabilizes the ETO/HDAC interactions. Indeed, recent data indicate that Gfi1 interacts with the histone lysine methyltransferase G9a and recruits G9a and HDAC1 to its target genes in order to repress transcription through histone H3-K9 dimethylation. The methylation of H3-K9 accumulates after Gfi1 binding and may be required for recruitment of HDAC1 [102]. The relationship between ETO, HDACs and G9a in Gfi1-mediated development and oncogenesis remains to be determined.
4. Gfi1 regulates hematopoietic stem cell (HSC) proliferation
In strict pathogen-free animal colonies, the inability of Gfi1−/− HSC to sustain long term hematopoiesis appears to be the dominant defect resulting in a short life span. Gfi1−/− mice show a dramatic decrease in Kit+Lin−Sca+ (KLS)Flt3+ HSC [5], and die within a few months after birth due to bone marrow failure [5,6]. Transplantation of large numbers of Gfi1−/− bone marrow or purified KLS HSC is capable of reconstitution of irradiated mice and sustained hematopoiesis for over 3 months. The same Gfi1−/− bone marrow cells are apparently impaired in competitive engraftment, even when excess numbers of Gfi1−/− bone marrow cells are used in relation to wild type marrow [5,6]. The failure of Gfi1−/− HSC is due to intrinsic HSC defects and not due to an altered microenvironment or homing ability of transplanted cells. Gfi1−/− HSC can home properly after transplantation as indicated by the short term reconstitution ability [5,6]. Since wild type bone marrow efficiently reconstitutes lethally irradiated Gfi1−/− mice, a defect in microenvironment due to Gfi1 absence can be excluded [6]. Moreover, chimeras generated with Gfi1−/− embryonic cells (ES) and wild type blastocysts demonstrated that the progeny of the Gfi1−/− ES cells are initially found throughout the developing hematopoietic system [5]; however, 2-month-old chimeras specifically lack bone marrow cells from Gfi1−/− ES cells. These data indicate that an intrinsic defect in early stem cell function results in a failure to sustain hematopoiesis in the chimeric animals [5].
Gfi1 is an inhibitor of HSC proliferation. HSC must balance self renewal with the production of proliferating daughter cells. Analysis of gene expression profiles of HSC as they proliferate and return to quiescence in vivo revealed that Gfi1 and Gfi1b steady state mRNA levels are depressed during proliferation and are dramatically induced when the cells return to quiescence [44]. While loss of Gfi1b is embryonic lethal due to a failure in erythropoiesis [45], Gfi1b−/− fetal liver HSC competitively repopulate and serial transplant [5]. Thus, the defect in Gfi1−/− HSC is unique to Gfi1. Cell cycle analysis of Gfi1−/− HSC showed marked differences when compared to wild type HSC. Significantly more Gfi1−/− HSC actively proliferate in comparison to wild type HSC [5,6]. The excessive proliferation of Gfi1−/− HSC most likely exhausts the rare HSC pool and thus compromises their hematopoietic function. In sharp contrast to the stem/progenitor cells, the thymocytes of the Gfi1−/− mouse show a mild decrease in cell cycle progression [5]. While the latter result is consistent with the oncogenic activity of Gfi1 in T cells, together these findings establish contrasting roles for Gfi1 in proliferation depending on the cell type and the developmental stage.
Several cell cycle genes have been suggested as Gfi1 targets including E2F5, E2F6 and p21waf1 [27,46]. Analysis of these genes in the KLS FLT3− Gfi1−/− HSC showed transcriptional down regulation of the cell cycle inhibitor p21waf1 in one case [5]. However, analysis of total KLS Gfi1−/− HSC did not also show transcriptional effects on p21waf1. Instead, the level of p21waf1 protein was significantly lower in Gfi1−/− bone marrow lysates [6]. The difference in the steady state levels of p21waf1 mRNA might be explained by a lack of KLS FLT3+ cells in the knockout animals, which would skew the Lin− populations. Interestingly, p21waf1 is a target gene for repression by Gfi1b [27]. Gfi1 regulates the transcription of Gfi1b [47] and vice versa [48,49] (elaborated below). In agreement with this, Gfi1b steady state mRNA levels were increased two-fold in Gfi1−/− marrow [5]. Therefore, it is possible that the defect in Gfi1−/− HSC is partly through induction of Gfi1b, which represses p21waf1. Since p21waf1 has been shown to regulate HSC self renewal [50], the deregulated expression of p21waf1 may partially explain the increased proliferative ability of the Gfi1−/− HSC [5,6].
5. Gfi1 controls lymphopoiesis
Gfi1 plays a unique role in T cell activation. In normal activated primary T lymphocytes, Gfi1 expression is induced considerably after that of IL2 [12]; suggesting a role for Gfi1 in transducing IL2-receptor signaling. Intriguingly, putative Gfi1 binding sites are found in the promoters of several genes encoding cytokine or cytokine receptors implicated in T cell activation [37]. The signal transducer and activator of transcription-3 (STAT3) transcription factor conveys signals from the IL-6 receptor to stimulate T cell proliferation upon antigenic stimulation [51,52]. Protein inhibitor of activated STAT3 (PIAS3) directly binds activated STAT3 and blocks STAT3-induced transcriptional activation [53]. Forced expression of Gfi1 augments IL-6-dependent T cell proliferation, perhaps through binding and sequestering PIAS3 [54]. However, no differences in STAT3 dependent electrophoretic mobility shift could be detected between control and Gfi1-overexpressing nuclear extracts [54]. Constitutively activated STAT3 is an oncoprotein [55]. Consequently, a portion of the oncogenic effect of Gfi1 may result from its role as a positive regulator of signal transduction through STAT3 [54].
Independent of IL-6, transgenic overexpression of Gfi1 in T cells increased antigen-induced T cell entry into S phase [22,54]. High level Gfi1 expression in Jurkat T cell lines induced an increase in the proliferation rate and a reduction in levels of p21waf1, p27Kip1 and Rb1 compared to control cell lines. Enforced Gfi1 expression reduced activation-induced apoptosis and phorbol ester-generated G1 arrest in Jurkat T cells [36]. Thus, Gfi1 may control survival and cell cycle checkpoints during T cell proliferation.
In activated naïve T cells, Gfi1 expression is increased by IL-4 in a STAT6-dependent process. IL-4 drives the differentiation and expansion of activated cells to a T-helper type 2 (Th2) phenotype. Retroviral vector mediated expression of Gfi1 in activated CD4+ cells resulted in preferential expansion of Th2 cells through enhancement of proliferation and reduction of apoptosis [56]. Similar to oncogenic studies [13], expression of a P2A SNAG-domain-mutant Gfi1 had no effect [56] indicating that the biological effects of Gfi1 in this context is SNAG domain dependent. Gfi1 is not induced in quiescent CD4+ cells by IL-4, and Gfi1 alone cannot drive Th2 differentiation. Another IL4/STAT6 target, the Gata3 transcription factor, controls Th2 differentiation. Together, GATA3 and Gfi1 dramatically synergize to expand and polarize activated CD4+ cells [56].
Gfi1 is required for early T cell development. Transgenic mice constitutively expressing Gfi1 or Pim1 in T cells reveal the involvement of both proteins in T lymphopoiesis. CD4−CD8− (double negative; DN) thymocytes that productively rearrange the β-chain of the T cell receptor are selected for expansion and differentiation to CD4+CD8+ (double positive; DP) cells. Forced overexpression of Gfi1 interferes with the selection of β-chain-expressing cells, and therefore, blocks pre-T cell development. In contrast, Pim1 promotes β selection. Moreover, forced Pim1 expression can rescue the Gfi1-induced block in Gfi1/Pim1 bitransgenic mice [57]. Notably, both proteins also collaborate in MoMLV transformation of T cells. These data indicate that there may be a desirable stoichiometry between Pim and Gfi1 in order to facilitate normal T cell development.
