


Abstract
Dr1 (also known as NC2β) was identified as a repressor of RNA polymerase (pol) II transcription. It was subsequently shown to inhibit pol III transcription when expressed at high levels in vitro or in yeast cells. However, endogenous Dr1 was not detected at pol III-transcribed genes in growing yeast. In contrast, we demonstrate that endogenous Dr1 is present at pol III templates in human cells, as is its dimerization partner DRAP1 (also called NC2α). Expression of tRNA by pol III is selectively enhanced by RNAi-mediated depletion of endogenous human Dr1, but we found no evidence that DRAP1 influences pol III output in vivo. A stable association was detected between endogenous Dr1 and the pol III-specific transcription factor Brf1. This interaction may recruit Dr1 to pol III templates in vivo, as crosslinking to these sites increases following Brf1 induction. On the basis of these data, we conclude that the physiological functions of human Dr1 include regulation of pol III transcription.
INTRODUCTION
Dr1 (down-regulator of transcription 1, also known as NC2β—negative cofactor 2β) was first identified in HeLa nuclear extracts as an activity that binds TBP and represses pol II transcription (1,2). Later, it was recognized that Dr1 dimerizes with the cofactor DRAP1 (Dr1-associated protein 1, also known as NC2α) (3,4). Human Dr1/DRAP1 can repress pol II transcription in vitro and in vivo from a range of promoters (1–5). Dr1 and DRAP1 are also found in Saccharomyces cerevisiae, where they are required for viability and seem to function in a similar manner to the mammalian proteins (4,6–9). Indeed, the human Dr1 and DRAP1 genes can substitute for their yeast equivalents (10). Dr1 can exclude TFIIA and TFIIB from DNA-bound TBP, suggesting a model in which it binds TBP at the promoter and disrupts formation of a functional pre-initiation complex (1–4,11). In support of this, a crystal structure revealed the Dr1/DRAP1 heterodimer as a molecular clamp gripping the upper and lower surfaces of the TBP/DNA complex and thereby blocking binding by TFIIB and TFIIA (12). The heterodimer can also mobilize TBP along DNA (13) and interact with the largest subunit of elongating pol II (14). Despite targeting the general transcription machinery, only a subset of mRNAs responds to Dr1/DRAP1 in vivo. For example, only 17% of pol II-transcribed genes show changes in expression when DRAP1 is inactivated in yeast (15,16). Furthermore, many mRNAs are induced by Dr1/DRAP1, in contrast to original expectations (15–20).
The fact that TBP is required for transcription by pols I and III raised the possibility that these systems might respond to Dr1/DRAP1. Indeed, purified or recombinant human Dr1 was found to repress transcription of VA and tRNA genes in vitro by pol III, although transcription by pol I from an rRNA gene promoter did not respond (21). The same pattern was observed when Dr1 was overproduced in S. cerevisiae, with tRNA expression by pol III repressed, but rRNA expression by pol I unchanged (7). A mechanistic explanation for pol III control was suggested by binding assays with recombinant proteins, which showed that excess Dr1 can disrupt the interaction of TBP with Brf1, a polypeptide that recruits pol III to its templates (21). This is consistent with Brf1 bearing strong homology to TFIIB in its TBP-binding domain (22). However, the evidence for Dr1 as a regulator of pol III transcription was obtained when it was present at elevated levels (7,21). In contrast, ChIP experiments revealed minimal crosslinking of endogenous Dr1 or DRAP1 to tRNA genes in growing wild-type yeast (15,20), raising doubts concerning the physiological relevance of the overexpression data. Here, we have addressed this issue by RNAi and ChIP approaches in human cells. We show that endogenous Dr1 and DRAP1 associate with pol III-transcribed genes when present at natural levels in growing HeLa cells and that tRNA synthesis is suppressed by Dr1 in this situation. Our data provide clear evidence that the physiological functions of human Dr1 include tRNA gene regulation.
MATERIALS AND METHODS
Cell lines and culture
HeLa and HEK293T cells were cultured at 37°C and 5% CO2 in DMEM supplemented with 10% FCS, 2 mM l-glutamine, penicillin (100 U/ml) and streptomycin (100 U/ml). Brf1-inducible Chinese hamster ovary (CHO) Tet-Off cells (23) were cultured in DMEM supplemented with 10% doxycycline-free FCS (Clontech), 2 mM l-glutamine, 100 U/ml penicillin, 100 U/ml streptomycin, 100 μg/ml G418 sulphate and 2 μg/ml doxycycline. For Brf1 induction, cells were washed twice with PBS and cultured for 48 h without doxycycline. HeLa cells were subjected to heat shock by incubating at 45°C for 30 min. Cells were then either harvested immediately or left to recover at 37°C for 2, 4 or 8 h before harvesting. Control cells were not subjected to heat shock.
RNAi
HeLa cells were transfected with a shRNA pSUPER vector, encoding an insert with the sequence 5′-GAAGAAAGGCCAGTTCTCG-3′, targeting the mRNA of the second exon of the Dr1 gene or empty pSUPER vector (24), or with siRNA targeting the first (5′-GACUCUUCCUAAUGUCCGG-3′, Ambion) or third (5′-GCCCUAUAUGAAUUAACUG-3′, Ambion) exons of human Dr1. For the DRAP1 RNAi experiments, HEK293T cells were transfected with siRNA (Ambion) targeting the second exon (5′-CGGACGAAGAGAUUGGGAA-3′), third exon (5′-CUAGAGUCGCUGUUGAAGA-3′) or fifth exon (5′-CGGUGGAUGGGAACGAAA-3′) of human DRAP1. A validated, non-targeting siRNA (AM4390844, Ambion) was used as control. Transfections were performed using the Nucleofector system (Amaxa Biosystems) for the shRNA or lipofectamine 2000 (Invitrogen) for the siRNAs, according to the manufacturers’ recommendations. Cells were harvested 48 h after transfection.
