

Abstract
Glycogen synthase kinase 3 beta (GSK-3β) is constantly active in cells and its activity increases after serum deprivation, indicating that GSK-3β might play a major role in cell survival under serum starvation. In this study, we attempted to determine how GSK-3β promotes cell survival after serum depletion. Under full culture conditions (10% FBS), GSK-3β inhibition with chemical inhibitors or siRNAs failed to induce cell death in human prostate cancer cells. By contrast, under conditions of serum starvation, a profound necrotic cell death was observed as evidenced by cellular morphologic features and biochemical markers. Further analysis revealed that GSK-3β-inhibition-induced cell death was in parallel with an extensive autophagic response. Interestingly, blocking the autophagic response switched GSK-3β-inhibition-induced necrosis to apoptotic cell death. Finally, GSK-3β inhibition resulted in a remarkable elevation of Bif-1 protein levels, and silencing Bif-1 expression abrogated GSK-3β-inhibition-induced autophagic response and cell death. Taken together, our study suggests that GSK-3β promotes cell survival by modulating Bif-1-dependent autophagic response and cell death.
Keywords: Bif-1, GSK-3β, Necrosis, Apoptosis, Autophagy
Introduction
Autophagy is a lysosome-based catalytic process mainly activated in cells under metabolic stress (Klionsky and Emr, 2000). In the early phase, autophagy generates additional energy supplies for starved cells, which maintains cell survival, while sustained cellular digestion through the process of autophagic cycling eventually results in cell death, known as autophagy-dependent cell death (Kourtis and Tavernarakis, 2009). Recently, a large body of literature demonstrated that there are multiple interchanges at different molecular levels between autophagy and apoptosis pathways (Maiuri et al., 2007).
Currently, little is known about the genetic control of necrotic cell death. Traditionally, necrosis was considered as a passive form of cell death resulting from physical damage or toxic insults (Walker et al., 1988). However, new evidence that has emerged in the past few years has shown that necrosis might not be an accidental form of cell death, but a programmed cell death with evolutionarily design and a distinctive biochemical cascade (Hitomi et al., 2008). In certain experimental settings where apoptotic machinery is blocked by pharmacological or genetic means, apoptotic stimuli induce necrotic cell death by activating intracellular signaling pathways (Chan et al., 2003; Zong et al., 2004; Okada et al., 2004; Degterev et al., 2005). In an elegant model system of atg1-deficient Dictyostelium cells in which the machinery for both apoptosis and autophagy is destroyed, irreversible lysosomal permeabilization was identified as the causal factor for necrotic cell death after mitochondrial uncoupling (Giusti et al., 2009). Consistently, in a genetically modified C. elegans model, the requirement of a lysosomal-dependent autophagic response was confirmed for necrotic cell death (Samara et al., 2008). These data suggested that lysosomal damage plays a critical role in necrotic cell death in addition to autophagy and apoptosis (Boya and Kroemer, 2008). However, it is not fully understood which molecular switches determine cell fate among the different types of cell death after a death inducing signal.
Glycogen synthase kinase-3 (GSK-3) is an ancient protein with a diverse range of cellular functions (reviewed by Forde and Dale, 2007). There are two isoforms of GSK-3 in mammals, GSK-3α and GSK-3β. Although these two isoforms are expressed ubiquitously and share over 98% identity within their kinase domains, they are not redundant in vivo. For example, in mice Gsk3b knockout resulted in embryonic death due to hepatocyte apoptosis, indicating that GSK-3β loss is not compensated by GSK-3α (Hoeflich et al., 2000). Unlike other protein kinases, GSK-3β is constantly active and its activity is even higher under serum starvation, indicating that GSK-3β plays a role in cell survival under resting conditions. Currently, it is not fully clear how GSK-3β controls cell survival under serum-free conditions.
Bax interacting factor 1 (Bif-1; also known as SH3GLB1) was initially cloned as a binding partner of the pro-apoptotic Bax protein (Cuddeback et al., 2001; Pierrat et al., 2001). Later, Bif-1 was shown to promote Bax conformational change under the control of Src kinase in apoptotic cells (Takahashi et al., 2005; Yamaguchi et al., 2008). Most interestingly, Bif-1 was recently found to modulate autophagy by interacting with beclin-1—VPS34 complex through the UVRAG protein (Takahashi et al., 2007). Moreover, a recent study suggested that Bif-1 might act as a driving force for autophagosome formation (Etxebarria et al., 2009) (reviewed by Takahashi et al., 2009). In this study, we discovered that Bif-1 is required for GSK-3 inhibitor-induced autophagic response, necrosis and apoptosis. Under serum-free conditions, suppressing GSK-3 activity induced a Bif-1-dependent autophagic response and subsequent massive necrotic cell death. Suppressing autophagy at either the early phase or later stage did not rescue cells but redirected necrotic cell death to apoptosis through a mechanism involving Bif-1-dependent Bax conformational change.
Results
GSK-3β suppression results in cell death under serum starvation
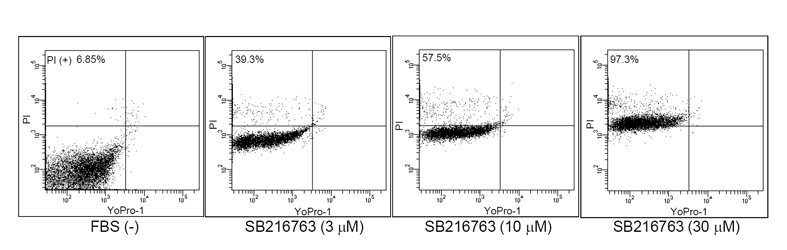
Although GSK-3β is proposed to promote cell survival (Forde and Dale, 2007), the mechanism is not fully clear. We recently reported that inhibition of GSK-3β activity by lithium chloride (LiCl) suppressed human cancer cell proliferation without affecting cell survival under full culture (10% FBS) conditions (Sun et al., 2007). To explore the functional role of GSK-3β in cell survival after serum withdrawal, we utilized multiple GSK-3β inhibitors, one of them is a pseudo-substrate peptide L803-mts (Plotkin et al., 2003) and another one is a non-ATP competitive small chemical TDZD8 (Martinez et al., 2002). Consistent with our previous report (Sun et al., 2007), treatment of human cancer PC-3 cells with these inhibitors in serum-containing medium did not induce obvious cell death although cell proliferation was significantly suppressed compared with the control (Fig. 1A). Surprisingly, under serum-free conditions, a significant cell death was found in both L803-mts- and TDZD8-treated cells compared with the solvent control. In addition to L803-mts and TDZD8, two other structurally unrelated GSK-3β inhibitors, AR-A014418 (Bhat et al., 2003) and SB216763 (Carmichael et al., 2002) also induced a dose-dependent cell death (supplementary material Fig. S1).