Like Gfi1 transgenics, Gfi1 deficient mice exhibit diminished thymic cellularity. Gfi1−/− defects result in blocked differentiation and proliferation as well as increased cell death of early thymic DN cells [4]. Within the double negative thymocyte pool, the transition between DN1 to DN2 marks a significant commitment to the T cell lineage. Notably, the level of Gfi1 mRNA increases by a log from DN1 to DN2 [22]. Thus, Gfi1−/− thymocytes may have difficulty commiting to the earliest stages of the T lymphopoiesis program. However, some cells escape this bottleneck and differentiate. In fact, Gfi1−/− DP thymocytes appear skewed towards development of CD8+ cells [4]. An increase in CD8+ cells also points to a critical role for Gfi1 in the CD4/CD8 lineage decision. Indeed, in mice which report the transcriptional activity of the Gfi1 promoter via knockin of the green fluorescent protein (GFP), the GFP signal in DP cells is highest in CD4hiCD8lo cells, which are poised to commit to either the CD4+ or CD8+ lineage [49].
Both naïve CD4+ and CD8+ T cells are dependent on IL-7 survival signals. IL-7 is limiting for T cell homeostasis. It was recently shown that IL-7 and other pro-survival cytokines repress IL7Rα expression in these cells as a possible mechanism to maximize the number of T cells receiving IL-7 stimulation [58]. Gfi1 is involved in transducing this signal in CD8+ T cells. Gfi1−/− CD8+ T cells, but not CD4+ T cells, have increased expression of IL7Rα. Restoration of Gfi1 expression in Gfi1−/− CD8+ T cells reduces IL7Rα expression. The IL7Rα gene has two potential Gfi1 binding sites, and chromatin immunoprecipitation experiments demonstrate Gfi1 binding to the IL7Rα gene in living cells [58]. It is not clear why the IL-7-induced repression of the IL7Rα gene in CD4+ cells does not require Gfi1.
Gfi1B is a homolog of Gfi1 with a 97% identical zinc finger DNA binding domain, although the homology of the remainder of the protein is limited to the N-terminal SNAG repressor domain. Gfi1B functions as a transcriptional repressor specific for a consensus DNA binding site virtually indistinguishable from that of Gfi1 [27]. While Gfi1 is expressed primarily in the bone marrow and thymus, Gfi1B is found predominately in splenocytes and in the bone marrow. One defined target of Gfi1B repression is p21waf1, a cyclin-dependent kinase inhibitor with a Gfi1/1B binding sites in the promoter [27]. Overexpression of Gfi1B inhibits both IL-6-induced G1 arrest and differentiation of the M1 murine myelomonocytic cell line [27]. Both Gfi1 and Gfi1B are expressed in T cell precursors; however, Gfi1B expression is up-regulated concurrent with T cell activation [22]. As stated earlier, enforced Gfi1 expression promotes T cell activation. In contrast, over-expression of Gfi1B reduces T cell activation [22]. Transgenic mice expressing Gfi1B in T cells display an increased number of CD4+ cells and severely reduced number of CD8+ thymocytes. Most probably, a striking reduction of IL7Rα expression in Gfi1B transgenic thymocytes reduces the survival of developing CD8+ thymocytes. Notably, the defect in generating CD8+ thymocytes can be rescued by the expression of Bcl2, a pro-survival IL-7R target [22]. These data compliment the above mentioned data indicating an increase in CD8+ cells in Gfi1−/− mice, which additionally show an increase in IL7Rα expression. Thus, repression of IL7Rα expression in Gfi1b transgenics reduced the generation of CD8+ thymocytes; whereas deletion of Gfi1 increased IL7Rα expression and the generation of CD8+ thymocytes.
Gfi1 is autoregulated in lymphoid, but not myeloid cells. Putative Gfi1/1B consensus binding sites are found in homologous regions of the human, mouse and rat Gfi1 promoters. Chromatin immunoprecipitation (ChIP) assays demonstrate that Gfi1 binds to its own promoter in vivo, and EMSA indicates multiple functional Gfi1 binding sites in the Gfi1 promoter. Ectopic expression of Gfi1 downregulates endogenous Gfi1 in murine thymocytes and transfected Jurkat T cells, but not in the myelo-monocytic U937 cell line [48]. Co-transfection with Gfi1 promoter/reporter constructs and a Gfi1 expression vector indicate that Gfi1 directly binds and represses the Gfi1 promoter. In the Gfi1B transgenic thymocytes described above, Gfi1 mRNA is undetectable. In fact, Gfi1B was also found to repress endogenous Gfi1. Moreover, Gfi1b binds to the same binding sites in the Gfi1 promoter as Gfi1, and represses the Gfi1 promoter. Thus, utilizing the same promoter binding sites, Gfi1 regulates the Gfi1 locus, and Gfi1B also potentially represses Gfi1 expression [48]. In support of this hypothesis, expression of a T cell targeted Gfi1 transgene in a Gfi1/GFP knock-in mouse lead to down-regulation of the Gfi1/GFP allele [49], as did expression of a Gfi1B transgene [47]. Interestingly, the Gfi1B promoter contains two adjacent Gfi1/Gfi1B binding sites that can be bound by both Gfi1 and Gfi1B [47]. In conclusion, both Gfi1 and Gfi1B are autoregulated and can cross regulate the other locus. To add complexity, the autoregulatory mechanisms appear to be cell type specific.
6. Gfi1 is essential for myelopoiesis, neutophil differentiation and function
Blood cells with specialized functions, such as neutrophils, are derived from multipotent hematopoietic stem cells by a process that is tightly controlled by transcription factors [59]. Failure to control hematopoietic stem cell fate decisions leads to neutropenia. Bone marrow myeloid progenitors normally switch between default monocytic differentiation, and granulopoiesis [60]. When monocyte production increases, neutrophil production decreases, and vice versa. Gfi1 deficient mice lack mature neutrophils and are especially susceptible to infections with Gram-positive bacteria. Interestingly, examination of the blood and the bone marrow of Gfi1−/− mice identified the accumulation of an undifferentiated immature cell population that express both granulocyte (Gr1+) and macrophage (Mac1+) markers; an indicator of a developmental block at the promyelocyte stage [2,3]. Both respiratory burst and phagocytic activity were not altered in Gfi1−/− cells; however, like immature promyelocytes, the abnormal Gfi1−/− cells express neutrophil primary granule proteins. A lack of secondary or tertiary granule proteins indicates that they are not mature [2]. Thus, Gfi1−/− mice completely lack neutrophils.
The developmental defects in neutrophil differentiation in Gfi1−/− mice are cell autonomous. Neither in vivo granulocyte colony-stimulating factor (G-CSF) administration nor ex vivo cytokine treatment in colony formation assays was sufficient for differentiation and the production of Gfi1−/− granulocytes [2,3]. G-CSF administration increased the accumulation of the abnormal Gr1+Mac1+ promyelocytes [2]. However, Gfi1−/− sorted granulocyte–monocyte progenitor (GMP) cells that were transduced with a Gfi1-expressing retrovirus vector could differentiate to neutrophils in response to G-CSF stimulation [2]. The rescue of the defect by reintroduction of Gfi1 in GMP also suggests that the developmental block to granulopoiesis occurs at or after this point.