RT–PCR
RNA was extracted using the TRI reagent (Sigma) according to manufacturer’s specifications. cDNA was synthesized by using 0.2 µg of RNA, random hexanucleotide mix (Roche) and Superscript III (Invitrogen), according to the manufacturers’ recommendations. RT–PCRs for ARPP P0, Brf1, TFIIB, 5S, U6, 7SL and tRNA transcripts were performed as previously described (25–29). RT–PCR for Alu RNA used primers 5′-CTTACACGTGTCATCCCAGC-3′ and 5′-GTAATTCTTTTGTAGAGACAGACTCAC-3′ to give a 113 bp product using the following cycling parameters: 95°C for 3 min, 27 cycles of (95°C for 30 s, 54°C for 30 s, 72°C for 30 s), 72°C for 5 min. RT–PCR for 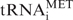 used primers 5′-AGCAGAGTGGCGCAGC-3′ and 5′-TTCGATCCATCGACCTCTG-3′ to give a 58 bp product using the following cycling parameters: 95°C for 3 min, 24 cycles of (95°C for 30 s, 54°C for 30 s, 72°C for 30 s), 72°C for 5 min. RT–PCR of Dr1 mRNA used primers 5′-AGAGCTGGTGGTGAACTGCT-3′ and 5′-CCAAGGTTTTCCAAACGAGA-3′ to give a 228 bp product using the following cycling parameters: 95°C for 3 min, 25 cycles of (95°C for 30 s, 58°C for 30 s, 72°C for 30 s), 72°C for 5 min. RT–PCR of DRAP1 mRNA used primers 5′-GGAACGAAAAGCAAGGACAA-3′ and 5′-CGTCCTCTTCATCAGGTGCT-3′ to give 226 bp product using the cycling parameters described above. Signal intensities were quantified by densitometry, normalized to the respective TFIIB or ARPP P0 signals and represented in graphs as the average fold increase or decrease, along with the standard deviations.
used primers 5′-AGCAGAGTGGCGCAGC-3′ and 5′-TTCGATCCATCGACCTCTG-3′ to give a 58 bp product using the following cycling parameters: 95°C for 3 min, 24 cycles of (95°C for 30 s, 54°C for 30 s, 72°C for 30 s), 72°C for 5 min. RT–PCR of Dr1 mRNA used primers 5′-AGAGCTGGTGGTGAACTGCT-3′ and 5′-CCAAGGTTTTCCAAACGAGA-3′ to give a 228 bp product using the following cycling parameters: 95°C for 3 min, 25 cycles of (95°C for 30 s, 58°C for 30 s, 72°C for 30 s), 72°C for 5 min. RT–PCR of DRAP1 mRNA used primers 5′-GGAACGAAAAGCAAGGACAA-3′ and 5′-CGTCCTCTTCATCAGGTGCT-3′ to give 226 bp product using the cycling parameters described above. Signal intensities were quantified by densitometry, normalized to the respective TFIIB or ARPP P0 signals and represented in graphs as the average fold increase or decrease, along with the standard deviations.
Western blotting
Whole cell extracts were prepared as previously (30). Western blotting was performed as described previously (31), using antibody sc-1615 against actin (Santa Cruz Biotechnology), antiserum 128 against Brf1 (23), antibodies D9390-01(US Biological) and sc-17272 (Santa Cruz Biotechnology) against DRAP1. Antisera 1162 and 1163 against Dr1 were raised by immunising rabbits with synthetic peptides ASSSGNDDDLTIPRA and SNQAESSQDEEDDDDI, corresponding to human Dr1 residues 2–16 and 161–176, respectively. Bands were quantified by densitometry, normalized to the respective actin signals and represented in graphs as the average fold increase or decrease, along with the standard deviations.
Co-immunoprecipitation
A total 300 μg of HeLa nuclear extract (Computer Cell Culture Centre) were incubated for 2 h rotating at 4°C with 5 μl anti-Dr1 (1162) and pre-immune serum in a final volume of 500 μl made up with microextraction buffer (150 mM NaCl, 50 mM NaF, 20 mM HEPES pH 7.8, 25% glycerol, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride, 0.2 mM EDTA, 40 μg/ml bestatin, 1 μg/ml trypsin inhibitor, 0.7 μg/ml pepstatin, 0.5 μg/ml aprotinin, 0.5 μg/ml leupeptin). Following that, 25 μl of packed protein G sepharose beads (Sigma) were used per immunoprecipitation and incubated rotating for 1 h at 4°C. The supernatant was then removed and the beads washed three times with 1 ml of PBS containing 0.05% Igepal (Sigma).
Brf1 was also in vitro transcribed and translated using the TNT reticulocyte lysate kit (Promega), according to the manufacturer’s instructions, and radiolabelled using 35S-methionine (Amersham Biosciences). HeLa nuclear extract and in vitro translated protein were pre-cleared and then incubated with the antibodies for 2 h rotating at 4°C, before 25 μl of protein A sepharose beads were added and the incubation continued for another hour. The beads were washed five times with TBS and the bound material was analyzed by SDS–PAGE and autoradiography.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (32), with the exception that a Bioruptor (UCD-200, Diagenode) sonicator was employed. The samples were sonicated at 4°C in two rounds of 15 min each (high power—320 W, 30 s on, 30 s off), with the water tank supplemented with crushed ice. Immunoprecipitations were carried out using antisera 1162 or 1163 or antibody Ab28185 (Abcam) against Dr1, antibodies TM-301A-55 (Austral Biologicals) and sc-17272 (Santa Cruz Biotechnology) against DRAP1, antibody MTBP-6 against TBP (33), antiserum 128 against Brf1 (34), antiserum 1900 against pol III (35). Pre-immune serum and normal goat (sc-2028), rabbit (sc-2027) or mouse (sc-2025) IgGs (all from Santa Cruz Biotechnology) were used as controls.
Immunoprecipitated DNA was analysed by PCR using primers and amplification conditions which have been previously described (26,32,36–39). For the amplification of Hsp70 DNA, the primers 5′-GGAGGTGCGGGAAGGTTCG-3′ and 5′-TTCTTGTCGGATGCTGGA-3′ were used to give a 187 bp product using the following cycling parameters: 95°C for 3 min, 28 cycles of (95°C for 30 s, 58°C for 30 s, 72°C for 30 s), 72°C for 5 min. Serial dilutions of input chromatin were used to establish that PCRs were in the linear range. Signal intensities were quantified by densitometry, normalized to input and represented in graphs as the average fold increase along with the standard deviations.