Fig. 1.

GSK-3β suppression leads to cell death under serum starvation. (A) PC-3 cells were plated in 12-well plates overnight and then treated with L803-mts (100 μM) or TDZD8 (10 μM) in FBS-supplied or serum-free medium for up to 3 days. At each time points as indicated, live cells were counted using the Trypan Blue exclusion assay as described in our previous publication (Liao et al., 2005). (B) PC-3 cells were transfected with GSK-3 siRNAs or a negative control siRNA mixture, as indicated, at a final concentration of 200 nM in serum-free medium. At each time point, as indicated, cells were counted as above. Inset: proteins were extracted from cells transfected with GSK-3 siRNA or the control siRNA for 3 days, and then subjected to SDS-PAGE and immunoblotting with anti-GSK-3 antibodies. (C) PC-3 cells were plated in 35 mm dishes overnight and then treated with the solvent, L803-mts alone (100 μM), 3-MA alone (10 mM) or L803-mts plus 3-MA in serum-supplied (10% FBS) or serum-free medium. Photomicrographs were taken 24 hours after treatment. Original magnification ×200. Black arrows indicate dead cells with cytolytic features. White arrows indicate collapsed cells without membrane rupture. (D) PC-3 cells were treated with SB216763 at the indicated doses for 3 days, and cell death was assessed by YoPro-1—PI-staining-based flow cytometry assay. In A, B and D data are means ± s.e.m. from three independent experiments. Asterisks indicate significant difference compared with the control (ANOVA analysis, P<0.05).
To determine the GSK-3β specificity of these inhibitors, siRNAs for GSK-3β and GSK-3α were transfected into PC-3 cells and cell death was monitored for up to a week under serum-free conditions. As shown in Fig. 1B, GSK-3β siRNAs but not GSK-3α siRNAs significantly induced cell death. These data indicate that GSK-3β activity is essential for cell survival under serum-free conditions.
To examine the characteristics of cell death induced by GSK-3β inhibition, we first used microscopy to examine cellular morphological change during L803-mts-induced cell death. As shown in Fig. 1Cd, a cytolytic event, as evidenced by cytoplasmic membrane rupture and cell lysis, was observed after L803-mts treatment under serum-free condition. By contrast, cells treated with L803-mts in the presence of serum or treated with the solvent in serum-free medium did not show any sign of cytolysis (Fig. 1Ca-c).
We then characterize the cell death with a flow cytometry-based YoPro-1—propidium iodine (PI) staining assay since the impermeant nuclear dye YoPro-1 stains apoptotic cells accurately. The combination of YoPro-1 with PI dye can provide a distinction between apoptotic (YoPro-1 positive) and non-apoptotic cell death (PI positive, YoPro-1 negative) (Idziorek et al., 1995). As shown in Fig. 1D, GSK-3β inhibition did not increase apoptotic death (YoPro-1 positive) but significantly increased non-apoptotic death (PI-positive, YoPro-1-negative) in a dose-dependent manner. Also, a clear dose-dependent transition from living (PI negative, YoPro-1 negative) to non-apoptotic death (PI positive, YoPro-1 negative) occurred after SB216763 treatment (supplementary material Fig. S2). These data indicate that GSK-3β inhibition resulted in a non-apoptotic cell death under serum starvation.
GSK-3β inhibitor-induced cell death is necrotic under serum starvation
Next, we confirmed the non-apoptotic cell death using two classical apoptosis markers, PARP cleavage and caspase-3 processing. A DNA damaging agent camptothecin (CPT) was included as a positive control for apoptotic cell death. As expected, CPT treatment induced a time-dependent caspase-3 processing and PARP cleavage in PC-3 cells. By contrast, L803-mts treatment failed to induce either event (Fig. 2). These data clearly indicate that GSK-3β-inhibition-induced cell death is a non-apoptotic event.
Fig. 2.
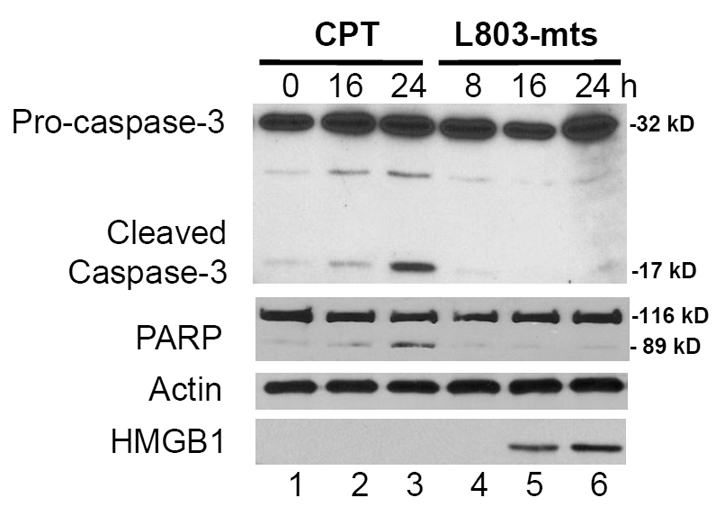
GSK-3β inhibitor induces necrotic cell death. PC-3 cells were plated in six-well plates and then treated with camptothecin (CPT; 3.0 μM) or L803-mts (100 μM) in serum-free medium. Equal amount of cellular proteins were used in western blotting to detect caspase processing and PARP cleavage as indicated on the left. Actin was used as a protein loading control. To analyze HMGB-1 release, cell culture media were collected and the supernatants were concentrated using a 3.0-kDa cutoff column. Equal amount of proteins from the concentrated supernatants were subjected to SDS-PAGE followed by immunoblotting with HMGB-1 antibody. The experiment was repeated twice.