Gfi1 is an essential transcription factor in the network of factors regulating granulopoiesis. Gfi1 has been shown to bind the promoters of several genes including CCAAT/enhancer-binding protein α (C/EBPα), C/EBPε, and PU.1 [46]. Both C/EBPα and C/EBPε are required for myelopoiesis, as deletion of either gene results in the absence of neutrophils and eosinophils [61,62]. Germline PU.1 deletion results in multiple defects including failure to produce granulocyte progenitors [63,64]. Northern analysis of RNA from Gfi1−/− whole bone marrow revealed reduced C/EBPε and increased PU.1 and C/EBPα mRNA steady state levels in total bone marrow [2]. Whether these changes reflect a direct effect of Gfi1 loss or an accumulation of abnormal promyelocytes remains to be determined. Interestingly, treatment of CD34+ cells with the thalidomide derivative CC-4047 inhibits erythroid colony formation and enhances the frequency of myeloid colonies. Microarray analysis of CC-4047 treated CD34+ cells showed induction of Gfi1 along with the transcription factors C/EBPα, C/EBPδ, C/EBPε and PU.1 [65]. While Gfi1 may not be critically required for the expression of C/EBPα, C/EBPδ, C/EBPε and PU.1, their co-regulation and possible modulation by Gfi1 suggest that Gfi1 may work with these factors to regulate granulopoiesis.
Gfi1 may regulate genes that are important for neutrophil function. The promoters of several genes encoding proteinases expressed in the myelomonocytic lineages such as azurocidin, and α1-antitrypsin were shown by chromatin immunoprecipitation to be bound by Gfi1 [46]. Moreover, Gfi1 binds to the promoter of IL8 and its receptor [46]. Both IL8 and its receptor are important for neutrophil activation and migration from peripheral blood into tissues [66]. Ultimately, characterization of these putative Gfi1 target genes will define the significance of Gfi1 regulation in the context of defined function.
Gfi1 is an essential regulator of human neutrophil development and function. A major proteinase of primary neutrophil granules is the neutrophil elastase encoded by Ela2. Mutations in Ela2 are a major cause of human severe congenital neutropenia [67,68]. Screening of SCN patients without Ela2 mutations identified mutations in Gfi1 that were associated with neutropenia. The Gfi1 mutant SCN patients were similar to Gfi1−/− mice in terms of their responsiveness to G-CSF, monocytosis, lymphopenia and the presence of an abnormal population of immature myeloid cells with both neutrophil and monocytic features. Importantly, the patients only had one mutant Gfi1 allele, suggesting that the mutant protein functions as a dominant negative. In contrast, murine Gfi1+/− heterozygotes appear to be normal [2,3]. Moreover, the heterozygous mutations in humans do not seem to induce defects associated with the inner ear, growth retardation and runting that are observed in Gfi1−/− mice [2,3,10]. It is possible that tissue specific autoregulation of Gfi1 may result in compensation by the wild type allele in non-myeloid tissues [48].
Gfi1 mutations in the fifth and sixth zinc finger have been identified in some SCN and non-immune chronic idiopathic neutropenic (NI-CINA) patients. The mutation in zinc finger five is a substitution of serine for the asparagine at position 382 (N382S) [10]. This mutation is critical because zinc finger five is involved in sequence-specific DNA binding [37]. The Asparagine at this position is hypothesized to contact an adenine in the “AATC” core of the consensus Gfi1 DNA-binding site [37]. The N382S mutation apparently abolishes proper DNA binding to this residue and thus subsequently disrupts Gfi1 transcriptional repression. The mutation in zinc finger six is a substitution of arginine for the lysine at position 403 (K403R). The disease associated with the K403R mutation is less severe than the disease associated with N382S zinc finger five mutation in SCN patients. Zinc finger six is dispensable for DNA binding [37]; however, K403R mutants were impaired in transcriptional repression assays with synthetic reporter constructs [10]. The decreased transcriptional repression and the phenotype associated with the mutation in zinc finger six suggests a role for this zinc finger in either the DNA binding or protein–protein interactions important for the transcriptional activity of Gfi1. It is also possible that K403 is important as a site of modification such as sumoylation or ubiquitination, and the K403R mutation prevents such modification. This change could affect the overall stability of Gfi1, or affect its interactions with the DNA or other proteins [10].
The defects in Gfi1 repression associated with the SCN mutations may help elucidate the mechanisms underlining the pathology of human neutropenic diseases. As mentioned earlier, in human myelomonocytic cell lines Ela2 is bound by Gfi1 [46]. Therefore, it is possible that a lack of efficient Gfi1 repression of the Ela2 gene results in overexpression of Ela2 in human SCN. Overproduction of neutrophil elastase could lead to mistargeting to cellular compartments other than the neutrophil granules, subsequently disrupting or altering its normal function [10]. Characterizing Gfi1 mutations and defining both their function and gene regulation will provide a valuable insight to the mechanisms and the pathways involved in neutrophil function and development.
Gfi1 is dispensable for macrophage development but required for activated macrophage function. Gfi1−/− mice accumulate abnormal promyelocytes with monocytic characteristics. Hock et al. [2] proposed that Gfi1 function is required to antagonize monocyte/macrophage differentiation during myelopoiesis. Therefore, Gfi1 most probably functions early in progenitor cell differentiation. Indeed, Gfi1−/− progenitors favor macrophage production upon cytokine stimulation, again suggesting a role for Gfi1 in antagonizing macrophage differentiation [2,3]. Likewise, Gfi1 expression is absent in mature macrophages. However, Gfi1 expression in macrophages is upregulated upon endotoxin-mediated activation, suggesting a requirement for macrophage function. Lipopolysaccharide (LPS) stimulation of Gfi1−/− macrophages resulted in increased levels of pro-inflammatory cytokine production (TNFα, IL-1β and IL10) when compared to wild type activated macrophages. These data suggest that Gfi1 modulates macrophage function through the regulation of cytokine production. Activated Gfi1−/− macrophages also show increased sensitivity to lower doses of LPS compared to wild type controls, implying an inherent defect in Gfi1−/− macrophages. Therefore, the increased sensitivity and cytokine production could be responsible for exaggerated inflammatory responses observed in the Gfi1−/− mice followed by a toxic shock [3].
Gfi1 is essential for dendritic cell development and function. Lack of Gfi1 expression appears to have profound effects on dendritic cell (DC) development and function. Gfi1−/− mice have reduced numbers of myeloid and lymphoid DC and increased epidermal Langerhans cells. The DC in Gfi1−/− mice showed defective maturation and, similar to activated Gfi1−/− macrophages [3], increased cytokine production upon stimulation [69]. Progenitor cells from Gfi1−/− mice were unable to differentiate into DC and instead differentiated into macrophages, supporting the hypothesis that Gfi1 antagonizes macrophage specification. The Gfi1−/− block to DC development is associated with decreased STAT3 activation. Rathinam et al. [69] suggested that Gfi1 control over STAT3 signaling through interaction with PIAS3 and regulation of SOCS genes might be essential for DC development. STAT3 has been described as an important mediator of DC differentiation [70]. The complex hierarchial molecular network controlling DC development, maturation and function is relatively unknown. Thus, the requirement for Gfi1 in the generation of DC should provide invaluable insights.