For sequential ChIP experiments, ∼5 × 107 cells per primary antibody were harvested and treated for ChIP as previously described (32). To reduce nonspecific binding, the primary antibodies were crosslinked to protein A beads (Sigma) with 10 mg/ml dimethyl pimelimidate •2 HCl (DMP). Specifically, 100 µl of protein A beads (50% slurry) were washed three times with 1% NP40/PBS, prior to incubating with ∼5 µg of antibody or control IgG (Sigma) for 2 h, rotating at 4°C. This was followed by three washes with 1% NP40/PBS and two washes with 100 mM HEPES–NaOH pH 8.5. The beads were then incubated with 10 mg/ml DMP in 100 mM HEPES–NaOH pH 8.5, for 1 h, rotating at room temperature, before being washed twice with 100 mM HEPES–NaOH pH 8.5 and incubated with 1 M glycine pH 7.5, for 30 min, rotating at room temperature. Following two washes with TE (10 mM Tris, 1 mM EDTA, pH 8.0), the beads were incubated overnight with the sonicated material, rotating at 4°C. After washes performed as previously (32), the immunoprecipitated material was eluted with 400 µl 1% SDS/TE, which was then 10-fold diluted with TE and subjected to another round of ChIP with the addition of ∼5 µg of secondary antibodies or the 1162 pre-immune serum (25 µl) and overnight incubation at 4°C.
RESULTS
Dr1 knock-down increases tRNA expression in human cells
Addition of a large excess of recombinant Dr1 to HeLa extracts was shown to inhibit transcription of pol III templates in vitro (21). We adopted an RNAi approach to test if endogenous human Dr1 influences expression of pol III transcripts in vivo. HeLa cells were transfected with siRNA targeting the first exon of Dr1 or a non-targeting control siRNA and harvested 48 h later. Western immunoblotting revealed that Dr1 protein levels were reduced by about 40% (Figure 1A and B), while the corresponding mRNA, as revealed by RT–PCR, was reduced by about 60% compared to the control (Figure 1C and D). Such treatment resulted in significant upregulation of each tRNA examined. This was the case for mature 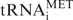 , as well as short-lived primary transcripts that reflect ongoing transcription (pre-tRNATyr and pre-tRNALeu). Alu RNA expression also increased, but 5S rRNA, U6 snRNA and 7SL RNA levels showed no significant response to the Dr1 siRNA (Figure 1C and D). Control experiments confirmed that assay conditions were not saturated and that changes in transcript levels gave a clear change in signal intensity for each primer set (Supplementary Figure S1).
, as well as short-lived primary transcripts that reflect ongoing transcription (pre-tRNATyr and pre-tRNALeu). Alu RNA expression also increased, but 5S rRNA, U6 snRNA and 7SL RNA levels showed no significant response to the Dr1 siRNA (Figure 1C and D). Control experiments confirmed that assay conditions were not saturated and that changes in transcript levels gave a clear change in signal intensity for each primer set (Supplementary Figure S1).
Figure 1.

Dr1 knock-down by RNAi results in upregulation of tRNA expression. HeLa cells were transfected with siRNA targeting the first exon of Dr1 and harvested 48 h later. A validated, non-targeting siRNA was used as control. (A) Western analysis for Dr1 from whole-cell extracts of control and Dr1 targeted cells. Actin was used as loading control. (B) Quantification of Dr1 western blotting signals from three independent experiments. (C) RT–PCR analysis of RNA expression. The pol II-transcribed ARPP P0 gene was used as control. (D) Quantification of the RT–PCR analysis signals from three independent experiments. 5S, LEU, TYR, MET and U6 refer to 5S rRNA, pre-tRNALeu, pre-tRNATyr, 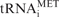 and U6 snRNA, respectively.
and U6 snRNA, respectively.
To validate these results, Dr1 was also depleted with a second siRNA and a shRNA (24) targeting exons 3 and 2, respectively. These gave comparable levels of Dr1 knockdown to the siRNA against exon 1 and produced identical effects on pol III transcript expression (Supplementary Figures S2 and S3). It is therefore unlikely that the observed responses reflect off-target effects, as they were obtained by targeting three independent regions of the Dr1 sequence. It is worth noting that the outcome was unchanged whether we normalized against ARPP P0, GAPDH or TFIIB mRNAs. Our data suggest that endogenous Dr1 is an inhibitor of tRNA expression, but does not show the more general pol III regulatory properties that we had expected. Thus, as found for pol II transcription (19,40), the effects of Dr1 on pol III output may be gene-selective in vivo.
Dr1 is found at pol III-transcribed genes in human cells
Although Dr1 and DRAP1 have been shown to be present at pol II-transcribed genes (15,41,42,43), they were not found at pol III templates in wild-type yeast (15,20). Nevertheless, ChIP experiments revealed the presence of endogenous human Dr1 at pol III-transcribed genes in HeLa cells (Figure 2A). Not only was Dr1 detected at tRNA genes, but it was also detected at 5S rRNA and U6 snRNA genes, expression of which was unperturbed by siRNAs and shRNA against Dr1. Binding is specific, as it is not observed further upstream of the U6 gene or at a site within the coding region of the ARPP P0 gene. It was confirmed with an alternative Dr1 antibody (Figure 2B) and in a different human cell type, the osteosarcoma line U2OS (data not shown). Furthermore, occupancy seems comparable to that detected at two pol II promoters that were used as positive controls, as judged by comparison with the TBP signal. Thus, the Dr1 ChIP signals were ∼50% and ∼70%, respectively, as strong as the TBP ChIP signals at the Hsp70 and U1 snRNA promoters, and the Dr1 signals at 5S, tRNA and U6 genes were ∼50% of the TBP signals (Supplementary Figure S4). Because the efficiencies of the antibodies may differ, such a comparison of ChIP signals does not reveal what proportion of TBP molecules are bound by Dr1 at these sites, but it does suggest that the pol III-transcribed genes examined are targeted as strongly by Dr1 as the Hsp70 promoter. We conclude that, in contrast to the situation in yeast (15,20), endogenous Dr1 can be readily detected at pol III-transcribed genes in human cells.
Figure 2.