Nuclear protein HMGB-1 is retained within the nuclear compartment in apoptotic cells but is released during necrotic cell death or autophagic response to cytotoxic reagents (Zong et al., 2004; Thorburn et al., 2009). Therefore, we determined if HMGB-1 was released during L803-mts-induced cell death. HMGB-1 levels in conditioned medium were assessed after treatment with L803-mts or CPT. As expected, CPT treatment did not induce HMGB-1 release (Fig. 2, bottom panel). Conversely, HMGB-1 release was detected at 16 h after L803-mts treatment. Taken together, these data suggest that GSK-3β inhibition induced necrotic cell death under serum-free conditions.
GSK-3β suppression promotes autophagy under serum starvation
To determine whether GSK-3β inhibition triggered autophagic response under serum starvation, we first assessed vacuole formation (vacuolization) with transmission electronic microscopy (TEM). As shown in Fig. 3A, L803-mts-treated PC-3 cells underwent a remarkable vacuolization at 12 hours post-treatment under serum-free conditions. Consistent with cell survival data, L803-mts did not induce notable vacuolization under full culture (10% FBS) conditions.
Fig. 3.

GSK-3β suppression induces strong autophagic response. (A) PC-3 cells were treated with L803-mts (100 μM) or the solvent as control for 6-12 hours under FBS-supplied or serum-free conditions. Transmission electron microscopy was used to evaluate the vacuolization. Note that massive vacuolization was observed in cells after L803-mts treatment under serum-free conditions. (B,C) PC-3 cells stably transfected with GFP-LC3 (PC3/GFP-LC3) were treated with the solvent or L803-mts (100 μM), TDZD8 (10 μM) for 12 hours under serum-supplied (10% FBS) or serum-free conditions. Photomicrographs were taken under a fluorescent microscope at a magnification of ×200. Quantitative data of the average GFP-positive loci per cell are summarized in C. (D,E) PC3/GFP-LC3 cells were transfected with GSK-3β siRNAs (siGSK-3β, 200 nM) or the control siRNA (200 nM) for 48 hours. Cells were then cultured in serum-supplied (10% FBS) or serum-free conditions, as indicated, for 12 hours. Quantitative data of the average GFP-positive loci per cell are summarized in E. (F,G) PC-3 cells were plated in six-well pates and treated with L803-mts (100 μM) under serum-free conditions. Cells were harvested at the indicated time points. Conditioned culture media were collected for HMGB-1 analysis as described earlier. Equal amount of cellular proteins were used in western blotting analysis for LC3 processing, p62 degradation and PARP cleavage. Actin served as a loading control. Quantitative data of band densities are summarized in G. Data are from three experiments and error bars indicate s.e.m. Asterisks indicate a significant difference compared with the control (ANOVA, P<0.01).
Next, we used another indicator of autophagic response, LC3 translocation. As an important component of the autophagy machinery, LC3 is processed from LC3-I to the membrane-bound form LC3-II by conjugating with phosphatidylethanolamine and subsequently accumulates in autophagosomes during the induction of autophagy (Kabeya et al., 2000). PC-3 cells with stably transfected GFP-LC3 were treated with or without GSK-3β inhibitors for 8 hours under serum-free or serum-supplied conditions. As shown in Fig. 3B,C, the defuse pattern of GFP-LC3 expression was not changed after treatment of cells with L803-mts or TDZD8 in serum-supplied (10% FBS) medium. By contrast, under serum-free conditions, L803-mts or TDZD8 treatment dramatically increased the punctate foci of GFP-LC3 expression, which was consistent with the TEM data (Fig. 3A). Then, GSK-3β siRNAs was used to confirm the specificity of the chemical inhibitor-induced GFP-LC3 translocation. As shown in Fig. 3D,E, GSK-3β siRNAs remarkably increased the formation of GFP-LC3 punctate foci under serum-free conditions compared with the control siRNAs or under the full culture (10% FBS) conditions.
To determine the correlation of the autophagic response and cell death induced by GSK-3β suppression, we analyzed LC3 processing, PARP cleavage and HMGB-1 release by western blotting. As shown in Fig. 3F,G, L803-mts dramatically enhanced LC3-I processing as evidenced by increased levels of the LC3-II fragment at 8 hours post-treatment. Meanwhile, PARP cleavage was not detected and HMGB-1 release was observed at 16 hours after L803-mts treatment. We also determined whether the autophagic flux induced by GSK-3β inhibition was completed by assessing p62 protein level, a marker for autophagy flux (Klionsky et al., 2008). As shown in Fig. 3F, p62 protein levels dramatically declined following L803-mts treatment. These data suggest that GSK-3β inhibition triggered a strong autophagic flux that was followed by a necrotic cell death.
Blocking autophagy switches GSK-3β inhibitor-induced necrosis to apoptosis
Since we observed GSK-3β-inhibition-induced autophagic response followed by necrotic cell death, we then asked if blocking autophagy would reduce GSK-3β-inhibition-induced cell death. To test this hypothesis, we treated cells with L803-mts plus the autophagy inhibitor 3-MA (Selgen et al., 1982) with or without serum supplement. Under serum-supplied (10% FBS) conditions, treatment with 3-MA alone had no obvious effect on cell survival (Fig. 1Ce,f), similar to the results when L803-mts was used alone (Fig. 1Cc). Unexpectedly, under serum-free conditions, a Trypan-Blue-based cell counting assay revealed that cell survival was similarly attenuated by 3-MA plus L803-mts treatment compared with L803-mts alone (Fig. 4A). In addition, a similar pattern of lactate dehydrogenase (LDH) release, the marker of cytotoxic effect that increases in both necrosis and apoptosis (Kroemer et al., 2009), was observed after treatment with either L803-mts alone or L803-mts plus 3-MA (Fig. 4B). However, microscopic examination revealed that cellular collapse but not plasma membrane rupture or cytolysis was induced after L803-mts plus 3-MA treatment (Fig. 1Ch) under serum-free conditions, indicating that 3-MA addition switched L803-mts-induced necrosis to a different type of cell death.