7. The role of Gfi1 in the peripheral nervous system (PNS)
Gfi1 is orthologous to Drosophila senseless and C. elegans Pag3. Both Senseless and Pag3 are essential for nervous system development and specification of sensory organs [1,71–74]. Drosophila peripheral nervous system development has been studied extensively and its conserved pathways may help to explain mammalian development. PNS development in Drosophila is regulated by proneural basic helix-loop-helix (bHLH) transcription factors atonal, scute, amos and achaete [1] (Fig. 2). These proneural bHLH transcription factors dimerize with daughterless proteins to bind E-box consensus DNA sequences and transactivate target genes. Proneural bHLH proteins act in direct antagonism to the Notch pathway. Conversely, activation of Notch signaling induces enhancer-of-split; a bHLH transcriptional repressor which antagonizes proneural bHLH factor expression and function. Proper function of proneural genes is required for the expression of senseless, which encodes a zinc-finger transcription factor (Senseless) [1]. PNS development requires senseless. Embryos lacking senseless specify PNS cells, but most of these cells die through apoptosis. In contrast, forced expression of Senseless is sufficient to mediate PNS development [1]. Thus, Senseless enforces the activity of the achaete and scute proneural bHLH transcription factors and antagonizes the activity of the Notch signaling pathway. Orthologous proteins for each member of this pathway can be found in mammalian neuronal and neuroendocrine systems.
Fig. 2.

Senseless and Gfi1 interaction with proneural basic-helix-loop-helix (bHLH) proteins in fly and human neurogenesis. (1) bHLH transcription factors of the achaete-scute complex (AS-C) regulate the development of the fly peripheral nervous system but are antagonized by Notch signaling. (2) AS-C encoded proteins are tissue specific, but bind the ubiquitous daughterless encoded proteins. These proneural bHLH dimers bind DNA sequences known as an E box (CANNTG) to transactivate the expression of senseless. (3) AS-C factors require Senseless for PNS development, because Senseless is a positive regulator of AS-C/Da function, probably through direct repression of a repressor of AS-C activity. (4) Senseless synergizes with the proneural bHLH factors to transactivate genes (including senseless), resulting in a positive-feed-back loop. Once critical levels of functional proneural bHLH dimers and Senseless are reached, Notch signals are antagonized within the cell. (5) Subsequent to inhibition of Notch signaling, neural cell fate is fixed. A similar sequence of events is proposed in human neurogenesis with orthologous proteins.
Gfi1−/− mice are deaf. Gfi1−/− mice show behavioral defects consistent with hearing loss and inner ear defects due to morphological and organizational defects of the inner hair cells and an eventual reduction in the numbers of cochlear neurons [7]. Examination of Gfi1 expression in late stage embryos revealed specific expression of Gfi1 mRNA and protein in the inner ear hair cells [7]. Cochlear neurons have Gfi1 mRNA but not detectable Gfi1 protein [7]. Initially, Gfi1−/− hair cells are specified but morphologically appear abnormal. In addition to their morphological defects, Gfi1−/− hair cells are disorganized, with aberrant cellular patterns both in the vestibule and cochlea as well as improper innervation [7]. The organizational and morphological defects, along with altered expression of neuronal markers in Gfi1−/− inner hair cells, represent a developmental block and emphasize a requirement for Gfi1. Furthermore, Gfi1−/− hair cells, at least in the cochlea, undergo rapid degeneration and are completely lost prior to or soon after birth [7]. This is very similar to Senseless−/− fruit fly PNS cells, which die of apoptosis shortly after they are specified [1]. While Gfi1−/− cochlear hair cells apoptose, the vestibular hair cells stay unorganized but do not disappear, suggesting a different site-specific requirement for Gfi1 function [7]. Following cochlear hair cell loss, Gfi1−/− cochlear neurons undergo a dramatic reduction in numbers by 5 months of age. It is likely that cochlear neuronal degeneration is a secondary effect to hair cell loss, as trophic support from hair cells is required for maintaining the neurons [7].
Inner ear hair cell specification requires bHLH and Gfi1 coexpression. The expression pattern of the Gfi1 protein was similar to Math1, the mammalian orthologue of the Drosophila bHLH protein atonal. Both Gfi1 and Math1 are coexpressed in inner ear hair cells. Math1 is necessary and sufficient for hair cell development and specification in vertebrates [75,76]. Based on the Drosophila model discussed above (Fig. 2) and the interplay between Senseless and atonal proteins, Math1 was suggested to be necessary for Gfi1 expression and vice versa. However, examination of Gfi1−/− mice indicates no obvious effects on Math1 expression. Therefore, in contrast to the observation in the fruit fly, Math1 expression is independent of Gfi1. Moreover, Math1 mutant mice do not exhibit altered Gfi1 mRNA expression, but there was a drastic reduction or even complete absence of Gfi1 protein expression in the otic epithelia. Thus, either Math1 is required for Gfi1 regulation on the translational level or the cells which express Gfi1 are absent [7]. Since Gfi1 mRNA is present in Math1 null hair cells, an upstream regulator of Gfi1 expression other than Math1 is suggested. In fact, the class IV Pou domain transcription factor, Pou4f3, an essential factor in the development of all hair cells in human and mouse, controls Gfi1 expression in hair cells [77–79].
The interplay between bHLH factors, class IV Pou domain containing factors and Gfi1 exists not only in inner ear hair cells but also in the mammalian retina [7,8,79]. Math5 is another mammalian orthologue of the Drosophila atonal. Math5 is essential, but not sufficient, for the differentiation of most retinal ganglion cells [8]. The fruit fly atonal bHLH protein is involved in the development of the R8 cells of the retina and acts in concert with Senseless in a positive feedback loop [72]. However, in the mammalian system Gfi1 appears to be downstream of Math5, as Gfi1 expression is detected after Math5 expression is turned off. Brn3b, also known as Pou4f2, is another member of the Pou domain family. Gfi1 expression is absent in the Brn3b−/− retina, indicating that Gfi1 is downstream of Brn3b. It is possible that Pou domain factors regulate Gfi1 expression in presumptive neurons to potentiate the action of bHLH factors [8].
The Gfi1 protein is expressed in specialized sensory cells of the PNS such as the hair cells of the inner ear, the eye, the presumptive Merkel cells, and rare pulmonary neuroendocrine cells [7,9,79]. Pulmonary neuroendocrine cells (PNEC) are a minor cell population of the airway epithelium that are present as single cells or in clusters referred to as neuroepithelial bodies (NEB). PNEC secrete amines and peptides that act to regulate vascular and bronchial tone [80]. NEB are highly innervated and are believed to act as intrapulmonary chemoreceptors. NEB are sensitive to hypoxia and perform several suggested “corpuscular” functions such as nociceptive signal transmission and mechanosensation [81–85]. Gfi1 expression in the PNEC persists both during embryonic development and in adult mice [9]. Ash1 (also referred to as hAsh1 or ASCL1, mouse homolog = Mash1) is the mammalian orthologue of Drosophila achaete and scute encoded proneural bHLH proteins. Ash1 is critical for murine PNEC development [86]. Analysis of bHLH Ash1 expression in Gfi1−/− mice showed no change in the number or the level of Ash1 expressing PNEC. Therefore, similar to the relationship of Gfi1 and bHLH in the hair cells of the inner ear and the retinal ganglion neurons, and unlike the Drosophila model, Gfi1 is not required for Ash1 expression in PNEC [1,7–9,72,79]. However, Gfi1−/− pulmonary Ash1-expressing cells showed a marked reduction in the expression of the PNEC neuroendocrine markers calcitonin gene related peptide (CGRP) and synaptophysin. These data indicate that, similar to the hair cells in the inner ear, PNEC were specified by Ash1 in the absence of Gfi1, but differentiation and proper maturation were impaired [9].