Dr1 is found at pol III-transcribed genes. HeLa cells were used for ChIP with Dr1 1162 (A) and Ab28185 (B) antibodies, together with the 1162 pre-immune (PI) serum and normal rabbit IgGs as negative controls. Brf1 and pol III antibodies were used as positive controls for pol III-transcribed genes and TBP as positive control for pol III- and pol II-transcribed genes. Input lanes show 10%, 2% and 0.4% of total input.
Endogenous Dr1 associates with TFIIIB
To be recruited to pol III promoters without TATA boxes, such as the 5S and tRNA genes examined here, TBP binds to Brf1, a TFIIB-homologous polypeptide that is anchored to promoters through the DNA-binding factor TFIIIC (22). A model was proposed by White et al. (21) in which Dr1 binding to TBP prevents its interaction with Brf1 and thereby blocks TBP recruitment to TATA-less pol III templates (Figure 3A). This model is contradicted by the clear ChIP signal that we detect for Dr1 at tRNA and 5S rRNA genes (Figure 2). We therefore examined a second prediction of the model, which was that Dr1 and Brf1 would not be present in the same complex (Figure 3A). However, when mixed with a HeLa cell extract, 35S-radiolabelled Brf1 can be co-immunoprecipitated with Dr1 (Figure 3B). Furthermore, a stable association of endogenous Dr1 and Brf1 from HeLa cells can also be detected (Figure 3C). To investigate the significance of this interaction, we used stably-transfected cells in which expression of Brf1 is controlled by a doxycycline-sensitive promoter (23). Induction of Brf1 by doxycycline withdrawal was found to increase crosslinking of TBP and Dr1, as well as Brf1 itself, to tRNA and 5S rRNA genes (Figure 3D). Specificity was established using preimmune serum and control IgG. Western blotting confirmed that the increased Dr1 ChIP signal following Brf1 induction is not due to an increase in its abundance (Figure 3E). These data suggest that Brf1 can mediate recruitment of Dr1 to tRNA and 5S rRNA genes in vivo, although they do not establish if this reflects a direct interaction. In support of Brf1-mediated recruitment, sequential ChIP (re-ChIP) experiments demonstrated simultaneous promoter association by Brf1 and Dr1, as well as by Dr1 and TBP (Figure 3F). As positive control, we confirmed the expected gene co-occupancy of pol III with Brf1 and TBP.
Figure 3.

Dr1 associates with Brf1. (A) Schematic model of how the pol III initiation complex was predicted to be disrupted by Dr1 (21). (B) Brf1 was in vitro translated and radio-labelled with 35S, mixed with HeLa nuclear extracts and then used in co-IP experiments with Dr1 1162 antiserum and pre-immune serum (PI). (C) Antiserum for Dr1 (1162) was used to co-immunoprecipitate endogenous Brf1 from HeLa nuclear extracts. 1162 pre-immune serum (PI) was used as control. (D) Overexpression of Brf1 results in increased Dr1 crosslinking at pol III-transcribed genes. CHO cells, stably transfected with an inducible Brf1 TET-OFF expression system (23), were induced to express Brf1 for 48 h before harvesting. ChIP experiments were performed using antibodies against TBP, Brf1 and Dr1 (1162). Pre-immune (PI) serum and rabbit IgGs were used as negative controls. The ‘+’ denotes induction of Brf1 compared to the uninduced ‘−’ control. (E) Western blot demonstrating that Dr1 protein levels do not increase after induction of Brf1 in the CHO cell Brf1 inducible system. (F) DNA recovered from ChIPs carried out using rabbit IgGs or antibodies against Brf1 and TBP was used for a second ChIP with preimmune serum or sera against Dr1 and pol III, as indicated. Input lanes show 10% of total input employed for the secondary ChIP.
Endogenous DRAP1 is present at pol III-transcribed genes
Since Dr1 is known to associate with DRAP1, we used ChIP to examine whether the latter can also be detected at pol III-transcribed genes in human cells. This was found to be the case, with endogenous DRAP1 crosslinking to 5S rRNA, tRNA and U6 snRNA genes in vivo (Figure 4A). The interaction appears specific, as it was not detected upstream of a U6 snRNA gene or within the coding region of the ARPP P0 gene. Moreover, it was confirmed using an alternative antibody against DRAP1 (Figure 4B). We conclude that endogenous DRAP1 is also present at pol III-transcribed genes in HeLa cells. A ChIP-on-chip study carried out previously with human B cells did not report DRAP1 at pol III-transcribed genes, but this can be readily explained by the fact that the study focused specifically on pol II promoter regions (44).
Figure 4.

DRAP1 occupancy at pol III-transcribed genes. HeLa cells were used for ChIP. (A) A polyclonal DRAP1 antibody (sc-17272) was used to test for DRAP1 presence at pol III-transcribed genes. TBP, Brf1 and pol III antibodies were used as positive controls. Goat IgGs and beads without antibody were used as negative controls. Input lanes show 10%, 2% and 0.4% of total input. (B) A monoclonal DRAP1 antibody (TM-301B-55) was used to confirm DRAP1 presence at pol III-transcribed genes, along with the Dr1 antibody 1162. TBP, Brf1 and pol III antibodies were used as positive controls; mouse IgGs and pre-immune (PI) serum were used as negative controls. Input lanes show 10%, 2% and 0.4% of total input.
RNAi was used to assess the influence of DRAP1 on pol III transcription. However, DRAP1 depletion (Figure 5A and B) had minimal effects on expression of pol III products, including the pre-tRNAs that responded clearly to knockdown of Dr1 (Figure 5C and D). The lack of response was not because assay conditions were saturated, since control experiments confirmed that changes in transcript levels gave a clear change in signal intensity for each primer set (Supplementary Figure S5). As a positive control, we examined expression of Bcl2 mRNA and confirmed that this responded to DRAP1 depletion in our assays, as previously reported (19). Three different siRNAs targeting three different exons of DRAP1 were analysed, but although DRAP1 protein levels were reduced by about 75%, only marginal changes in pre-tRNA and other pol III transcript levels were obtained, that were not consistent and did not reach statistical significance (Figure 5 and Supplementary Figures S6 and S7). Furthermore, a pool of three additional siRNAs and a shRNA targeting different regions of DRAP1 were also tested, resulting in similar outcomes (data not shown). After utilising seven different siRNA and shRNA in both HeLa and HEK293T cells, we conclude that, at least under the conditions used, DRAP1 depletion has minimal effect on the expression of pol III transcripts. This is reminiscent of the pol II-dependent Egr1 promoter, which responds to depletion of Dr1, but not of DRAP1 (19).
