

Abstract
Ebola virus causes rapidly progressive haemorrhagic fever, which is associated with severe immuosuppression. In infected dendritic cells (DCs), Ebola virus replicates efficiently and inhibits DC maturation without inducing cytokine expression, leading to impaired T-cell proliferation. However, the underlying mechanism remains unclear. In this study, we report that Ebola virus VP35 impairs the maturation of mouse DCs. When expressed in mouse immature DCs, Ebola virus VP35 prevents virus-stimulated expression of CD40, CD80, CD86 and major histocompatibility complex class II. Further, it suppresses the induction of cytokines such as interleukin (IL)-6, IL-12, tumour necrosis factor α and alpha/beta interferon (IFN-α/β). Notably, Ebola VP35 attenuates the ability of DCs to stimulate the activation of CD4+ T cells. Addition of type I IFN to mouse DCs only partially reverses the inhibitory effects of VP35. Moreover, VP35 perturbs mouse DC functions induced by lipopolysaccharide, an agonist of Toll-like receptor 4. Deletion of the amino terminus abolishes its activity, whereas a mutation in the RNA binding motif has no effect. Our work highlights a critical role of VP35 in viral interference in DC function with resultant deficiency in T-cell function, which may contribute to the profound virulence of Ebola virus infection.
INTRODUCTION
Ebola virus, a non-segmented negative-stranded RNA virus, belongs to the family Filoviridae (Mahanty & Bray, 2004). Infection of humans and non-human primates with Ebola virus results in a rapidly progressive severe illness characterized by bleeding, capillary leakage, multisystem dysfunction and a shock-like state culminating in death for 90 % of infected individuals (Mahanty & Bray, 2004). While many factors, including viraemia and severe immunosuppression (Baize et al., 1999; Ksiazek et al., 1999a, b; Towner et al., 2004), contribute to the lethal infection, viral control of innate immunity most probably plays a critical and direct role (Bray, 2001). In infected cells, Ebola virus impairs the expression of interferon (IFN)-stimulated genes induced by either IFN or double-stranded RNA (dsRNA) (Gibb et al., 2002, Harcourt et al., 1998, 1999). Ebola virus suppresses IFN production in monocyte, macrophage and dendritic (DC) cells (Bosio et al., 2003; Gupta et al., 2001; Mahanty et al., 2003). Further, Ebola virus is insensitive to IFN-α/β (Harcourt et al., 1999; Jahrling et al., 1999; Kash et al., 2006).
Recent studies demonstrate that knockdown of VP35 from Ebola virus inhibits viral replication and protects against lethal infection in animal models (Enterlein et al., 2006; Warfield et al., 2006). Ebola VP35 is an essential cofactor of RNA replication and transcription (Mühlberger et al., 1999), and can also suppress RNA silencing (Haasnoot et al., 2007). Additionally, Ebola VP35 functions as an IFN antagonist by binding to dsRNA or TANK-binding kinase 1, leading to the inhibition of IFN regulatory factor 3 (IRF3) that induces the expression of antiviral genes and cytokines (Basler et al., 2000, 2003; Cardenas et al., 2006; Hartman et al., 2006; Prins et al., 2009). Reverse genetic studies showed that an Ebola VP35 mutant, with the disrupted RNA binding domain, is highly attenuated both in cell culture and in vivo (Hartman et al., 2006, 2008a). This defect parallels with the inability of Ebola virus to suppress the expression of an array of host genes (Hartman et al., 2008b). We have noted that Ebola VP35 blocks the antiviral action of IFN-α/β mediated by dsRNA-dependent protein kinase PKR (Feng et al., 2007). Recently, PKR has been found as a host factor that can limit replication of Ebola virus (Strong et al., 2008). These studies suggest that the interaction of VP35 and PKR may dictate the outcome of Ebola virus infection.
Ebola virus initially infects macrophage cells and DCs, which link innate and adaptive immunity (Geisbert et al., 2003; Gibb et al., 2001; Mahanty et al., 2003). Immature DCs, which reside in almost all peripheral tissues, are able to capture and process viral antigens (Shortman & Liu, 2002). In response to virus infection, DCs migrate to lymph nodes and undergo maturation. Mature DCs display relatively higher levels of co-stimulatory molecules and release pro-inflammatory cytokines, which subsequently activate adaptive immune responses. Remarkably, Ebola virus replicates efficiently in DCs, where it blocks DC maturation (Bosio et al., 2003; Mahanty et al., 2003). The inactivated Ebola virus particle still retains this activity (Mahanty et al., 2003; Warfield et al., 2003). In this context, it is interesting that the Ebola virus-like particle, consisting of only matrix protein VP40 and glycoprotein GP, stimulates DC maturation and activates T cells, as well as B cells (Warfield et al., 2003, 2005). The GP mucin domain is required to stimulate DC maturation (Martinez et al., 2007). However, the viral component(s) that impairs DC function remains elusive. This study was designed to investigate the interaction of Ebola virus VP35 and DCs. We report that Ebola VP35 interferes with DC maturation induced by virus and Toll-like receptor (TLR) signalling, resulting in impaired T-cell activation.
RESULTS
Ebola virus VP35 suppresses virus-induced expression of co-stimulatory molecules on DCs
To explore the interaction of Ebola virus VP35 and DCs, we used a herpes simplex virus (HSV) vector expressing Ebola VP35 (Feng et al., 2007). This vector, derived from a defective HSV-1 lacking γ134.5 and thymidine kinase genes, stimulates innate immunity (Cheng et al., 2003; Verpooten et al., 2008). We first tested the infection efficiency in mouse DCs. Purified CD11c+ DCs, generated from bone marrow in the presence of granulocyte–macrophage colony-stimulating factor (GM-CSF), were exposed to virus. Infectivity was determined by FACS analysis. As shown in Fig. 1(a), HSV and HSV–VP35 infected immature DCs in a dose-dependent manner. At 5 p.f.u. per cell, they infected over 90 % of cells. Cell viability was 91 % in mock-infected cells and 85 % in virus-infected cells (Fig. 1b). HSV–VP35 and the HSV vector both infected mouse DCs efficiently, where HSV–VP35 expressed the VP35 protein (Fig. 1c).