8. Gfi1 is a small cell lung carcinoma (SCLC) oncoprotein
Lung tumors with a neuroendocrine phenotype include SCLCs, large cell neuroendocrine carcinomas (LCNE), and classical and atypical carcinoid tumors [87]. SCLC are aggressive lung tumors which are strongly associated with smoking, and for which there is no cure [87,88]. SCLC account for 15–20% of all human lung cancers [88]. The neuroendocrine phenotype of SCLC involves the production of neuropeptides which stimulate transformation and tumor growth through autocrine signaling [89]. Although direct mutations in neuroendocrine pathway genes have not been discovered, the neuroendocrine phenotype in SCLC is well accepted as the driving force behind SCLC paracrine and autocrine stimulation [86,90]. While it is clear that NE growth factors secreted by SCLC participate in tumorigenesis and tumor maintenance, the genetic and biochemical pathways underlying NE differentiation are not well understood. Subsequently, attempts at clinical intervention rescue few SCLC patients beyond 5 years [87].
Similar to PNEC, pulmonary neuroendocrine tumors such as SCLC express Ash1 bHLH proteins [91,92]. Human SCLC secrete neuropeptides that stimulate proliferation and transformation in autocrine and paracrine stimulatory loops [89]. The neuroendocrine phenotype is essential in maintaining these loops [86,90]. Gfi1 is expressed in murine PNEC and, like Ash1 and neuroendocrine markers, is expressed in cell lines and primary human biopsies of lung neuroendocrine tumors [9]. Gfi1 coexpression with Ash1 and neuroendocrine markers that are downstream targets of Ash1 in PNEC, as well as in human primary biopsy specimens, strongly suggest its involvement in the maintenance of the neuoendocrine phenotype of neuroendocrine lung tumors [9]. Indeed, forced Gfi1 expression in SCLC cell lines potentiated tumor formation in nude mice xenografts, suggesting that Gfi1 acts as an oncogene in SCLC [9].
It should be noted that Drosophila developmental cascades have illustrated “gatekeeper” pathways which form the molecular basis of our understanding of colorectal carcinoma (Wnt/beta catenin) and basal cell carcinoma (Hedgehog/Gli) [93]. These pathways are conserved between humans and Drosophila, and function in pattern formation in the Drosophila embryo. Moreover, they have both positive (oncogenes) and negative acting (tumor suppressor) components that regulate each other and terminate in the regulation of oncogenic transcription factors [93]. The illustrated conservation between Drosophila sensory organ precursor development and SCLC oncogenesis contains many of these features.
The mechanism through which Gfi1 enhances SCLC xenograft growth is unknown. It is possible that the sole oncogenic action of Gfi1 in SCLC is to maintain the Ash1/neuroendocrine phenotype, and that in non-neuroendocrine cells this activity is tumor suppressive. In the Drosophila model of sensory organ precursor development (Fig. 2), Notch signaling prevents cells from adopting a neuronal fate; whereas bHLH activity with Senseless antagonizes Notch signaling to induce sensory organ precursors. The orthologous proteins are active in human SCLC, which rely on Ash1 and neuroendocrine factors for survival and cell cycle progression. The non-neuroendocrine cells of the lung form Non-SCLC tumors. NSCLC express the Notch pathway target gene HES1 [86] and may thus be considered similar to the Drosophila cells that fail to become the sensory organ precursors. In fact, forced expression of an activated Notch protein in human neuroendocrine SCLC cell lines causes a G1 block in cell cycle progression and correlates to destabilization of Ash1 [94,95]. In a similar manner, forced expression of Gfi1 in non-neuroendocrine NSCLC cell lines results in G1 cell cycle arrest. Moreover, while SCLC tumors show the highest level of Gfi1 expression and have a poor prognosis, the few human biopsies from patients with NSLC that express Gfi1 show a trend towards better prognosis and longer survival (Gilks and Grimes, unpublished data). These data suggest that Notch and Ash1/Gfi1 play opposing roles in neuroendocrine versus non-neuroendocrine lung tumors. Each may be oncogenic or suppressive depending on the commitment of the cell to the neuroendocrine phenotype. Given the reduced cell cycle progression of Gfi1−/− thymocytes but increased cell cycle progression of Gfi1−/− bone marrow stem/progenitor cells, it is enticing to speculate that another Notch versus Gfi1 axis is at work in blood cells. Notably, Notch can regulate commitment to the T cell lineage, and stem cell self renewal [96]. However, it is likely that non-neuroendocrine pathway targets of Gfi1 are involved.
Repression of non-neuroendocrine pathway targets of Gfi1 in different tissues might also yield opposing biological outcomes. Non-neuroendocrine pathway targets might also be functional in the oncogenic action of Gfi1 in SCLC lines. 25-Hydroxyvitamin D 1-α hydrolase (CYP27B1) is one non-neuroendocrine target of Gfi1 in epithelial tumors [97]. In prostate cancer, 1,25-dihydroxyvitamin D (1,25D) is hypothesized to have potential cancer preventative capacity due to its anti-proliferative and pro-differentiation actions [98–100]. CYP27B1 regulates local 1,25D levels, and CYP27B1 is substantially repressed in prostate cancers [101]. Interestingly, Gfi1 is induced in prostate cancer (Gilks and Grimes, unpublished data) and Gfi1 binds to and represses the CYP27B1 promoter [97]. Gfi1 may therefore, be involved in epithelial tumor progression by focally repressing 1,25D levels. Tissues other than the prostate may be more or less sensitive to 1,25D antiproliferative action. Thus, like the neuroendocrine phenotype, the biological effect of CYP27B1 downregulation may be dependent on the target tissue.
Finally, it appears that Gfi1 can act as an oncogene in T-lymphocytes, SCLC and prostate cancer but is anti-proliferative and perhaps a tumor suppressor in hematopoietic stem cells, myeloid progenitors and NSCLC (Fig. 3). These observations advocate for common mechanistic pathways through which Gfi1 functions to exert it oncogenic or anti-proliferative activities, and suggest that examination of genes regulated by Gfi1 in each system should uncover common targets.
Fig. 3.

Gfi1 may function as either an oncogene or a tumor suppressor. Biological processes corresponding to either function are listed and discussed further in the text.
Biography
H. Leighton Grimes received his doctorate in molecular pathology and immunology from the University of Florida School of Medicine. He received postdoctoral training at the Fox Chase Cancer Center, where he was part of the team that first identified and characterized the Gfi1 and Gfi1b oncoproteins. He is currently a tenured associate professor in pediatrics at the Cincinnati Children’s Hospital Medical Center, and his scientific work focuses on the role of Gfi1 in hematopoiesis, leukemogenesis and lung tumorigenesis.
Footnotes
This work was partially supported by NIH grants CA112405, CA105152 and HL079574, and by a Leukemia and Lymphoma Society Scholar Award.