Figure 5.

DRAP1 knock-down by RNAi does not affect expression of pol III transcripts. HEK293T cells were transfected with siRNA agaist DRAP1 exon 2 and harvested 48 h later. A validated, non-coding siRNA was used as control. (A) Western analysis for DRAP1 from control and DRAP1-targeted cells. Actin was used as loading control. (B) Quantification of DRAP1 western blotting signals from three independent experiments. (C) RT–PCR analysis of RNA expression. The mRNAs encoding Bcl2, TFIIB and ARPP P0 were used as controls. (D) Quantification of the RT–PCR analysis signals from three independent experiments. 5S, LEU, TYR and U6 refer to 5S rRNA, pre-tRNALeu, pre-tRNATyr and U6 snRNA, respectively.
In addition to examining the effect of DRAP1 depletion, we also considered whether elevated DRAP1 levels might influence pol III output in vivo. We found that heat shock provokes a ∼5-fold increase in DRAP1 protein with minimal change in Dr1 levels (Figure 6A and B). This response appears to be post-transcriptional, as DRAP1 mRNA expression is almost unchanged (Figure 6C and D). Heat shock therefore provides an opportunity to examine whether a robust, physiological increase in DRAP1 levels is accompanied by inhibition of pol III activity. This was not the case. No decrease was detected in any of the pol III transcripts examined; indeed, expression of pre-tRNATyr increased ∼2-fold after heat shock, whilst 5S rRNA and pre-tRNALeu showed slight and transient induction (Figure 6C and E). Our data therefore provide no evidence that DRAP1 is a repressor of pol III transcription under the conditions we have studied. Such a role under other circumstances can clearly not be excluded.
Figure 6.

DRAP1 protein levels are upregulated after heat shock but do not repress the expression of pol III transcripts. HeLa cells were subjected to heat shock at 45°C for 30 min, and then either harvested immediately (0 h) or left to recover for 2, 4 or 8 h prior to harvesting. Control cells were not subjected to heat shock. (A) Western analysis for DRAP1 and Dr1 after heat-shock. Actin was used as loading control. (B) Quantification of DRAP1 and Dr1 western blotting signals from three independent experiments. (C) RT–PCR analysis for DRAP1 mRNA, Dr1 mRNA and pol III transcripts after heat-shock. ARPP P0 mRNA was used as control. (D) Quantification of the RT–PCR signals for DRAP1 and Dr1 mRNAs from three independent experiments. (E) Quantification of the RT–PCR signals for pol III transcripts from three independent experiments. 5S, LEU, TYR and U6 refer to 5S rRNA, pre-tRNALeu, pre-tRNATyr and U6 snRNA, respectively.
DISCUSSION
Recombinant human Dr1 was shown previously to inhibit tRNA gene transcription in vitro (21). The assays, however, required a large excess of Dr1 protein and therefore left open the question of physiological relevance. Indeed, Dr1 was found to suppress tRNA expression when produced at elevated levels in yeast (7), even though endogenous Dr1 is not detected at tRNA genes when wild-type yeast are grown under standard conditions (20). Nevertheless, our ChIP assays reveal that human tRNA genes are occupied by endogenous Dr1 in HeLa cells, without the need for overexpression. Association is specific and was confirmed using alternative antibodies and in a different human cell type. Quantitation of binding by qPCR confirms that it is highly significant (P < 0.0001; Supplementary Figure S8). Furthermore, RNAi reveals that transcription of these tRNA genes is suppressed ∼2-fold by physiological levels of Dr1. This response is unlikely to reflect off-target effects, as it was reproduced using shRNA and siRNAs directed against three different regions of the Dr1 sequence. We conclude that the functions of human Dr1 include the suppression of tRNA expression.
As all pol III-transcribed genes are thought to utilize TBP, we had expected them all to respond to Dr1. We were therefore surprised that expression of 5S rRNA, U6 snRNA and 7SL RNA was unchanged by Dr1 RNAi. The corresponding genes clearly associate with Dr1, as revealed by ChIP. It may be relevant that 5S, U6 and 7SL genes all have distinct promoter organization and more complex transcription factor requirements than the simpler tRNA and Alu genes (22), which might influence their responsiveness in some way. Promoter sequence has been shown to influence responsiveness of pol II promoters to Dr1/DRAP1 (18,19,40). Perhaps 5S, U6 and 7SL genes would respond to more complete depletion of Dr1 than we have been able to achieve. However, more extreme depletion is likely to increase secondary effects that would be difficult to interpret with confidence. It is also possible that transcriptional changes in fact occur, but are not reflected in the steady-state transcript levels. Because 5S, U6 and 7SL RNAs are relatively abundant and stable, their abundance is probably much less responsive to altered rates of transcription than short-lived transcripts, such as pre-tRNAs.
We cannot exclude the possibility that the effect of Dr1 on tRNA levels in vivo is mediated indirectly through its ability to regulate pol II transcription. Clearly, pol III transcription factors depend on pol II for their expression. However, the partial depletion of Dr1 in our experiments is not causing global changes to mRNA expression. We found no significant alteration to the levels of mRNAs encoding TFIIIB and TFIIIC subunits under RNAi conditions that are sufficient to induce tRNA (Supplementary Figure S9). This is consistent with the selectivity of the response, as altered expression of pol III or its factors might be expected to affect a broad range of pol III products (all the transcription machinery known to be required by tRNA genes is also required by 5S rRNA genes, for example). Although indirect effects may also occur, our data provide clear evidence that human Dr1 interacts with TFIIIB and pol III-transcribed genes in vivo and therefore support its function as a direct regulator of these targets, consistent with its pol III inhibitory effects in vitro with a partially-purified and reconstituted system (21).