Fig. 1.
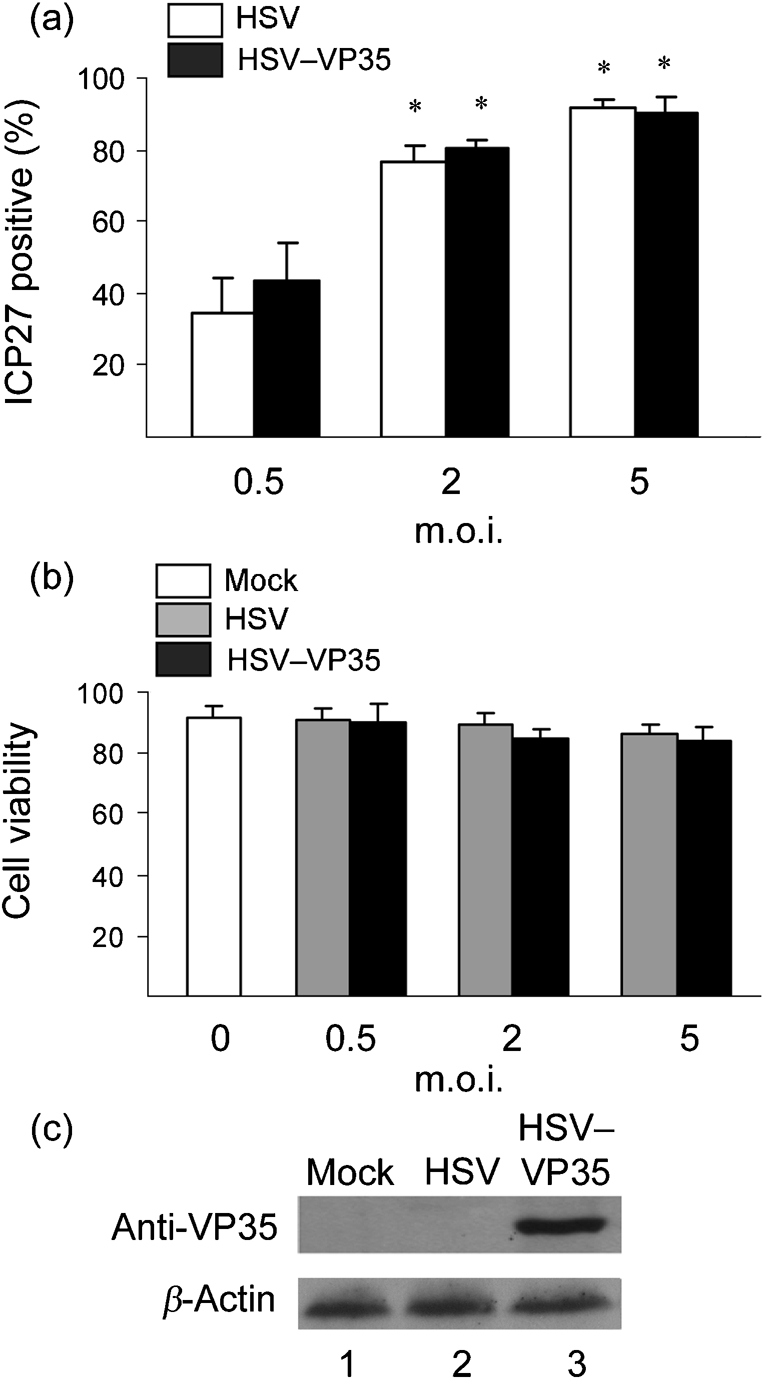
(a) Virus infection of immature CD11c+ DCs. Bone-marrow-derived DCs, grown in the presence of GM-CSF, were infected with an HSV vector or HSV–VP35 expressing Ebola VP35 at different m.o.i. values. At 12 h post-infection, cells were stained with a mouse mAb for an HSV-1 antigen ICP27 and an FITC-labelled secondary antibody. Expression of ICP27, as a measure of infectivity, was determined by flow cytometry. (b) Viability of virus-infected immature DCs. DCs were mock-infected or infected with viruses at different doses. Cell viability was measured by the trypan blue exclusion method at 12 h post-infection. (c) Expression of VP35. DCs were mock-infected or infected with the indicated viruses (5 p.f.u. per cell). At 12 h post-infection, cell lysates were subjected to Western immunoblot analysis with antibody against Ebola VP35. The data are representative of three independent experiments with triplicate samples. Asterisks denote statistical differences (P<0.05) between an infection group and mock group, and error bars indicate sem.
Based on the above analyses, we evaluated the impact of Ebola VP35 on mouse DC maturation. Immature CD11c+ DCs either mock-infected or infected with viruses (5 p.f.u. per cell) were subjected to FACS analysis 12 h after infection. As illustrated in Fig. 2, basal levels of major histocompatibility complex (MHC) class II, CD40, CD80 and CD86 were seen in mock-infected cells. Treatment with LPS, a prototype stimulator of DC maturation, dramatically enhanced these proteins. Infection of immature DCs with HSV showed responses similar to that induced by LPS; it enhanced the expression of cell surface molecules. In sharp contrast, HSV–VP35 did not enhance the expression of these cell surface markers; the expression of MHC class II, CD40, CD80 and CD86 remained at levels similar to those of mock-treated cells. These results suggest that VP35 suppresses virus-induced upregulation of co-stimulatory molecules in immature DCs.