Fig. 4.

Blocking autophagy flux redirects GSK-3β suppression-induced necrosis to apoptosis. (A) PC-3 cells were plated in 12-well plates and then treated with 3-MA (10 mM), L803-mts (100 μM) or 3-MA plus L803-mts in serum-free conditions for 24 hours. Live cells were counted after Trypan Blue staining, as described earlier. (B) PC-3 cells were plated in 24-well plates and then treated with L803-mts (100 μM) with or without 3-MA (10 mM) under serum-free conditions. Cell culture media were collected at the indicated time points and subjected to LDH assay as described in the text. Data in A and B are means from four independent experiments and error bars indicate s.e.m. (C) PC-3 cells were plated in six-well plates and then treated with L803-mts in serum-free medium in the presence of 3-MA (10 mM), CQ (5 μM) or the solvent for 48 hours. Cells were harvested and stained with YoPro-1 and PI dyes. Representative graphics of cell profiles from the flow cytometry analysis are shown. Note the pattern shift of cell distribution from necrosis (d) to apoptotic death (e and f). (D) PC-3 cells were treated with L803-mts (100 μM), AR-A011418 (3 μM), LiCl (10 mM), SB216763 (30 μM) in the presence of 3-MA (10 mM) or the solvent for 24 hours in serum-free medium. Cells were then stained with JC-1 from a mitochondrial membrane potential assay kit, as described in our previous publication (Liao et al., 2005). Original magnification ×200. (E) PC-3 cells were plated in six-well plates overnight and then treated with L803-mts (100 μM) in serum-free medium for different time periods, as indicated, in the presence or absence of 3-MA (10 mM). Cells were harvested and equal amounts of cellular proteins were used for immunoblotting with antibodies as indicated. HMGB-1 analysis was described earlier. The molecular masses of the bands are indicated on the right side of the blots. (F,G) PC-3 cells were transfected with Atg5 siRNAs (siAtg5; 200 nM) or the negative control siRNA (siControl; 200 nM) for 48 hours, and then left untreated (F) or treated with L803-mts (100 μM) with or without 3-MA (10 mM) in serum-free medium for 18 hours. Equal amounts of proteins from cell lysates were subjected to SDS-PAGE and immunoblotting with the antibodies indicated. Data in C-G are the results from two independent experiments each.
Then, we went on to determine if the cell death induced by L803-mts plus 3-MA is apoptotic. In addition to 3-MA, another autophagy blocker chloroquine (CQ), was included to block the autophagy flux. Cell death was examined with the YoPro-1—PI assay. As expected, 3-MA and CQ alone did not induce obvious cell death compared with the control (Fig. 4Cb and c vs a), but treatment with L803-mts alone resulted in a significant increase of PI-positive cells in the population, a sign of necrotic cell death (Fig. 4Cd). Interestingly, addition of 3-MA or CQ to L803-mts treatment resulted in a dramatic increase in YoPro-1-positive, PI-positive cell population (Fig. 4Ce,f), suggesting that blocking autophagy flux redirected necrosis to apoptotic cell death.
Next, we used a fluorescent dye JC-1, an indicator of mitochondrial damage due to apoptosis (Smiley et al., 1991), to ascertain the shift of apoptotic cell death. As shown in Fig. 4D, JC-1 staining of mitochondria (red color, evidence of functional mitochondria) was not changed after treatment with GSK-3β inhibitors alone. Conversely, treatment with GSK-3β inhibitors plus 3-MA resulted in green JC-1 staining, an indication of severe damage of mitochondrial membrane potential and apoptotic cell death.
Thirdly, we examined this pattern change of cell death with two classical apoptotic hallmarks, caspase-3 processing and PARP cleavage. As shown in Fig. 4E, 3-MA addition to L803-mts treatment dramatically reduced L803-mts-induced LC3 processing, p62 degradation and HMGB-1 release but increased PARP cleavage compared with L803-mts single treatment.
Finally, a gene silencing approach was used to confirm the effect of pharmacological blockage of autophagy flux on the switch from necrosis to apoptosis. Atg5, a critical component of the autophagy machinery (Kuma et al., 2004), was silenced with a pooled siRNA mixture. As shown in Fig. 4F, compared with the control siRNAs, transfection with Atg5 siRNAs largely reduced Atg5 protein levels. As expected, Atg5 siRNA dramatically suppressed serum starvation-induced LC3 processing (Fig. 4F lane 3 vs lane 2), confirming the blockade of autophagy. Consistently, L803-mts treatment-induced LC3 processing and p62 degradation were largely attenuated in Atg5 siRNA-transfected cells (Fig. 4G, lane 4 vs lane 2), similar to 3-MA treatment (Fig. 4G, lane 3). Meanwhile, PARP cleavage was detected in Atg5-silenced cells but not in the control siRNA-transfected cells after L803-mts treatment. Taken together, these data demonstrated that blocking autophagy flux switches GSK-3β-inhibition-induced necrosis to apoptotic cell death.
Bif-1 is required for GSK-3β inhibitor-induced cell death under serum starvation
As described so far, GSK-3β inhibition induced a strong autophagic response and subsequent necrotic cell death that was redirected to apoptosis when autophagy was blocked. To determine the mechanisms involved in GSK-3β-inhibition-induced autophagy flux and cell death, we assessed the protein levels of several autophagy- and apoptosis-related genes in response to L803-mts treatment with or without 3-MA. As shown in Fig. 5A, a remarkable increase of Bif-1 protein levels was found after L803-mts treatment with or without 3-MA addition. No dramatic change was observed for Bcl-2, Bax, beclin-1 and VPS34 proteins. Under serum-free conditions, the increase of Bif-1 protein is in a L803-mts dose- and time-dependent manner (Fig. 5A,C). By contrast, under culture conditions with 10% FBS, Bif-1 protein levels remained unchanged after GSK-3β inhibition (Fig. 5D), which is consistent with the cytotoxicity data. These results indicate that Bif-1 is involved in GSK-3β-inhibition-induced cell death.