References
- [1].Nolo R, Abbott LA, Bellen HJ. Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell. 2000;102:349–62. doi: 10.1016/s0092-8674(00)00040-4. [DOI] [PubMed] [Google Scholar]
- [2].Hock H, Hamblen MJ, Rooke HM, et al. Intrinsic requirement for zinc finger transcription factor gfi-1 in neutrophil differentiation. Immunity. 2003;18:109–20. doi: 10.1016/s1074-7613(02)00501-0. [DOI] [PubMed] [Google Scholar]
- [3].Karsunky H, Zeng H, Schmidt T, et al. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet. 2002;30:295–300. doi: 10.1038/ng831. [DOI] [PubMed] [Google Scholar]
- [4].Yucel R, Karsunky H, Klein-Hitpass L, Moroy T. The transcriptional repressor Gfi1 affects development of early, uncommitted c-Kit+ T cell progenitors and CD4/CD8 lineage decision in the thymus. J Exp Med. 2003;197:831–44. doi: 10.1084/jem.20021417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hock H, Hamblen MJ, Rooke HM, et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 2004;431:1002–7. doi: 10.1038/nature02994. [DOI] [PubMed] [Google Scholar]
- [6].Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 2004;23:4116–25. doi: 10.1038/sj.emboj.7600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wallis D, Hamblen M, Zhou Y, et al. The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development. 2003;130:221–32. doi: 10.1242/dev.00190. [DOI] [PubMed] [Google Scholar]
- [8].Yang Z, Ding K, Pan L, Deng M, Gan L. Math5 determines the competence state of retinal ganglion cell progenitors. Dev Biol. 2003;264:240–54. doi: 10.1016/j.ydbio.2003.08.005. [DOI] [PubMed] [Google Scholar]
- [9].Kazanjian A, Wallis D, Au N, et al. Growth factor independence-1 is expressed in primary human neuroendocrine lung carcinomas and mediates the differentiation of murine pulmonary neuroendocrine cells. Cancer Res. 2004;64:6874–82. doi: 10.1158/0008-5472.CAN-04-0633. [DOI] [PubMed] [Google Scholar]
- [10].Person RE, Li FQ, Duan Z, et al. Gfi1 proto-oncogene mutation causes human neutropenia and targets neutrophil elastase. Nat Genet. 2003;34:308–12. doi: 10.1038/ng1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lazo PA, Klein-Szanto AJ, Tsichlis PN. T-cell lymphoma lines derived from rat thymomas induced by Moloney murine leukemia virus: phenotypic diversity and its implications. J Virol. 1990;64:3948–59. doi: 10.1128/jvi.64.8.3948-3959.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gilks CB, Bear SE, Grimes HL, Tsichlis PN. Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol Cell Biol. 1993;13:1759–68. doi: 10.1128/mcb.13.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grimes HL, Chan TO, Zweidler-McKay PA, Tong B, Tsichlis PN. The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol. 1996;16:6263–72. doi: 10.1128/mcb.16.11.6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zornig M, Schmidt T, Karsunky H, Grzeschiczek A, Moroy T. Zinc finger protein GFI-1 cooperates with myc and pim-1 in T-cell lymphomagenesis by reducing the requirements for IL-2. Oncogene. 1996;12:1789–801. [PubMed] [Google Scholar]
- [15].Scheijen B, Jonkers J, Acton D, Berns A. Characterization of pal-1, a common proviral insertion site in murine leukemia virus-induced lymphomas of c-myc and Pim-1 transgenic mice. J Virol. 1997;71:9–16. doi: 10.1128/jvi.71.1.9-16.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schmidt T, Zornig M, Beneke R, Moroy T. MoMuLV proviral integrations identified by Sup-F selection in tumors from infected myc/pim bitransgenic mice correlate with activation of the gfi-1 gene. Nucleic Acids Res. 1996;24:2528–34. doi: 10.1093/nar/24.13.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liao X, Tang Y, Chattopadhyay SK, Hartley JW, Morse HC. Upregulation of Gfi-1, a gene involved in IL-2-independent growth of T cells, in a murine retrovirus-induced immunodeficiency syndrome. In Vivo. 1997;11:9–12. [PubMed] [Google Scholar]
- [18].Berns A, van der Lugt N, Alkema M, et al. Mouse model systems to study multistep tumorigenesis. Cold Spring Harb Symp Quant Biol. 1994;59:435–47. doi: 10.1101/sqb.1994.059.01.049. [DOI] [PubMed] [Google Scholar]
- [19].Liao X, Buchberg AM, Jenkins NA, Copeland NG. Evi-5, a common site of retroviral integration in AKXD T-cell lymphomas, maps near Gfi-1 on mouse chromosome 5. J Virol. 1995;69:7132–7. doi: 10.1128/jvi.69.11.7132-7137.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32:D523–7. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shin MS, Fredrickson TN, Hartley JW, Suzuki T, Agaki K, Morse HC., III High-throughput retroviral tagging for identification of genes involved in initiation and progression of mouse splenic marginal zone lymphomas. Cancer Res. 2004;64:4419–27. doi: 10.1158/0008-5472.CAN-03-3885. [DOI] [PubMed] [Google Scholar]
- [22].Doan LL, Kitay MK, Yu Q, et al. Growth factor independence-1B expression leads to defects in T cell activation, IL-7 receptor alpha expression, and T cell lineage commitment. J Immunol. 2003;170:2356–66. doi: 10.4049/jimmunol.170.5.2356. [DOI] [PubMed] [Google Scholar]
- [23].Schmidt T, Karsunky H, Gau E, Zevnik B, Elsasser HP, Moroy T. Zinc finger protein GFI-1 has low oncogenic potential but cooperates strongly with pim and myc genes in T-cell lymphomagenesis. Oncogene. 1998;17:2661–7. doi: 10.1038/sj.onc.1202191. [DOI] [PubMed] [Google Scholar]
- [24].Bell DW, Taguchi T, Jenkins NA, et al. Chromosomal localization of a gene, GF1, encoding a novel zinc finger protein reveals a new syntenic region between man and rodents. Cytogenet Cell Genet. 1995;70:263–7. doi: 10.1159/000134048. [DOI] [PubMed] [Google Scholar]
- [25].Roberts T, Cowell JK. Cloning of the human Gfi-1 gene and its mapping to chromosome region 1p22. Oncogene. 1997;14:1003–5. doi: 10.1038/sj.onc.1200910. [DOI] [PubMed] [Google Scholar]
- [26].Berns A, Mikkers H, Krimpenfort P, Allen J, Scheijen B, Jonkers J. Identification and characterization of collaborating oncogenes in compound mutant mice. Cancer Res. 1999;59:1773s–7s. [PubMed] [Google Scholar]
- [27].Tong B, Grimes HL, Yang TY, et al. The Gfi-1B proto-oncoprotein represses p21WAF1 and inhibits myeloid cell differentiation. Mol Cell Biol. 1998;18:2462–73. doi: 10.1128/mcb.18.5.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Alkema MJ, Jacobs H, van LM, Berns A. Pertubation of B and T cell development and predisposition to lymphomagenesis in Emu Bmi1 transgenic mice require the Bmi1 RING finger. Oncogene. 1997;15:899–910. doi: 10.1038/sj.onc.1201262. [DOI] [PubMed] [Google Scholar]
- [29].van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–52. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- [30].Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753–63. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- [31].Alkema MJ, van der Lugt NM, Bobeldijk RC, Berns A, van LM. Transformation of axial skeleton due to overexpression of bmi-1 in transgenic mice. Nature. 1995;374:724–7. doi: 10.1038/374724a0. [DOI] [PubMed] [Google Scholar]