Fig. 2.

Effects of VP35 on the expression of cell surface molecules. Immature DCs were mock-infected or infected with an HSV vector or HSV–VP35 (5 p.f.u. per cell). At 12 h post-infection, cells were stained with PE-labelled antibodies against MHC class II, CD40, CD80 and CD86, respectively. Samples were subjected to FACS analysis. LPS (0.5 μg ml−1) was used as a positive control. Filled areas indicate isotype controls; open areas indicate test samples. The data are representative of three independent experiments.
VP35 inhibits viral induction of proinflammatory cytokines in DCs
One of the properties of mature DCs is the secretion of an array of proinflammatory cytokines. We carried out a cytokine analysis after exposure of DCs to different stimuli by intracellular staining followed by flow cytometry. As shown in Fig. 3(a), interleukin (IL)-6 expression remained at basal levels (8 %) in mock-infected cells. However, 6 h after the HSV vector infection, IL-6 expression increased from 21 to 45 % as infection progressed to 24 h. Notably, HSV–VP35 failed to affect this change at all time points tested, with IL-6 expression retained at levels seen in mock-infected cells. A similar profile of IL-12 expression was also seen after mock- or virus-infection (Fig. 3b). Expression of tumour necrosis factor (TNF)-α displayed a different pattern (Fig. 3c). Its expression was low (7 %) in mock-infected cells but drastically increased to 46 % in cells infected with HSV at 24 h. However, expression of TNF-α increased modestly in cells infected with HSV–VP35 over the course of infection (22 %).
Fig. 3.

Effects of Ebola VP35 on cytokine expression. Immature DCs were mock-infected or infected with an HSV vector or HSV–VP35 (5 p.f.u. per cell). (a–e) At 6, 12 and 24 h post-infection, cells were subjected to intracellular staining with antibodies against IL-6 (a), IL-12 (b), TNF-α (c), IFN-α (d) and IFN-β (e). Samples were then subjected to FACS analysis. (f) Type I IFN secretion from virus-infected DCs. Cell culture media from infected cells (described above) were assayed for IFN-β by ELISA. The data are representative of three independent experiments with triplicate samples. Asterisks denote statistical differences (P<0.05) between the HSV- and HSV–VP35-infected groups, and error bars indicate sem.
Next, we analysed the expression of type I IFN in DCs. As indicated in Fig. 3(d, e), IFN-α/β expression was low in mock-infected cells, whereas its expression increased markedly at 12 and 24 h in cells infected with HSV. However, IFN-α/β expression remained low in cells infected with HSV–VP35 at all time points examined. Thus, unlike HSV, HSV–VP35 inhibited IFN-α/β expression in DCs. We further determined the IFN-α/β secretion by ELISA assays. Type I IFN was barely detectable in media derived from mock-infected or HSV–VP35-infected cells (Fig. 3f). In contrast, a significant amount of type I IFN was detected at 6 h in medium from HSV-infected cells (65 pg ml−1) and its level increased drastically at 12 and 24 h (300 pg ml−1). We conclude that Ebola VP35 blocks virus-induced cytokine production in DCs.
Suppression of type I IFN production by VP35 inhibits the induction of cell surface molecules
Since VP35 negatively modulated the expression of co-stimulatory molecules, we asked whether this was related to IFN production. To address this issue, we assessed the effect of exogenous type I IFN on DC maturation. Immature DCs, pretreated or untreated with IFN-β, were infected with viruses and the expression of cell surface molecules was determined. The results in Fig. 4 show that IFN-β alone induced upregulation of MHC class II, CD86, CD80 and CD40 in mock-infected cells. Similarly, HSV stimulated the expression of these molecules to a greater magnitude. Exposure of DCs, pretreated with IFN-β, to HSV further enhanced the expression of MHC class II, CD86, CD80 and CD40. As noted before, HSV–VP35 prevented enhanced expression of MHC class II and co-stimulatory molecules, and addition of IFN-β only partially reversed this inhibitory effect. These data suggest that VP35 suppresses the expression of MHC class II and CD86, CD80 and CD40 via both type I IFN-dependent and type I IFN-independent pathways.
Fig. 4.

Effects of exogenously added IFN on the expression of cell surface markers. Immature DCs were pretreated overnight with mouse IFN-β (800 U ml−1); cells were then mock-infected or infected with an HSV vector or HSV–VP35 (5 p.f.u. per cell). At 12 h post-infection, cells were stained with PE-labelled antibodies against MHC class II (a), CD86 (b), CD80 (c) and CD40 (d), and subjected to FACS analysis. The data are representative of three independent experiments with triplicate samples. Asterisks denote statistical differences (P<0.05) between the HSV vector- and HSV–VP35-infected groups, and error bars indicate sem.
VP35 attenuates the capacity of DCs to stimulate T-cell activation
As antigen presentation by mature DCs is essential to initiate adaptive immune responses, we examined the effect of Ebola VP35 on T-cell activation. Immature DCs were mock-infected or infected with viruses for 12 h and then treated with UV light to inactivate viruses. These cells were cultured with allogeneic CD4+ naive T cells and analysed for T-cell activation by flow cytometry. We first measured the cell surface expression of CD44, CD69 and CD62L on CD4+ T cells. As shown in Fig. 5(a), naive CD4+ T cells incubated with mock-infected DCs had low levels of CD44 and CD69 with mean fluorescent intensities of 9.8 and 8.3, respectively. These cells, however, had a higher level of CD62L, with a mean fluorescent intensity of 908. DCs infected with HSV induced a drastic increase in CD44 and CD69 and a decrease in CD62L on CD4+ T cells, reaching mean fluorescent intensities of 122, 96 and 23, respectively, indicative of T-cell activation. In contrast, DCs infected with HSV–VP35 reversed these effects on CD44, CD69 and CD62L substantially, showing mean fluorescent intensities of 64, 26 and 129, respectively.