Fig. 5.

GSK-3β suppression causes Bif-1 protein accumulation. (A,B) PC-3 cells were left untreated or treated with L803-mts at indicated doses (μM) in the presence or absence of 3-MA (10 mM) for 18 hours in serum-free medium. After harvesting, equal amounts of cellular proteins were subjected to SDS-PAGE and immunoblotting with the indicated antibodies. The relative band densities were normalized against the anti-actin blot and summarized in B. Data are from two separate experiments and error bars indicate the s.e.m. Asterisks indicate significant difference compared with the control (ANOVA, P<0.05). (C) PC-3 cells were treated with the solvent or L803-mts (100 μM) in serum-free medium. Cells were harvested at the indicated time points and Bif-1 protein levels were assessed by western blotting. (D) PC-3 cells were treated with the solvent, L803-mts (100 μM) or LiCl (10 mM) in serum-supplied (FBS, 10%) medium. Cells were harvested at the indicated time points and western blotting was conducted with the primary antibodies as indicated. The anti-actin blot served as a protein loading control.
Recent reports have shown that Bif-1 participates in both the autophagic pathway (Takahashi et al., 2007) and apoptotic cell death (Takahashi et al., 2005). Thus, we reasoned that Bif-1 might act as a molecular switch between GSK-3β-inhibition-induced apoptosis and necrosis. To test this hypothesis, we established a stable subline of PC-3 cells in which Bif-1 expression was silenced (PC-3/shBif-1). An empty-vector-transfected cell subline (PC-3/Puro) was established as a control. These cells were treated with L803-mts with or without 3-MA. Similar to the parental PC-3 cells, in PC-3/Puro cells, L803-mts treatment significantly reduced cell survival (Fig. 6A), enhanced LC3 processing and p62 degradation, as well as caused HMGB-1 release (Fig. 6B). Addition of 3-MA did not attenuate L803-mts-induced cell death (Fig. 6A) but resulted in PARP cleavage and abolished HMGB-1 release (Fig. 6B). In PC-3/shBif-1 cells, in a sharp contrast, L803-mts treatment did not induced cell death, and all of LC-3 processing, p62 degradation, HMGB-1 release and PARP cleavage were abolished, indicating that Bif-1 is required for GSK-3β-inhibition-induced autophagy flux and cell death.
Fig. 6.

Bif-1 is required for GSK-3β-suppression-induced autophagic response and cell death. (A) PC-3/Puro and PC-3/shBif-1 cells were treated with L803-mts (100 μM) with or without 3-MA (10 mM) in serum-free medium for 2 days. Live cells were counted after Trypan Blue staining, as described previously. Inset: exponentially grown PC-3/Puro and PC-3/shBif-1 cells were harvested for western blotting with Bif-1 antibodies. The membrane was re-probed with anti-actin antibody as a loading control. (B,C) PC-3/Puro and PC-3/shBif-1 cells were treated with the solvent or L803-mts (100 μM) plus 3-MA (10 mM) for 16 hours. Cell extracts were subjected to immunoblotting with the antibodies as indicated. To detect Bax conformational change, cellular extracts were prepared in Chaps buffer and then subject to immunoprecipitation with Bax 6A7 antibodies, followed by immunoblotting with regular Bax antibody. Relative band densities were normalized against the anti-actin blot and summarized in C. (D,E) PC-3 cells were treated with L803-mts (100 μM) plus or minus 3-MA (10 mM) in serum-free medium for 18 hours and cellular proteins were subjected to anti-beclin-1 immunoprecipitation and the eluted immunocomplexes were analyzed by western blotting with Bif-1 and VPS34 antibodies, as indicated. Data are from two independent experiments. Relative band densities were shown in E. Asterisks indicate significant difference compared with the control (ANOVA, P<0.05).
Because Bif-1 is required for apoptotic cell death induced by L803-mts plus 3-MA, we then determined whether Bax was activated during apoptotic cell death. In PC-3/Puro cells, the level of active Bax protein, determined with the conformational specific Bax antibody 6A7 (Hsu et al., 1997), remained largely unchanged after L803-mts treatment, compared with the control. However, addition of 3-MA to the L803-mts treatment dramatically increased the levels of the active Bax form (Fig. 6B, lane 3 vs 2). Most interestingly, Bax activation was abrogated in PC-3/shBif-1 cells after L803-mts treatment with or without addition of 3-MA (Fig. 6B, lanes 4 and 5), which was in agreement with the cell survival data as shown in Fig. 6A. Taken together, these results clearly indicate that Bif-1 is required for GSK-3β-inhibition-induced autophagic response and cell death, and blocking autophagy flux redirects necrosis to apoptosis, which is associated with Bif-1-dependent Bax conformational change and/or activation.
It has been shown that in response to metabolic stress, Bif-1 interacts with a beclin-1—VPS34 complex, leading to VPS34 activation and autophagy induction (Takahashi et al., 2007). Thus, we determined whether GSK-3β inhibition increases Bif-1 interaction with the beclin-1—VPS34 complex. As shown in Fig. 6D, L803-mts treatment largely increased beclin-1 interaction with both Bif-1 and VPS34 (lane 2). However, addition of 3-MA to the L803-mts treatment reduced Bif-1 interaction with beclin-1 but enhanced beclin-1—VPS34 interaction (lane 4). These data suggest that increased interaction between Bif-1 and the beclin-1—VPS34 complex may be involved in the GSK-3β-inhibition-induced autophagic response. Addition of 3-MA prevented the interaction between Bif-1 and beclin-1, leading to Bif-1-dependent Bax activation and apoptotic cell death. However, the significance of increased interaction of VPS34 with beclin-1 after 3-MA addition needs further investigation.