Structural considerations predict strongly that Dr1 will obstruct binding of Brf1 to TBP. Two separate domains of Brf1 interact with TBP, one composed of cyclin folds and the other referred to as ‘homology II’ (45). Mutagenesis, crosslinking and modelling suggest that the cyclin fold domain of Brf1 grasps the C-terminal stirrup of TBP in a manner similar to the cyclin folds of TFIIB, with which it bears homology (45). Crystallography shows that this interaction between TFIIB and TBP is blocked by the penultimate helix of Dr1 (12). The adjacent helix of Dr1 traverses the convex surface of TBP and is likely to cross the region occupied by Brf1 homology domain II (46). This is supported by the ability of recombinant Dr1 to displace TBP from a Brf1 fragment containing homology domain II (21). These observations predict that Dr1-bound TBP should dissociate from Brf1 and be released from TATA-less promoters, where TBP is anchored by Brf1. Our current data, however, show otherwise. Perhaps interactions with the third TFIIIB subunit, Bdp1, allow Dr1-bound TBP to be retained at the promoter despite a disrupted interaction with Brf1 (Figure 7A). Binding to Brf1 itself might also explain the presence of Dr1 at pol III-transcribed genes. A putative interaction with Brf1 could occur that does not disrupt the interface with TBP or inhibit expression. Repression might follow the transfer of Dr1 to TBP, perhaps triggered by phosphorylation changes to Brf1, TBP and/or Dr1 (Figure 7B). Ample precedent exists for the presence of Dr1 at active pol II promoters (15,16,19,20,41), even though its documented interaction with TBP is incompatible with current models of pol II transcription. Further structural studies will be required to elucidate the disposition of Dr1 in active transcription complexes.
Figure 7.

Models to explain the presence of Dr1 at TATA-less pol III templates. (A) The TFIIIB subunit Bdp1 might provide interactions with TBP that allow it to be retained at promoters when Dr1 disrupts its interface with Brf1. (B) Dr1 might be able to bind Brf1 in a manner that is compatible with transcription. Transfer of Dr1 to TBP might disrupt the TBP/Brf1 interface and repress transcription. Such transfer might be triggered by changes in post-translational modification; a hypothetical dephosphosphorylation of Dr1 is illustrated as an example. P denotes protein phosphorylation.
The Dr1/DRAP1 complex (NC2) can mobilize TBP on DNA, an effect that may have positive or negative transcriptional effects, depending on context (13). This activity is not displayed by Dr1 or DRAP1 individually (13). Endogenous DRAP1 is clearly detected at pol III templates (P = 0.004; Supplementary Figure S8), presumably due to its dimerization with Dr1. However, DRAP1 depletion by RNAi did not reproducibly affect expression of pol III products. Furthermore, these transcripts were not repressed when heat shock triggered a ∼5-fold increase in DRAP1 protein levels. DRAP1 may therefore be a passive partner of Dr1 at pol III-transcribed genes, with no influence on their expression. This would imply that TBP mobilization is not required for Dr1 to regulate pol III transcription. Dr1 is known to be able to function independently of DRAP1 in vivo (7,41). Nevertheless, a role for DRAP1 in pol III control has not been excluded and might become apparent under different experimental or physiological conditions. Precedent is provided in yeast, where Dr1/DRAP1 represses certain pol II promoters after diauxic shift, but not during exponential growth (9,41).
In contrast to our findings in HeLa cells, little or no crosslinking of endogenous Dr1 or DRAP1 to tRNA genes has been detected in S. cerevisiae (15,20). However, Dr1 is capable of repressing tRNA expression when overexpressed in this yeast (7). A potential explanation is provided by the observations that Mot1 is found at yeast pol III-transcribed genes and has been shown to inhibit promoter association by Dr1 (47). Natural levels of Dr1 might therefore be excluded from these sites by Mot1, whereas overexpressed Dr1 may compete more effectively. Indeed, although the ChIP signal for Dr1 and DRAP1 is close to background at a tRNA gene in wild-type yeast, it increases significantly in a mot1 mutant strain (47).
Pol III output is strongly linked with the growth state of cells (48,49). Indeed, elevated expression of 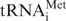 in immortalized murine fibroblasts is sufficient to stimulate proliferation and induce oncogenic transformation (23). Our data show that levels of this tRNA increase in response to Dr1 depletion. It is therefore conceivable that pol III control may provide a link between Dr1 and the proliferative and oncogenic status of mammalian cells. Dr1 expression was found to be significantly compromised in 74% of the 58 cases examined of neuroblastoma, usually due to genomic deletion (50). Little or no change in DRAP1 was detected in any of the samples (50). Our data suggest that the reduced Dr1 levels in these early childhood tumours may result in elevated tRNA expression.
in immortalized murine fibroblasts is sufficient to stimulate proliferation and induce oncogenic transformation (23). Our data show that levels of this tRNA increase in response to Dr1 depletion. It is therefore conceivable that pol III control may provide a link between Dr1 and the proliferative and oncogenic status of mammalian cells. Dr1 expression was found to be significantly compromised in 74% of the 58 cases examined of neuroblastoma, usually due to genomic deletion (50). Little or no change in DRAP1 was detected in any of the samples (50). Our data suggest that the reduced Dr1 levels in these early childhood tumours may result in elevated tRNA expression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wellcome Trust (081977/Z/07/Z); Wellcome Trust PhD program Molecular Functions in Disease (to T.K.); Cancer Research UK (to R.J.W.). Funding for open access charge: Wellcome Trust (081977/Z/07/Z).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Stefan Roberts for helpful discussion of this work.