Fig. 5.

Activation of naive CD4 T cells by virus-infected DCs. Immature DCs were mock-infected or infected with an HSV vector or HSV–VP35 (5 p.f.u. per cell). At 12 h post-infection, cells were UV-irradiated (0.25 J cm−2) to inactivate viruses and co-cultured with allogeneic CD4+ T cells in vitro for 48 h. (a) Expression of CD4+ T-cell activation markers. CD4+ T cells were stained with antibodies against CD44, CD69 and CD62L, respectively. (b, c) Samples were subjected to flow cytometry analysis; IFN-γ (b) and IL-10 (c) production in CD4+ T cells was determined. The data are representative of three independent experiments with triplicate samples.
Next, we determined IFN-γ production in CD4+ T cells. Mock-infected DCs induced a background level of IFN-γ-producing cells (9.28 %) in naive CD4+ T cells (Fig. 5b). DCs infected with HSV stimulated IFN-γ-producing cells in CD4+ T cells (26.55 %), whereas DCs infected with HSV–VP35 did not (7.66 %), suggesting Ebola VP35 inhibited DCs from providing the necessary signals for IFN-γ production by CD4+ T cells. Lastly, we measured IL-10 expression in CD4+ T cells. While 12.72 % of CD4+ T cells, incubated with mock-infected DCs, expressed IL-10, only 2.03 % of CD4+ T cells incubated with DCs infected with HSV expressed IL-10 (Fig. 5c). When incubated with DCs infected with HSV–VP35, a significant population of CD4+ T cells (23.73 %) produced IL-10. We conclude from these experiments that the VP35 protein attenuates the capacity of DCs to stimulate CD4+ T-cell activation.
VP35 alone inhibits DC maturation stimulated by LPS
To further examine VP35, we evaluated whether VP35 alone had any impact on DC maturation induced by TLR signalling. Specifically, immature DCs were transduced with a retrovirus vector or vectors expressing VP35. Cells were then treated with LPS and assayed for the expression of CD80, CD86, IL-6, IL-12 and IFN-β, respectively. As shown in Fig. 6(a, b), in the absence of LPS stimulation, all cells displayed a basal level of CD80 and CD86 expression. The addition of LPS greatly stimulated the expression of these co-stimulatory molecules in cells mock-transduced or transduced with a retroviral vector, reaching a mean fluorescent intensity of approximately 1170. However, this effect was suppressed in cells transduced with wild-type VP35 or a mutant R312A which fails to bind dsRNA. Notably, a mutant dN190, which lacks amino acids 1–190, was unable to suppress CD80 and CD86 expression. Western blot analysis showed that VP35 variants expressed these molecules at comparable levels (Fig. 6c).
Fig. 6.

Inhibition of TLR-induced DC maturation by Ebola VP35. Immature DCs were mock-transduced or transduced with retroviral vectors expressing VP35 variants. Five days after transduction, the GFP-positive cells were stimulated with LPS (500 ng ml−1) and subjected to FACS analysis with PE-labelled antibodies against CD86 (a) and CD80 (b). In parallel, lysates of cells were prepared and subjected to Western blot analysis with antibodies against VP35 and anti-β-actin (c). Cell supernatants were also assayed for the production of IL-6 (d), IL-12 (e) and IFN-β (f). The data are representative of three independent experiments with triplicate samples. Asterisks denote statistical differences (P<0.05) of samples compared with mock- and vector-transduced groups, and error bars indicate sem.
In parallel experiments, we also examined cytokine production. Addition of LPS similarly stimulated the expression of IL-6, IL-12 and IFN-β in cells mock-transduced or transduced with a retroviral vector (Fig. 6d–f). Both wild-type VP35 and the mutant R312A were able to suppress cytokine production induced by LPS. It appeared that wild-type VP35 and R312A inhibited IFN-β expression more efficiently than IL-6 and IL-12. Under these conditions, the mutant dN190 was unable to exert any inhibitory effect. These results suggested that VP35 is able to suppress DC maturation induced by TLR4 signalling. This activity requires the amino-terminal domain but not the dsRNA binding domain.
DISCUSSION
Ebola virus replicates robustly and inhibits DC maturation without inducing cytokine expression, which affects T-cell proliferation (Bosio et al., 2003; Mahanty et al., 2003). In this study, we provide evidence that Ebola VP35 disrupts the critical biological functions of DCs. When expressed in immature DCs, Ebola VP35 inhibited virus-induced expression of co-stimulatory molecules and proinflammatory cytokines. Furthermore, Ebola VP35 inhibited the ability of DCs to stimulate T-cell activation as illustrated by the levels of expression of CD44, CD69, CD62L, IFN-γ and IL-10 in CD4+ T cells. DCs play a crucial role in different aspects of immunity, including cytokine production, antigen presentation, antibody production and interaction with natural killer cells (Bosio et al., 2004; Shortman & Liu, 2002). In this context, our results suggest that VP35 can have a broad impact on DC maturation and function, with a consequent negative impact on adaptive immune responses.