- [32].Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- [33].Jacobs JJ, Scheijen B, Voncken JW, Kieboom K, Berns A, van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc- induced apoptosis via INK4a/ARF. Genes Dev. 1999;13:2678–90. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Juin P, Hueber AO, Littlewood T, Evan G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999;13:1367–81. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Grimes HL, Gilks CB, Chan TO, Porter S, Tsichlis PN. The Gfi-1 protooncoprotein represses Bax expression and inhibits T-cell death. Proc Natl Acad Sci USA. 1996;93:14569–73. doi: 10.1073/pnas.93.25.14569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Karsunky H, Mende I, Schmidt T, Moroy T. High levels of the onco-protein Gfi-1 accelerate T-cell proliferation and inhibit activation induced T-cell death in Jurkat T-cells. Oncogene. 2002;21:1571–9. doi: 10.1038/sj.onc.1205216. [DOI] [PubMed] [Google Scholar]
- [37].Zweidler-McKay PA, Grimes HL, Flubacher MM, Tsichlis PN. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol. 1996;16:4024–34. doi: 10.1128/mcb.16.8.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Batlle E, Sancho E, Franci C, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- [39].Nakakura EK, Watkins DN, Schuebel KE, et al. Mammalian Scratch: a neural-specific Snail family transcriptional repressor. Proc Natl Acad Sci USA. 2001;98:4010–5. doi: 10.1073/pnas.051014098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Breslin MB, Zhu M, Notkins AL, Lan MS. Neuroendocrine differentiation factor, IA-1, is a transcriptional repressor and contains a specific DNA-binding domain: identification of consensus IA-1 binding sequence. Nucleic Acids Res. 2002;30:1038–45. doi: 10.1093/nar/30.4.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ayyanathan K, Fredericks WJ, Berking C, et al. Hormone-dependent tumor regression in vivo by an inducible transcriptional repressor directed at the PAX3-FKHR oncogene. Cancer Res. 2000;60:5803–14. [PubMed] [Google Scholar]
- [42].McGhee L, Bryan J, Elliot L, et al. Gfi1 attaches to the nuclear matrix, associates with ETO (MTG8) and histone deacetylase proteins and represses transcription using a TSA-sensitive mcehanism. J Cell Biochem. 2003;89:1005–18. doi: 10.1002/jcb.10548. [DOI] [PubMed] [Google Scholar]
- [43].Amann JM, Nip J, Strom DK, et al. ETO, a target of t(8; 21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21:6470–83. doi: 10.1128/MCB.21.19.6470-6483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Venezia TA, Merchant AA, Ramos CA, et al. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2:e301. doi: 10.1371/journal.pbio.0020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Saleque S, Cameron S, Orkin SH. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 2002;16:301–6. doi: 10.1101/gad.959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Duan Z, Horwitz M. Targets of the transcriptional repressor onco-protein Gfi-1. Proc Natl Acad Sci USA. 2003;100:5932–7. doi: 10.1073/pnas.1031694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vassen L, Fiolka K, Mahlmann S, Moroy T. Direct transcriptional repression of the genes encoding the zinc-finger proteins Gfi1b and Gfi1 by Gfi1b. Nucleic Acids Res. 2005;33:987–98. doi: 10.1093/nar/gki243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Doan LL, Porter SD, Duan Z, et al. Targeted transcriptional repression of Gfi1 by GFI1 and GFI1B in lymphoid cells. Nucleic Acids Res. 2004;32:2508–19. doi: 10.1093/nar/gkh570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yucel R, Kosan C, Heyd F, Moroy T. Gfi1:GFP knock-in mutant reveals differential expression and auto-regulation of the gene growth factor independence 1 (Gfi1) during lymphocyte development. J Biol Chem. 2004;279:40906–17. doi: 10.1074/jbc.M400808200. [DOI] [PubMed] [Google Scholar]
- [50].Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–8. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- [51].Akaishi H, Takeda K, Kaisho T, et al. Defective IL-2-mediated IL-2 receptor alpha chain expression in Stat3-deficient T lymphocytes. Int Immunol. 1998;10:1747–51. doi: 10.1093/intimm/10.11.1747. [DOI] [PubMed] [Google Scholar]
- [52].Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–60. [PubMed] [Google Scholar]
- [53].Chung CD, Liao J, Liu B, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- [54].Rodel B, Tavassoli K, Karsunky H, et al. The zinc finger protein Gfi-1 can enhance STAT3 signaling by interacting with the STAT3 inhibitor PIAS3. EMBO J. 2000;19:5845–55. doi: 10.1093/emboj/19.21.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- [56].Zhu J, Guo L, Min B, et al. Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity. 2002;16:733–44. doi: 10.1016/s1074-7613(02)00317-5. [DOI] [PubMed] [Google Scholar]
- [57].Schmidt T, Karsunky H, Rodel B, Zevnik B, Elsasser HP, Moroy T. Evidence implicating Gfi-1 and Pim-1 in pre-T-cell differentiation steps associated with beta-selection. EMBO J. 1998;17:5349–59. doi: 10.1093/emboj/17.18.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Park JH, Yu Q, Erman B, et al. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 2004;21:289–302. doi: 10.1016/j.immuni.2004.07.016. [DOI] [PubMed] [Google Scholar]
- [59].Friedman AD. Transcriptional regulation of granulocyte and monocyte development. Oncogene. 2002;21:3377–90. doi: 10.1038/sj.onc.1205324. [DOI] [PubMed] [Google Scholar]
- [60].Horwitz M, Benson KF, Duan Z, et al. Role of neutrophil elastase in bone marrow failure syndromes: molecular genetic revival of the chalone hypothesis. Curr Opin Hematol. 2003;10:49–54. doi: 10.1097/00062752-200301000-00008. [DOI] [PubMed] [Google Scholar]
- [61].Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–74. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yamanaka R, Barlow C, Lekstrom-Himes J, et al. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc Natl Acad Sci USA. 1997;94:13187–92. doi: 10.1073/pnas.94.24.13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor Pu.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–7. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- [64].McKercher SR, Torbett BE, Anderson KL, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–58. [PMC free article] [PubMed] [Google Scholar]
- [65].Koh KR, Janz M, Mapara MY, et al. Immunomodulatory derivative of thalidomide (IMiD CC-4047) induces a shift in lineage commitment by suppressing erythropoiesis and promoting myelopoiesis. Blood. 2004;105:3833–40. doi: 10.1182/blood-2004-03-0828. [DOI] [PubMed] [Google Scholar]
- [66].Modi WS, Dean M, Seuanez HN, Mukaida N, Matsushima K, O’Brien SJ. Monocyte-derived neutrophil chemotactic factor (MDNCF/IL-8) resides in a gene cluster along with several other members of the platelet factor 4 gene superfamily. Hum Genet. 1990;84:185–7. doi: 10.1007/BF00208938. [DOI] [PubMed] [Google Scholar]
- [67].Dale DC, Person RE, Bolyard AA, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317–22. [PubMed] [Google Scholar]
- [68].Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet. 1999;23:433–6. doi: 10.1038/70544. [DOI] [PubMed] [Google Scholar]