Discussion
In this study, we examined the functional role of GSK-3β in cell survival under serum-free conditions and the mechanisms involved in GSK-3β-inhibition-induced cell death. We demonstrated that inhibition of GSK-3β activity results in a strong autophagic response and subsequent necrotic cell death, suggesting a critical role of GSK-3β in fine-tuning autophagic response and in promoting cell survival after serum deprivation. Most importantly, we identified that Bif-1 plays a key role in the autophagic response and necrotic cell death after GSK-3β inhibition. Blockage of autophagy flux using pharmacological reagents and genetic means redirected Bif-1-dependent necrosis to apoptosis, which was associated with a conformational change in Bax. Therefore, we propose that Bif-1 is a part of the cell death determinant that is modulated by GSK-3β (Fig. 7).
Fig. 7.
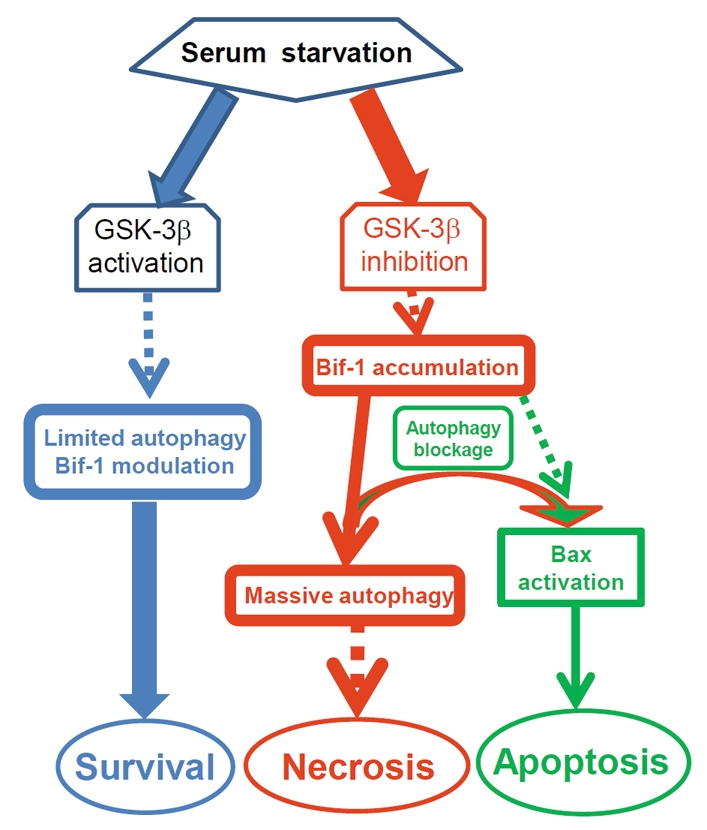
Proposed mechanism of GSK-3β modulation of Bif-1-dependent autophagy and cell death. Blue: under serum starvation conditions, GSK-3β maintains a limited autophagic response for cell survival by modulating the level of Bif-1 protein. Red: once GSK-3β activity is suppressed, Bif-1 protein is increased, resulting in a massive autophagy response and necrotic cell death. Green: if autophagy is blocked, the cell death route is redirected to apoptotic cell death through a mechanism associated with Bif-1-dependent Bax activation.
In recent years, autophagy has been the subject of considerable attention because of its important role in cell fate determination and human diseases (Klionsky and Emr, 2000). It has been shown that autophagy suppression had different effects, either triggering or preventing apoptotic cell death, depending on the death stimulus (Boya et al., 2005; Takacs-Vellai et al., 2005; Wang et al., 2008). However, under certain experimental settings, blunting the apoptotic pathway leads to increased autophagic response and necrotic cell death (Raymond et al., 2003; Cao et al., 2006; Ullman et al., 2008), and simultaneous blocking of both autophagy and apoptosis induces necrotic cell death (Degenhardt et al., 2006). Therefore, there may be interactions among different cell death routes, of which the autophagy-related mechanism might be the one to activate for cell death fate (Mora and Regnier-Vigouroux, 2009). However, it is not entirely clear, at the molecular level, which mechanism determines the cell death route following a given death stimulus.
In this study, we demonstrated that Bif-1, the molecule associated with both autophagy and apoptosis, was required for both necrotic and apoptotic cell death induced by GSK-3β inhibition. We showed that Bif-1 was upregulated at the protein level and was required for the autophagic response and cell death (either necrotic or apoptotic death). Based on these data, we propose that under serum-free conditions or possibly other metabolic stresses, the autophagic response is activated in cells seeking survival. As a survival factor, GSK-3β acts as a modulator to fine-tune Bif-1 protein levels and the associated autophagy process in order to avoid excessive self-digestion and cell death. Suppression of GSK-3β activity diminishes the tight modulation of Bif-1 levels, resulting in Bif-1 accumulation and profound autophagic response, which eventually leads to necrotic cell death. If the autophagic response is blocked, then Bif-1 turns on Bax-dependent apoptotic machinery (Fig. 7).
Although necrosis has been recently considered as one type of programmed cell death (Zong and Thompson, 2006; Golstein and Kroemer, 2007), there is a controversy regarding the interplay among different death pathways. Frequently, necrosis inducers, such as ischemia or nutrient stress, also stimulate autophagic responses as a survival mechanism in the early stage towards necrosis, however, blocking autophagy suppresses necrosis, indicating that autophagy is critical for necrosis (Samara et al., 2008). Conversely, in apoptosis-deficient mammalian cells, blocking autophagy induces necrotic death under conditions of nutrient limitation (Degenhardt et al., 2006). In this study, we found that blocking autophagy flux redirects GSK-3β-inhibition-induced necrosis to apoptosis under serum-free conditions. Therefore, the interchange between different death routes may be based on the death stimulus and the genetic background of cellular models.