The VP35 gene of Ebola virus is essential to promote viral pathogenesis (Enterlein et al., 2006; Warfield et al., 2006). Previous studies demonstrated that VP35 inhibits the expression of IFN-α/β (Basler et al., 2000, 2003). In this process, it blocks activation of IRF3 in both RNA-dependent and RNA-independent manners (Cardenas et al., 2006; Hartman et al., 2004). Unlike wild-type virus, an Ebola VP35 mutant that fails to inhibit IRF3 activation is greatly reduced in viral virulence (Hartman et al., 2008a). As type I IFN is an activator of DCs (Gallucci et al., 1999; Luft et al., 1998; Santini et al., 2000), it is reasonable to assume that VP35 most probably modulates DC functions by disrupting IFN-mediated effects on DCs. We noted that expression of VP35 in DCs led to the inhibition of IFN-α/β production and reduced expression of MHC class II, CD86, CD80 and CD40. Addition of type I IFN to DCs relieved this inhibitory effect to some extent, which was evident for MHC class II, CD86 and CD80. Altogether, these results suggest that Ebola VP35 impairs DC maturation, in part, by blocking of IFN production, which probably depends on the ability of VP35 to disrupt IRF3 activation. Herein, our work established a link between VP35 and impaired DC maturation probably related to IFN production.
Our work highlights a critical role of Ebola VP35 in viral interference in DC maturation. Although this involves type I IFN, it does not fully account for the observed defects in DC functions. We speculate that VP35 may work through additional mechanisms. Two lines of evidence support this prediction. Firstly, we observed that exogenous IFN-β did not fully overcome the inhibitory effect of VP35 on MHC class II, CD40, CD80 and CD86. Secondly, VP35 drastically suppressed virus-induced expression of IL-6, IL-12 and TNF-α. Although these cytokines are known to contribute to DC maturation, their expression is not regulated by type I IFN (Lopez et al., 2003). Accordingly, a key question arises as to how VP35 functions at the molecular level. Ebola VP35 has the capacity to bind dsRNA, which is involved in the inhibition of IRF3 activation and RNA silencing (Basler et al., 2003; Haasnoot et al., 2007). Additionally it inhibits PKR, a component integrating innate pathways leading to phosphorylation of eIF-2α and activation of NF-κB, MAP kinase and TLR responses (Feng et al., 2007; Goh et al., 2000; Jiang et al., 2003; Oganesyan et al., 2006). Thus, this would predict that perturbation of DC functions by VP35 may depend on its ability to modulate RNA silencing or PKR activities. Our mutational analysis revealed a site-specific mutation in the RNA binding motif of VP35 did not impair its ability to block DC maturation. Interestingly, deletion of the amino-terminal domain, which is required to inhibit PKR, disrupted its activity in blocking DC maturation. Additional work is needed to understand the underlying mechanism.
Activation of TLR pathways leads to DC maturation (Honda et al., 2003). In this regard, we observed that VP35 blocked DC maturation initiated by TLR4 signalling. In immature DCs, VP35 effectively suppressed the induction of CD80, CD86, IL-6, IL-12 and IFN-β by LPS. TLR4 signalling activates a number of transcription factors, such as IRF3, NF-κB and AP1, which requires the engagement of the adaptors TRIF and MyD88 (Takeda & Akira, 2004). Therefore, the inhibitory effects on cell surface molecules and cytokines cannot be explained solely by the ability of VP35 to inactivate IRF3. An intriguing possibility is that VP35 may target an additional component(s) in the innate immune pathways. Work is in progress to address this issue.
Previous studies revealed that, like live Ebola virus, inactivated Ebola virus retains its ability to suppress DC maturation and cytokine production (Bosio et al., 2003; Mahanty et al., 2003). Because Ebola VP35 is a structural protein (Mahanty & Bray, 2004), a simple interpretation is that it may be sufficient to inhibit DC function. The data in the present study are in line with this hypothesis. Notably, when expressed in DCs, VP35 blocked DC functions. Interestingly, analysis with virus-like particles showed that glycoprotein GP is required to stimulate DC maturation (Bosio et al., 2004; Martinez et al., 2007; Warfield et al., 2003; Ye et al., 2006). Surprisingly, virus-like particles containing VP35 as well as VP24 failed to suppress DC maturation (Martinez et al., 2007), which could be due to their low abundance. While we have not assessed VP24, we found that VP35 inhibited the induction of MHC class II, co-stimulatory molecules and proinflammatory cytokines when expressed in surrogate systems. These phenotypes replicated those seen in DCs infected with Ebola virus (Bosio et al., 2003; Mahanty et al., 2003), implicating VP35 as a suppressor of DC maturation.
In response to virus infection, DCs generate and integrate signals, which are vital to prime naive T cells. Our results show that, when expressed in immature DCs, VP35 interrupted this process. Notably, VP35 suppressed virus-induced IFN-γ production in CD4+ T cells. Additionally, VP35 inhibited virus-induced expression of CD44 and CD69 and relieved viral-mediated suppression of CD62L. VP35 also stimulated the expression of IL-10, an anti-inflammatory cytokine. Collectively, these results suggest that the expression of VP35 in immature DCs has a profound effect on different aspects of CD4+ T-cell activation, which may, in part, explain why Ebola VP35 is essential in viral virulence. Work is in progress to understand the mechanism(s) by which VP35 modulates the engagement of DCs with T-cell proliferation.
METHODS
Mice.
BALB/c and C57BL/6 mice were purchased from Harlan Sprague–Dawley and housed under specific-pathogen-free conditions in a biosafety level 2 containment. Groups of 5-week-old mice were used in this study, which was carried out in accordance with the guidelines of the Animal Care Use Committee of the University of Illinois at Chicago.
Cells and viruses.