- [69].Rathinam C, Geffers R, Yucel R, et al. The transcriptional repressor Gfi1 controls STAT3-dependent dendritic cell development and function. Immunity. 2005;22:717–28. doi: 10.1016/j.immuni.2005.04.007. [DOI] [PubMed] [Google Scholar]
- [70].Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19:903–12. doi: 10.1016/s1074-7613(03)00332-7. [DOI] [PubMed] [Google Scholar]
- [71].Cameron S, Clark SG, McDermott JB, Aamodt E, Horvitz HR. PAG-3, a Zn-finger transcription factor, determines neuroblast fate in C. elegans. Development. 2002;129:1763–74. doi: 10.1242/dev.129.7.1763. [DOI] [PubMed] [Google Scholar]
- [72].Frankfort BJ, Nolo R, Zhang Z, Bellen H, Mardon G. Senseless repression of rough is required for R8 photoreceptor differentiation in the developing Drosophila eye. Neuron. 2001;32:403–14. doi: 10.1016/s0896-6273(01)00480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jafar-Nejad H, Acar M, Nolo R, et al. Senseless acts as a binary switch during sensory organ precursor selection. Genes Dev. 2003;17:2966–78. doi: 10.1101/gad.1122403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jia Y, Xie G, Aamodt E. pag-3, a Caenorhabditis elegans gene involved in touch neuron gene expression and coordinated movement. Genetics. 1996;142:141–7. doi: 10.1093/genetics/142.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bermingham NA, Hassan BA, Price SD, et al. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–41. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- [76].Chen ZY, Corey DP. An inner ear gene expression database. J Assoc Res Otolaryngol. 2002;3:140–8. doi: 10.1007/s101620020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Erkman L, McEvilly RJ, Luo L, et al. Role of transcription factors Brn-3.1 and Brn-3. 2 in auditory and visual system development. Nature. 1996;381:603–6. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- [78].Xiang M, Gan L, Li D, et al. Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc Natl Acad Sci USA. 1997;94:9445–50. doi: 10.1073/pnas.94.17.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hertzano R, Montcouquiol M, Rashi-Elkeles S, et al. Transcription profiling of inner ears from Pou4f3(ddl/ddl) identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum Mol Genet. 2004;13:2143–53. doi: 10.1093/hmg/ddh218. [DOI] [PubMed] [Google Scholar]
- [80].Ito T. Differentiation and proliferation of pulmonary neuroendocrine cells. Prog Histochem Cytochem. 1999;34:247–322. doi: 10.1016/s0079-6336(99)80001-1. [DOI] [PubMed] [Google Scholar]
- [81].Lauweryns JM, Van RL. Immunocytochemical localization of aromatic L-amino acid decarboxylase in human, rat, and mouse bronchopulmonary and gastrointestinal endocrine cells. J Histochem Cytochem. 1988;36:1181–6. doi: 10.1177/36.9.2900264. [DOI] [PubMed] [Google Scholar]
- [82].Fu XW, Nurse CA, Wong V, Cutz E. Hypoxia-induced secretion of serotonin from intact pulmonary neuroepithelial bodies in neonatal rabbit. J Physiol. 2002;539:503–10. doi: 10.1113/jphysiol.2001.013071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cutz E, Jackson A. Neuroepithelial bodies as airway oxygen sensors. Respir Physiol. 1999;115:201–14. doi: 10.1016/s0034-5687(99)00018-3. [DOI] [PubMed] [Google Scholar]
- [84].Neuhuber WL. Lung sensors: complex functions require complex structures. Am J Respir Cell Mol Biol. 2003;28:265–6. doi: 10.1165/rcmb.F261. [DOI] [PubMed] [Google Scholar]
- [85].Ito T, Udaka N, Yazawa T, et al. Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development. 2000;127:3913–21. doi: 10.1242/dev.127.18.3913. [DOI] [PubMed] [Google Scholar]
- [86].Borges M, Linnoila RI, van de Velde HJ, et al. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature. 1997;386:852–5. doi: 10.1038/386852a0. [DOI] [PubMed] [Google Scholar]
- [87].Younossian AB, Brundler MA, Totsch M. Feasibility of the new WHO classification of pulmonary neuroendocrine tumours. Swiss Med Wkly. 2002;132:535–40. doi: 10.4414/smw.2002.09880. [DOI] [PubMed] [Google Scholar]
- [88].Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol. 2001;28:3–13. [PubMed] [Google Scholar]
- [89].Koutsami MK, Doussis-Anagnostopoulou I, Papavassiliou AG, Gorgoulis VG. Genetic and molecular coordinates of neuroendocrine lung tumors, with emphasis on small-cell lung carcinomas. Mol Med. 2002;8:419–36. [PMC free article] [PubMed] [Google Scholar]
- [90].Linnoila RI, Zhao B, DeMayo JL, et al. Constitutive achaete-scute homologue-1 promotes airway dysplasia and lung neuroendocrine tumors in transgenic mice. Cancer Res. 2000;60:4005–9. [PubMed] [Google Scholar]
- [91].Ito T, Udaka N, Ikeda M, Yazawa T, Kageyama R, Kitamura H. Significance of proneural basic helix-loop-helix transcription factors in neuroendocrine differentiation of fetal lung epithelial cells and lung carcinoma cells. Histol Histopathol. 2001;16:335–43. doi: 10.14670/HH-16.335. [DOI] [PubMed] [Google Scholar]
- [92].Chen H, Biel MA, Borges MW, et al. Tissue-specific expression of human achaete-scute homologue-1 in neuroendocrine tumors: transcriptional regulation by dual inhibitory regions. Cell Growth Differ. 1997;8:677–86. [PubMed] [Google Scholar]
- [93].Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- [94].Sriuranpong V, Borges MW, Ravi RK, et al. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61:3200–5. [PubMed] [Google Scholar]
- [95].Sriuranpong V, Borges MW, Strock CL, et al. Notch signaling induces rapid degradation of achaete-scute homolog 1. Mol Cell Biol. 2002;22:3129–39. doi: 10.1128/MCB.22.9.3129-3139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Pear WS, Radtke F. Notch signaling in lymphopoiesis. Semin Immunol. 2003;15:69–79. doi: 10.1016/s1044-5323(03)00003-4. [DOI] [PubMed] [Google Scholar]
- [97].Dwivedi PP, Anderson PH, Omdahl JL, Grimes HL, Morris HA, May BK. Identification of growth factor independent-1 (GFI1) as a repressor of 25-hydroxyvitamin D 1-alpha hydroxylase (CYP27B1) gene expression in human prostate cancer cells. Endocr Relat Cancer. 2005;12:351–65. doi: 10.1677/erc.1.00920. [DOI] [PubMed] [Google Scholar]
- [98].Zhuang SH, Burnstein KL. Antiproliferative effect of 1alpha, 25-dihydroxyvitamin D3 in human prostate cancer cell line LNCaP involves reduction of cyclin-dependent kinase 2 activity and persistent G1 accumulation. Endocrinology. 1998;139:1197–207. doi: 10.1210/endo.139.3.5770. [DOI] [PubMed] [Google Scholar]
- [99].Blutt SE, McDonnell TJ, Polek TC, Weigel NL. Calcitriol-induced apoptosis in LNCaP cells is blocked by overexpression of Bcl-2. Endocrinology. 2000;141:10–7. doi: 10.1210/endo.141.1.7289. [DOI] [PubMed] [Google Scholar]
- [100].Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78:1193–231. doi: 10.1152/physrev.1998.78.4.1193. [DOI] [PubMed] [Google Scholar]
- [101].Hsu JY, Feldman D, McNeal JE, Peehl DM. Reduced 1alpha-hydroxylase activity in human prostate cancer cells correlates with decreased susceptibility to 25-hydroxyvitamin D3-induced growth inhibition. Cancer Res. 2001;61:2852–6. [PubMed] [Google Scholar]
- [102].Duan Z, Zarebski A, Montoya-Durango D, Grimes HL, Horwitz M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol. 2005;25:10338–51. doi: 10.1128/MCB.25.23.10338-10351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]