Currently, it is largely unknown how necrotic cell death and its interplay with other types of cell death mechanisms are regulated, although lysosomal biogenesis and mitochondrial dysfunction were found to be critical for induction of necrosis in D. discoideum or C. elegans (Artal-Sanz et al., 2006; Giusti et al., 2009). In mammalian cells, the death domain kinase receptor interacting protein 1 and 3 (RIP1 and 3) and BH3-only Bcl-2 modifying factor (Bmf) were found to regulate the canonical signal-induced necroptosis, a type of necrotic cell death when apoptosis is blocked (Holler et al., 2000; Hitomi et al., 2008; Cho et al., 2009; He et al., 2009; Zhang et al., 2009). In this study, we found that a strong autophagic response proceeded necrotic cell death after GSK-3β inhibition. Most importantly, our data revealed that Bif-1, a binding partner of the pro-apoptotic Bax protein, plays a key role in the autophagic response and cell death. It has been demonstrated that Bif-1 forms a complex with beclin-1 through UVRAG and knockout or knockdown of Bif-1 reduces the number of autophagosomes induced by nutrient starvation (Takahashi et al., 2007). However, precisely how Bif-1 regulates autophagy remains to be investigated. It is possible that the SH3 domain of Bif-1 binds to and suppresses UVRAG-mediated autophagosome maturation, whereas the N-BAR domain of Bif-1 is involved in the generation of autophagosomal membranes independently of its interaction with the beclin-1 complex.
Currently, it is not clear how, after GSK-3β inhibition, blockade of the autophagic response, either in the early stage by 3-MA or in the late stage by CQ, redirects Bif-1 to activate Bax. One plausible mechanism is that blocking autophagy reduces Bif-1 interaction with the beclin-1—VPS34 complex (Fig. 6D), which subsequently leads to increased Bif-1-dependent Bax conformational change and/or activation for apoptosis induction (Fig. 7). Notably, a recent publication showed that Bif-1 was not detected in a beclin-1-containing complex isolated from mouse brain or liver tissues (Zhong et al., 2009). Since we and others (Takahashi et al., 2007) reproducibly found that Bif-1 interacts with the beclin-1—VPS34 complex after GSK-3β inhibition or during nutrient starvation, it is hypothesized that Bif-1 interaction with the beclin-1—VPS34 complex is not steady but transient or dynamic in response to extracellular stimuli or metabolic stress.
In summary, our study revealed that GSK-3β promotes cell survival by modulating Bif-1-dependent autophagic response after serum starvation. Inhibition of GSK-3β activity diminishes the GSK-3β control over Bif-1-dependent autophagic response, leading to an enhanced autophagic response and eventually necrotic cell death. In addition, Bif-1 is possibly involved in a necrosis-apoptosis switch if autophagy is inhibited. Further investigation is under way to elucidate the mechanism involved in GSK-3β-mediated regulation of Bif-1 expression under serum-free conditions.
Materials and Methods
Antibodies and chemicals
Antibodies for PARP, caspase-3, VPS34, beclin-1, Bax, Bcl-2 and Atg5 were from Cell Signaling Inc (Danvers, MA). Anti-Bif-1 antibody was from Imgenex Co. (San Diego, CA). Anti-HMGB-1 and anti-LC3 antibodies were from GeneTex (Irvine, CA). Anti-Bax clone 6A7 and p62/SQSTM1 clone D-3 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). LiCl and 3-MA were from Sigma-Aldrich (St Louis, MO). TDZD8, AR-A014418 and Chloroquine (CQ) were purchased from Calbiochem (San Diego, CA). SB216763 was obtained from Biomol (Plymouth Meeting, PA). L803-mts [N-myristol-GKEAPPAPPQS(P)P] was synthesized by Genemed Synthesis (San Antonio, TX) as previously described (Plotkin et al., 2003), and dissolved in DMSO at more than 1000-fold the final concentration used in cell culture.
Cell culture and transfection
PC-3 cells were cultured in RPMI 1640 medium supplemented with 10% FBS (fetal bovine serum), 100 μg/ml streptomycin and 100 IU/ml penicillin. All the siRNAs were obtained from Santa Cruz Biotechnology and transfected into cells at the indicated concentration using the OligoFectamine reagent (Invitrogen, Carlsbad, CA). GFP-LC3 and Bif-1 shRNA expression constructs were described previously (Takahashi et al., 2007). Cells were transfected with plasmid DNA using LipoFectamin (Invitrogen). To establish a stable cell line expressing GFP-LC3 or Bif-1 shRNA, cells were co-transfected with the expression vector and pBabe-puro vector. After 24 hours transfection, single colonies were selected in the puromycin-containing medium as described in our previous publication (Liao et al., 2005).
Cytotoxicity assays, JC-1 staining and flow cytometry
Cell viability was assessed with Trypan Blue exclusion assay. Mitochondrial membrane potential was determined using JC-1 staining kit and the protocol obtained from the manufacturer (Invitrogen) was followed as described previously (Liao et al., 2005). Cell death was determined using a FACScalibur flow cytometer (BD Biosciences) using a YoPro-1 Apoptosis Assay Kit (Invitrogen) according to the manufacturer's protocol.
Immunoprecipitation and immunoblotting analysis
Cells were harvested, rinsed with PBS, and lysed on ice in RIPA buffer (Sigma-Aldrich). Equal amount of proteins from each lysate was loaded onto SDS-PAGE gels and immunoblotted (IB) with antibodies as indicated in the figures. For detection of Bax conformational change, total cell lysates were prepared in Chaps-containing cell extract buffer (Cell Signaling Technology), and subjected to anti-Bax 6A7 immunoprecipitation (IP) followed by immunoblotting (IB) analysis with regular Bax antibody. For beclin-1 IP, cell lysates were prepared with RIPA buffer. Anti-beclin-1 (Cell Signaling Technology) antibodies were used in the IP assay overnight and thereafter with protein A and/or G plus agarose (Santa Cruz Biotechnology) for 2 hours. Antibodies-bound beads were collected by centrifugation and washed five times with lysis buffer. Immunoprecipitates were eluted in sample buffer and subjected to western blot analysis. Band densities on the immunoblots were scanned and the relative band densities were normalized against anti-actin blots. The band densities from the control was set as a value of 1.
HMGB-1 and LDH release assay
For the HMGB-1 release assay, cell culture medium was collected and spun at 1000 g for 10 minutes and the supernatant was then concentrated through Millipore centrifugal filter devices YM-3. Equal amount of the concentrated supernatants were subjected to western blotting with anti-HMGB-1 antibody. For the LDH release assay, culture medium was collected and LDH activity was assessed using a LDH cytotoxicity assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's protocol.