Vero cells were obtained from the ATCC and propagated in Dulbecco's modified Eagle's medium supplemented with 10 % fetal bovine serum (FBS). Myeloid DCs were generated as described previously (Inaba et al., 1992). Briefly, bone marrow cells were removed from the tibia and femur of BALB/c mice. Following red blood cell lysis and washing, progenitor cells were plated in RPMI 1640 medium (Invitrogen) supplemented with 10 % FBS, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate and 20 ng GM-CSF ml−1 (Biosource) in six-well plates at 4×106 cells per well. Cells were supplemented with 2 ml fresh medium every other day. On day 8, DCs were positively selected for surface CD11c expression using magnetic beads (Miltenyi Biotech) to give a >97 % pure population of CD11c+ MHCII+ cells. DCs displayed low levels of CD40, CD80, CD86 and MHC class II molecules, characteristic of immature DCs. Purified CD11c+ DCs were cultured in fresh medium with FBS and GM-CSF and used in subsequent experiments.
The recombinant HSV-1 vector (KY0234) lacks γ134.5 and the thymidine kinase genes (Cheng et al., 2003). The recombinant virus HSV–VP35 (MC0201) has the γ134.5 gene of HSV replaced by the Ebola VP35 gene and lacks the thymidine kinase gene (Feng et al., 2007).
Viral infection.
Purified CD11c+ DCs were plated in 12-well plates (5×105 cells per well) and infected with viruses at the indicated m.o.i. After 2 h incubation, cells were washed with PBS and resuspended in RPMI 1640 supplemented with 10 % FBS and 20 ng GM-CSF ml−1. Cells were harvested and analysed at different times after infection. Where indicated, mouse IFN-β (800 U ml−1; Sigma), neutralizing antibodies (30 μg ml−1) specific to mouse IFNα/β or isotype control antibodies (PBL Laboratories) were added to media before virus infection. For TLR stimulation, LPS (1 μg ml−1; Sigma) was added to DCs after virus infection.
T-cell activation assay.
Spleens were harvested from C57BL/6 mice and single cell suspensions were prepared. After red blood cell depletion, CD4+ T cells were purified by using micro-beads (Miltenyi Biotech) according to the manufacturer's instructions and were used as the responder cells. After virus infection, bone-marrow-derived DCs from BALB/c mice were treated with UV light and served as stimulator cells. The responder cells (1×106) were labelled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) and co-cultured with the DC stimulator cells (2×105) in 2 ml media. After 48 h incubation, cells were stained with antibodies against CD44-PE, CD69-PE, CD62L-PE, IL-10 and IFN-γ (eBioscience). CD4+ T cells were analysed by gating of CFSE-positive cells.
Flow cytometry.
Cells were stained with FITC- or phycoerythrin (PE)-linked mAbs according to the manufacturer's instructions. Briefly, cells were blocked with Fcγ mAb (0.5 μg ml−1) for 30 min at 4 °C. After washing with PBS, cells were stained with anti-CD11c–PE, anti-MHCII–FITC, anti-CD40–FITC anti-CD80–FITC, anti-CD86–FITC or isotype-matched control antibodies for 30 min on ice with gentle shaking (eBioscience). Samples were analysed by using FACS Calibur and data were analysed with CellQuest Pro software (BD).
Flow cytometry of intracellular cytokine production of IL-6, IL-12, TNF-α, IFN-α and IFN-β in cells was performed as follows. A single-cell suspension was treated with brefeldin A for 6 h. After washing twice with PBS, cells were blocked with 1 μl Fcγ mAb (0.5 μg ml−1) for 30 min at 4 °C and fixed with 4 % paraformaldehyde at 4 °C for 15 min before treating with permeabilizing buffer (eBioscience) at 4 °C for 10 min. After washing once with PBS, cells were stained with appropriate isotype controls, anti-IL-6–FITC, anti-IL-12–FITC, anti-TNF-α–FITC, anti-IFN-α–FITC and IFN-β–FITC antibodies (PBL Laboratories). Samples were analysed using FACS Calibur and data were analysed with CellQuest Pro software.
To determine viral infectivity, virus-infected DCs were fixed in 4 % paraformaldehyde (Sigma) and permeabilized as described by the manufacturer (eBioscience). Cells were blocked with 5 % normal mouse serum (Sigma), incubated with a mAb against HSV-1 ICP27 (Virusys) and reacted with a goat anti-mouse FITC-conjugated antibody (Santa Cruz Biotech). ICP27 expression was determined by flow cytometry.
Transduction and ELISA.
pSIN-Ova–GFP, a dual-promoter human immunodeficiency virus type 1 vector, was a gift from Mary Collins (University College London, UK) (Rowe et al., 2009). PCR fragments of wild-type VP35 or VP35 mutants were cloned into the BamHI and NotI sites, resulting in plasmids VP35, R312A and dN190. These plasmids were co-transfected along with HIVtrans and VSV-G into 293T cells using Lipofectamine 2000 (Invitrogen) as described previously (Feng et al., 2007). At 48 h after transfection, supernatant was collected and the titres were determined by GFP expression. Immature DCs were transduced with retroviral constructs and cells were fed with fresh RPMI 1640 medium containing GM-CSF (20 ng ml−1) every 2 days. On day 5, GFP-positive DCs were sorted by FACS. For ELISA, supernatants of cell culture were collected and levels of IL-6, IL-12 and IFN-β were quantified using kits from R&D systems and PBL laboratories according to the manufacturers’ instructions.
Acknowledgments
We acknowledge membership within and support from the Region V ‘Great Lakes’ Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (NIH Award 1-U54-AI-057153). This work was partially supported by the NIH grant AI058190 (B. S. P). We thank Hans-Dieter Klenk, Peter Palese, Christopher F. Basler and Mary Collins for valuable reagents. We are grateful to Chenthamarakshan Vasu for helpful discussion and suggestions.