REFERENCES
- 1.Inostroza JA, Mermelstein FH, Ha I, Lane WS, Reinberg D. Dr1, a TATA-binding protein-associated phosphoprotein and inhibitor of class II gene transcription. Cell. 1992;70:477–489. doi: 10.1016/0092-8674(92)90172-9. [DOI] [PubMed] [Google Scholar]
- 2.Meisterernst M, Roeder RG. Family of proteins that interact with TFIID and regulate promoter activity. Cell. 1991;67:557–567. doi: 10.1016/0092-8674(91)90530-c. [DOI] [PubMed] [Google Scholar]
- 3.Mermelstein F, Yeung K, Cao J, Inostroza JA, Erdjument-Bromage H, Eagelson K, Landsman D, Levitt P, Tempst P, Reinberg D. Requirement of a corepressor for Dr1-mediated repression of transcription. Genes Dev. 1996;10:1033–1048. doi: 10.1101/gad.10.8.1033. [DOI] [PubMed] [Google Scholar]
- 4.Goppelt A, Meisterernst M. Characterization of the basal inhibitor of class II transcription NC2 from Saccharomyces cerevisiae. Nucleic Acids Res. 1996;24:4450–4455. doi: 10.1093/nar/24.22.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraus VB, Inostroza JA, Yeung K, Reinberg D, Nevins JR. Interaction of the Dr1 inhibitory factor with the TATA binding protein is disrupted by adenovirus E1A. Proc. Natl Acad. Sci. USA. 1994;91:6279–6282. doi: 10.1073/pnas.91.14.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prelich G. Saccharomyces cerevisiae BUR6 encodes a DRAP1/NC2alpha homolog that has both positive and negative roles in transcription in vivo. Mol. Cell Biol. 1997;17:2057–2065. doi: 10.1128/mcb.17.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim S, Na JG, Hampsey M, Reinberg D. The Dr1/DRAP1 heterodimer is a global repressor of transcription in vivo. Proc. Natl Acad. Sci. USA. 1997;94:820–825. doi: 10.1073/pnas.94.3.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gadbois EL, Chao DM, Reese JC, Green MR, Young RA. Functional antagonism between RNA polymerase II holoenzyme and global negative regulator NC2 in vivo. Proc. Natl Acad. Sci. USA. 1997;94:3145–3150. doi: 10.1073/pnas.94.7.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemaire M, Xie J, Meisterernst M, Collart MA. The NC2 repressor is dispensable in yeast mutated for the Sin4p component of the holoenzyme and plays roles similar to Mot1p in vivo. Mol. Microbiol. 2000;36:163–173. doi: 10.1046/j.1365-2958.2000.01839.x. [DOI] [PubMed] [Google Scholar]
- 10.Yeung KC, Inostroza JA, Mermelstein FH, Kannabiran C, Reinberg D. Structure-function analysis of the TBP-binding protein Dr1 reveals a mechanism for repression of class II gene transcription. Genes Dev. 1994;8:2097–2109. doi: 10.1101/gad.8.17.2097. [DOI] [PubMed] [Google Scholar]
- 11.Yeung K, Kim S, Reinberg D. Functional dissection of a human Dr1-DRAP1 repressor complex. Mol. Cell Biol. 1997;17:36–45. doi: 10.1128/mcb.17.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamada K, Shu F, Chen H, Malik S, Stelzer G, Roeder RG, Meisterernst M, Burley SK. Crystal structure of negative cofactor 2 recognizing the TBP-DNA transcription complex. Cell. 2001;106:71–81. doi: 10.1016/s0092-8674(01)00417-2. [DOI] [PubMed] [Google Scholar]
- 13.Schluesche P, Stelzer G, Piaia E, Lamb DC, Meisterernst M. NC2 mobilizes TBP on core promoter TATA boxes. Nat. Struct. Mol. Biol. 2007;14:1196–1201. doi: 10.1038/nsmb1328. [DOI] [PubMed] [Google Scholar]
- 14.Castano E, Gross P, Wang Z, Roeder RG, Oelgeschlager T. The C-terminal domain-phosphorylated IIO form of RNA polymerase II is associated with the transcription repressor NC2 (Dr1/DRAP1) and is required for transcription activation in human nuclear extracts. Proc. Natl Acad. Sci. USA. 2000;97:7184–7189. doi: 10.1073/pnas.140202297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geisberg JV, Holstege FC, Young RA, Struhl K. Yeast NC2 associates with the RNA polymerase II preinitiation complex and selectively affects transcription in vivo. Mol. Cell Biol. 2001;21:2736–2742. doi: 10.1128/MCB.21.8.2736-2742.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cang Y, Prelich G. Direct stimulation of transcription by negative cofactor 2 (NC2) through TATA-binding protein (TBP) Proc. Natl Acad. Sci. USA. 2002;99:12727–12732. doi: 10.1073/pnas.202236699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu JY, Juven-Gershon T, Marr MT, 2nd, Wright KJ, Tjian R, Kadonaga JT. TBP, Mot1, and NC2 establish a regulatory circuit that controls DPE-dependent versus TATA-dependent transcription. Genes Dev. 2008;22:2353–2358. doi: 10.1101/gad.1681808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willy PJ, Kobayashi R, Kadonaga JT. A basal transcription factor that activates or represses transcription. Science. 2000;290:982–985. doi: 10.1126/science.290.5493.982. [DOI] [PubMed] [Google Scholar]
- 19.Deng W, Malecova B, Oelgeschlager T, Roberts SG. TFIIB recognition elements control the TFIIA-NC2 axis in transcriptional regulation. Mol. Cell Biol. 2009;29:1389–1400. doi: 10.1128/MCB.01346-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Werven FJ, van Bakel H, van Teeffelen HA, Altelaar AF, Koerkamp MG, Heck AJ, Holstege FC, Timmers HT. Cooperative action of NC2 and Mot1p to regulate TATA-binding protein function across the genome. Genes Dev. 2008;22:2359–2369. doi: 10.1101/gad.1682308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White RJ, Khoo BC, Inostroza JA, Reinberg D, Jackson SP. Differential regulation of RNA polymerases I, II, and III by the TBP-binding repressor Dr1. Science. 1994;266:448–450. doi: 10.1126/science.7939686. [DOI] [PubMed] [Google Scholar]
- 22.Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002;16:2593–2620. doi: 10.1101/gad.1018902. [DOI] [PubMed] [Google Scholar]