References
- Baize, S., Leroy, E. M., Georges-Courbot, M. C., Capron, M., Lansoud-Soukate, J., Debre, P., Fisher-Hoch, S. P., McCormick, J. B. & Georges, A. J. (1999). Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat Med 5, 423–426. [DOI] [PubMed] [Google Scholar]
- Basler, C. F., Wang, X., Mühlberger, E., Volchkov, V., Paragas, J., Klenk, H. D., García-Sastre, A. & Palese, P. (2000). The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc Natl Acad Sci U S A 97, 12289–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler, C. F., Mikulasova, A., Martinez-Sobrido, L., Paragas, J., Mühlberger, E., Bray, M., Klenk, H. D., Palese, P. & García-Sastre, A. (2003). The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol 77, 7945–7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio, C. M., Aman, M. J., Grogan, C., Hogan, R., Ruthel, G., Negley, D., Mohamadzadeh, M., Bavari, S. & Schmaljohn, A. (2003). Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J Infect Dis 188, 1630–1638. [DOI] [PubMed] [Google Scholar]
- Bosio, C. M., Moore, B. D., Warfield, K. L., Ruthel, G., Mohamadzadeh, M., Aman, M. J. & Bavari, S. (2004). Ebola and Marburg virus-like particles activate human myeloid dendritic cells. Virology 326, 280–287. [DOI] [PubMed] [Google Scholar]
- Bray, M. (2001). The role of the Type I interferon response in the resistance of mice to filovirus infection. J Gen Virol 82, 1365–1373. [DOI] [PubMed] [Google Scholar]
- Cardenas, W. B., Loo, Y. M., Gale, M., Jr, Hartman, A. L., Kimberlin, C. R., Martínez-Sobrido, L., Saphire, E. O. & Basler, C. F. (2006). Ebola virus VP35 protein binds double-stranded RNA and inhibits α/β interferon production induced by RIG-I signaling. J Virol 80, 5168–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, G., Yang, K. & He, B. (2003). Dephosphorylation of eIF-2α mediated by the γ134.5 protein of herpes simplex virus type 1 is required for viral response to interferon but is not sufficient for efficient viral replication. J Virol 77, 10154–10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enterlein, S., Warfield, K. L., Swenson, D. L., Stein, D. A., Smith, J. L., Gamble, C. S., Kroeker, A. D., Iversen, P. L., Bavari, S. & Mühlberger, E. (2006). VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob Agents Chemother 50, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z., Cerveny, M., Yan, Z. P. & He, B. (2007). The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA dependent protein kinase PKR. J Virol 81, 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci, S., Lolkema, M. & Matzinger, P. (1999). Natural adjuvants: endogenous activators of dendritic cells. Nat Med 5, 1249–1255. [DOI] [PubMed] [Google Scholar]
- Geisbert, T. W., Hensley, L. E., Larsen, T., Young, H. A., Reed, D. S., Geisbert, J. B., Scott, D. P., Kagan, E., Jahrling, P. B. & Davis, K. J. (2003). Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol 163, 2347–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb, T. R., Bray, M., Geisbert, T. W., Steele, K. E., Kell, W. M., Davis, K. J. & Jaax, N. K. (2001). Pathogenesis of experimental Ebola Zaire virus infection in BALB/c mice. J Comp Pathol 125, 233–242. [DOI] [PubMed] [Google Scholar]
- Gibb, T. R., Norwood, D. A., Jr, Woollen, N. & Henchal, E. A. (2002). Viral replication and host gene expression in alveolar macrophages infected with Ebola virus (Zaire strain). Clin Diagn Lab Immunol 9, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh, K. C., deVeer, M. J. & Williams, B. R. (2000). The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J 19, 4292–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, M., Mahanty, S., Ahmed, R. & Rollin, P. E. (2001). Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with Ebola virus secrete MIP-1α and TNF-α and inhibit poly-IC-induced IFN-α in vitro. Virology 284, 20–25. [DOI] [PubMed] [Google Scholar]
- Haasnoot, J., de Vries, W., Geutjes, E. J., Prins, M., de Haan, P. & Berkhout, B. (2007). The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog 3, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt, B. H., Sanchez, A. & Offermann, M. K. (1998). Ebola virus inhibits induction of genes by double-stranded RNA in endothelial cells. Virology 252, 179–188. [DOI] [PubMed] [Google Scholar]
- Harcourt, B. H., Sanchez, A. & Offermann, M. K. (1999). Ebola virus selectively inhibits responses to interferons, but not to interleukin-1β, in endothelial cells. J Virol 73, 3491–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, A. L., Towner, J. S. & Nichol, S. T. (2004). A C-terminal basic amino acid motif of Zaire Ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 328, 177–184. [DOI] [PubMed] [Google Scholar]
- Hartman, A. L., Dover, J. E., Towner, J. S. & Nichol, S. T. (2006). Reverse genetic generation of recombinant Zaire Ebola viruses containing disrupted IRF-3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF-3 activation without inhibiting viral transcription or replication. J Virol 80, 6430–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, A. L., Bird, B. H., Towner, J. S., Antoniadou, Z. A., Zaki, S. R. & Nichol, S. T. (2008a). Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of Ebola virus. J Virol 82, 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, A. L., Ling, L., Nichol, S. T. & Hibberd, M. L. (2008b). Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J Virol 82, 5348–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda, K., Sakaguchi, S., Nakajima, C., Watanabe, A., Yanai, H., Matsumoto, M., Ohteki, T., Kaisho, T., Takaoka, A. & other authors (2003). Selective contribution of IFN-α/β signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A 100, 10872–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba, K., Inaba, M., Romani, N., Aya, H., Deguchi, M., Ikehara, S., Muramatsu, S. & Steinman, R. M. (1992). Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med 176, 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling, P. B., Geisbert, T. W., Geisbert, J. B., Swearengen, J. R., Bray, M., Jaax, N. K., Huggins, J. W., LeDuc, J. W. & Peters, C. J. (1999). Evaluation of immune globulin and recombinant interferon-α2b for treatment of experimental Ebola virus infections. J Infect Dis 179 (Suppl. 1), S224–S234. [DOI] [PubMed] [Google Scholar]