- 23.Marshall L, Kenneth NS, White RJ. Elevated tRNA(iMet) synthesis can drive cell proliferation and oncogenic transformation. Cell. 2008;133:78–89. doi: 10.1016/j.cell.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 24.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 25.Larminie CG, Sutcliffe JE, Tosh K, Winter AG, Felton-Edkins ZA, White RJ. Activation of RNA polymerase III transcription in cells transformed by simian virus 40. Mol. Cell Biol. 1999;19:4927–4934. doi: 10.1128/mcb.19.7.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winter AG, Sourvinos G, Allison SJ, Tosh K, Scott PH, Spandidos DA, White RJ. RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc. Natl Acad. Sci. USA. 2000;97:12619–12624. doi: 10.1073/pnas.230224097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daly NL, Arvanitis DA, Fairley JA, Gomez-Roman N, Morton JP, Graham SV, Spandidos DA, White RJ. Deregulation of RNA polymerase III transcription in cervical epithelium in response to high-risk human papillomavirus. Oncogene. 2005;24:880–888. doi: 10.1038/sj.onc.1208031. [DOI] [PubMed] [Google Scholar]
- 28.Felton-Edkins ZA, White RJ. Multiple mechanisms contribute to the activation of RNA polymerase III transcription in cells transformed by papovaviruses. J. Biol. Chem. 2002;277:48182–48191. doi: 10.1074/jbc.M201333200. [DOI] [PubMed] [Google Scholar]
- 29.Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 30.Innes F, Ramsbottom B, White RJ. A test of the model that RNA polymerase III transcription is regulated by selective induction of the 110 kDa subunit of TFIIIC. Nucleic Acids Res. 2006;34:3399–3407. doi: 10.1093/nar/gkl432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White RJ, Gottlieb TM, Downes CS, Jackson SP. Mitotic regulation of a TATA-binding-protein-containing complex. Mol. Cell Biol. 1995;15:1983–1992. doi: 10.1128/mcb.15.4.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421:290–294. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- 33.Pruzan R, Chatterjee PK, Flint SJ. Specific transcription from the adenovirus E2E promoter by RNA polymerase III requires a subpopulation of TFIID. Nucleic Acids Res. 1992;20:5705–5712. doi: 10.1093/nar/20.21.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cairns CA, White RJ. p53 is a general repressor of RNA polymerase III transcription. EMBO J. 1998;17:3112–3123. doi: 10.1093/emboj/17.11.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fairley JA, Scott PH, White RJ. TFIIIB is phosphorylated, disrupted and selectively released from tRNA promoters during mitosis in vivo. EMBO J. 2003;22:5841–5850. doi: 10.1093/emboj/cdg544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fairley JA, Kantidakis T, Kenneth NS, Intine RV, Maraia RJ, White RJ. Human La is found at RNA polymerase III-transcribed genes in vivo. Proc. Natl Acad. Sci. USA. 2005;102:18350–18355. doi: 10.1073/pnas.0506415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodfellow SJ, Graham EL, Kantidakis T, Marshall L, Coppins BA, Oficjalska-Pham D, Gerard M, Lefebvre O, White RJ. Regulation of RNA polymerase III transcription by Maf1 in mammalian cells. J. Mol. Biol. 2008;378:481–491. doi: 10.1016/j.jmb.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 38.Schramm L, Pendergrast PS, Sun Y, Hernandez N. Different human TFIIIB activities direct RNA polymerase III transcription from TATA-containing and TATA-less promoters. Genes Dev. 2000;14:2650–2663. doi: 10.1101/gad.836400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan CC, Zhao X, Florens L, Swanson SK, Washburn MP, Hernandez N. CHD8 associates with human Staf and contributes to efficient U6 RNA polymerase III transcription. Mol. Cell Biol. 2007;27:8729–8738. doi: 10.1128/MCB.00846-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malecova B, Gross P, Boyer-Guittaut M, Yavuz S, Oelgeschlager T. The initiator core promoter element antagonizes repression of TATA-directed transcription by negative cofactor NC2. J. Biol. Chem. 2007;282:24767–24776. doi: 10.1074/jbc.M702776200. [DOI] [PubMed] [Google Scholar]
- 41.Creton S, Svejstrup JQ, Collart MA. The NC2 alpha and beta subunits play different roles in vivo. Genes Dev. 2002;16:3265–3276. doi: 10.1101/gad.234002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masson P, Leimgruber E, Creton S, Collart MA. The dual control of TFIIB recruitment by NC2 is gene specific. Nucleic Acids Res. 2008;36:539–549. doi: 10.1093/nar/gkm1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christova R, Oelgeschlager T. Association of human TFIID-promoter complexes with silenced mitotic chromatin in vivo. Nat. Cell Biol. 2002;4:79–82. doi: 10.1038/ncb733. [DOI] [PubMed] [Google Scholar]
- 44.Albert TK, Grote K, Boeing S, Stelzer G, Schepers A, Meisterernst M. Global distribution of negative cofactor 2 subunit-alpha on human promoters. Proc. Natl Acad. Sci. USA. 2007;104:10000–10005. doi: 10.1073/pnas.0703490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kassavetis GA, Geiduschek EP. Transcription factor TFIIIB and transcription by RNA polymerase III. Biochem. Soc. Trans. 2006;34:1082–1087. doi: 10.1042/BST0341082. [DOI] [PubMed] [Google Scholar]
- 46.Juo ZS, Kassavetis GA, Wang J, Geiduschek EP, Sigler PB. Crystal structure of a transcription factor IIIB core interface ternary complex. Nature. 2003;422:534–539. doi: 10.1038/nature01534. [DOI] [PubMed] [Google Scholar]
- 47.Geisberg JV, Moqtaderi Z, Kuras L, Struhl K. Mot1 associates with transcriptionally active promoters and inhibits association of NC2 in Saccharomyces cerevisiae. Mol. Cell Biol. 2002;22:8122–8134. doi: 10.1128/MCB.22.23.8122-8134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marshall L, White RJ. Non-coding RNA production by RNA polymerase III is implicated in cancer. Nat. Rev. Cancer. 2008;8:911–914. doi: 10.1038/nrc2539. [DOI] [PubMed] [Google Scholar]
- 49.Johnson DL, Johnson SA. Cell biology. RNA metabolism and oncogenesis. Science. 2008;320:461–462. doi: 10.1126/science.1158680. [DOI] [PubMed] [Google Scholar]
- 50.Di Pietro C, Ragusa M, Barbagallo D, Duro LR, Guglielmino MR, Majorana A, Giunta V, Rapisarda A, Tricarichi E, Miceli M, et al. Involvement of GTA protein NC2β in neuroblastoma pathogenesis suggests that it physiologically participates in the regulation of cell proliferation. Mol. Cancer. 2008;7:52. doi: 10.1186/1476-4598-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.