- Jiang, Z., Zamanian-Daryoush, M., Nie, H., Silva, A. M., Williams, B. R. & Li, X. (2003). Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem 278, 16713–16719. [DOI] [PubMed] [Google Scholar]
- Kash, J. C., Mühlberger, E., Carter, V., Grosch, M., Perwitasari, O., Proll, S. C., Thomas, M. J., Weber, F., Klenk, H. D. & Katze, M. G. (2006). Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol 80, 3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek, T. G., Rollin, P. E., Williams, A. J., Bressler, D. S., Martin, M. L., Swanepoel, R., Burt, F. J., Leman, P. A., Khan, A. S. & other authors (1999a). Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis 179 (Suppl 1), S177–S187. [DOI] [PubMed] [Google Scholar]
- Ksiazek, T. G., West, C. P., Rollin, P. E., Jahrling, P. B. & Peters, C. J. (1999b). ELISA for the detection of antibodies to Ebola viruses. J Infect Dis 179 (Suppl 1), S192–S198. [DOI] [PubMed] [Google Scholar]
- Lopez, C. B., Garcia-Sastre, A., Williams, B. R. & Moran, T. M. (2003). Type I interferon induction pathway, but not released interferon, participates in the maturation of dendritic cells induced by negative-strand RNA viruses. J Infect Dis 187, 1126–1136. [DOI] [PubMed] [Google Scholar]
- Luft, T., Pang, K. C., Thomas, E., Hertzog, P., Hart, D. N., Trapani, J. & Cebon, J. (1998). Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol 161, 1947–1953. [PubMed] [Google Scholar]
- Mahanty, S. & Bray, M. (2004). Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis 4, 487–498. [DOI] [PubMed] [Google Scholar]
- Mahanty, S., Hutchinson, K., Agarwal, S., McRae, M., Rollin, P. E. & Pulendran, B. (2003). Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J Immunol 170, 2797–2801. [DOI] [PubMed] [Google Scholar]
- Martinez, O., Valmas, C. & Basler, C. F. (2007). Ebola virus-like particle-induced activation of NF-κB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology 364, 342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlberger, E., Weik, M., Volchkov, V. E., Klenk, H. D. & Becker, S. (1999). Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73, 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan, G., Saha, S. K., Guo, B., He, J. Q., Shahangian, A., Zarnegar, B., Perry, A. & Cheng, G. (2006). Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439, 208–211. [DOI] [PubMed] [Google Scholar]
- Prins, K. C., Cardenas, W. B. & Basler, C. F. (2009). Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKε and TBK-1. J Virol 83, 3069–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe, H. M., Lopes, L., Brown, N., Efklidou, S., Smallie, T., Karrar, S., Kaye, P. M. & Collins, M. K. (2009). Expression of vFLIP in a lentiviral vaccine vector activates NF-κB, matures dendritic cells, and increases CD8+ T-cell responses. J Virol 83, 1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini, S. M., Lapenta, C., Logozzi, M., Parlato, S., Spada, M., Di Pucchio, T. & Belardelli, F. (2000). Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J Exp Med 191, 1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman, K. & Liu, Y. J. (2002). Mouse and human dendritic cell subtypes. Nat Rev Immunol 2, 151–161. [DOI] [PubMed] [Google Scholar]
- Strong, J. E., Wong, G., Jones, S. E., Groller, A., Theriault, S., Koninger, G. P. & Feldmann, H. (2008). Stimulation of Ebola virus production from persistent infection through activation of the Ras/MAPK pathway. Proc Natl Acad Sci U S A 105, 17982–17987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda, K. & Akira, S. (2004). TLR signaling pathways. Semin Immunol 16, 3–9. [DOI] [PubMed] [Google Scholar]
- Towner, J. S., Rollin, P. E., Bausch, D. G., Sanchez, A., Crary, S. M., Vincent, M., Lee, W. F., Spiropoulou, C. F., Ksiazek, T. G. & other authors (2004). Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription-PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome. J Virol 78, 4330–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpooten, D., Ma, Y. J., Hou, S. W., Yan, Z. P. & He, B. (2008). Control of TANK binding kinase mediated signaling by the γ134.5 protein of herpes simplex virus 1. J Biol Chem 284, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield, K. L., Bosio, C. M., Welcher, B. C., Deal, E. M., Mohamadzadeh, M., Schmaljohn, A., Aman, M. J. & Bavari, S. (2003). Ebola virus-like particles protect from lethal Ebola virus infection. Proc Natl Acad Sci U S A 100, 15889–15894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield, K. L., Olinger, G., Deal, E. M., Swenson, D. L., Bailey, M., Negley, D. L., Hart, M. K. & Bavari, S. (2005). Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J Immunol 175, 1184–1191. [DOI] [PubMed] [Google Scholar]
- Warfield, K. L., Swenson, D. L., Olinger, G. G., Nichols, D. K., Pratt, W. D., Blouch, R., Stein, D. A., Aman, M. J., Iversen, P. L. & Bavari, S. (2006). Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog 2, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, L., Lin, J., Sun, Y., Bennouna, S., Lo, M., Wu, Q., Bu, Z., Pulendran, B., Compans, R. W. & Yang, C. (2006). Ebola virus-like particles produced in insect cells exhibit dendritic cell stimulating activity and induce neutralizing antibodies. Virology 351, 260–270. [DOI] [PubMed] [Google Scholar]