Phase-contrast, fluorescence and electron microscopy
For phase-contrast microscopy, PC-3 cells were plated in 35 mm dishes and then were treated with the solvent L803-mts at 100 μM or L803-mts (100 μM) plus 3-MA at 10 mM in serum-free medium. For fluorescence microscopy, cells were grown on eight-well chamber slides. After fluorescent dye loading, slides were mounted with Vectashield fluorescent mounting medium (Vector Laboratories, Burlingame, CA). Photomicrographs were taken with a QImaging Retiga 4000R digital camera driven by QCapture Pro 5.1 image capture software under a Olympus 1X71 inverted microscope. For electron microscopy, cells were fixed in 2.5% glutaraldehyde overnight at 4°C and post-fixed in 0.5% osmium tetroxide for 30 minutes. Specimens were stained with 2% uranyl acetate in 50% ethanol. The cells were then dehydrated in a graded ethanol series, infiltrated with plastic, embedded in Epon-812 and polymerized at 60°C for 2 days. Sections were cut at thickness of 80 nm and stained with lead citrate, and then photographed on JEOL 100CX II transmission electron microscope.
Statistical analysis
Images of western blots and immunostaining are from a representative experiment. The mean and standard error of the mean (s.e.m.) from cell counting, autophagosome, GFP-loci counts, band densities of immunoblots and flow cytometry experiments are shown. The significance of the differences between treatment and control was analyzed using SPSS software (SPSS, Chicago, IL).
Supplementary Material
Acknowledgments
We thank Barbara Fegley for her excellent assistance in electronic microscopy. We also thank Joyce Slusser, Director of KUMC Flow Cytometry Core Facility that is funded by an NIH COBRE program from the National Center for Research Resources (P20 RR016443), for her technical support. We are also grateful to the Imaging Core Facility of the Center for Reproductive Sciences at KUMC for image processing. This study was partially supported by the KU William L. Valk Endowment, grants from Kansas Mason's Foundation, Department of Defense PCRP program (W81XWH-04-1-0214 and W81XWH-07-1-0021) to B.L., and NIH (CA82197 and CA129682) to H.-G.W. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/6/861/DC1
References
- Artal-Sanz M., Samara C., Syntichaki P., Tavernarakis N. (2006). Lysosomal biogenesis and function is critical for necrotic cell death in Caenorhabditis elegans. J. Cell Biol. 173, 231-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R., Xue Y., Berg S., Hellberg S., Ormö M., Nilsson Y., Radesäter A. C., Jerning E., Markgren P. O., Borgegård T., et al. (2003). Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 278, 45937-45945 [DOI] [PubMed] [Google Scholar]
- Boya P., Kroemer G. (2008). Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434-6451 [DOI] [PubMed] [Google Scholar]
- Boya P., González-Polo R. A., Casares N., Perfettini J. L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T., et al. (2005). Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25, 1025-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C., Subhawong T., Albert J. M., Kim K. W., Geng L., Sekhar K. R., Gi Y. J., Lu B. (2006). Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 66, 10040-10047 [DOI] [PubMed] [Google Scholar]
- Carmichael J., Sugars K. L., Bao Y. P., Rubinsztein D. C. (2002). Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington's disease mutation. J. Biol. Chem. 277, 33791-33798 [DOI] [PubMed] [Google Scholar]
- Chan F. K., Shisler J., Bixby J. G., Felices M., Zheng L., Appel M., Orenstein J., Moss B., Lenardo M. J. (2003). A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 278, 51613-51621 [DOI] [PubMed] [Google Scholar]
- Cho Y. S., Challa S., Moquin D., Genga R., Ray T. D., Guildford M., Chan F. K. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112-1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddeback S. M., Yamaguchi H., Komatsu K., Miyashita T., Yamada M., Wu C., Singh S., Wang H. G. (2001). Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J. Biol. Chem. 276, 20559-20565 [DOI] [PubMed] [Google Scholar]
- Degenhardt K., Mathew R., Beaudoin B., Bray K., Anderson D., Chen G., Mukherjee C., Shi Y., Gélinas C., Fan Y., et al. (2006). Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., Yuan J. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112-119 [DOI] [PubMed] [Google Scholar]
- Etxebarria A., Terrones O., Yamaguchi H., Landajuela A., Landeta O., Antonsson B., Wang H. G., Basañez G. (2009). Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large scale membrane morphological rearrangements. J. Biol. Chem. 284, 4200-4212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde J. E., Dale T. C. (2007). Glycogen synthase kinase 3, a key regulator of cellular fate. Cell Mol. Life Sci. 64, 1930-1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giusti C., Luciani M. F., Klein G., Aubry L., Tresse E., Kosta A., Golstein P. (2009). Necrotic cell death: From reversible mitochondrial uncoupling to irreversible lysosomal permeabilization. Exp. Cell Res. 315, 26-38 [DOI] [PubMed] [Google Scholar]
- Golstein P., Kroemer G. (2007). Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32, 37-43 [DOI] [PubMed] [Google Scholar]
- He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100-1111 [DOI] [PubMed] [Google Scholar]
- Hitomi J, Christofferson D. E., Ng A., Yao J., Degterev A., Xavier R. J., Yuan J. (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311-1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich K. P., Luo J., Rubie E. A., Tsao M. S., Jin O., Woodgett J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86-90 [DOI] [PubMed] [Google Scholar]
- Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J. L., Schneider P., Seed B., Tschopp J. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489-495 [DOI] [PubMed] [Google Scholar]
- Hsu Y. T., Youle R. J. (1997). Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272, 13829-13834 [DOI] [PubMed] [Google Scholar]
- Idziorek T., Estaquier J., De Bels F., Ameisen J. C. (1995). YOPRO-1 permits cytofluorometric analysis of programmed cell death (apoptosis) without interfering with cell viability. J. Immunol. Methods 185, 249-258 [DOI] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO. J. 19, 5720-5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J., Emr S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717-1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J., Abeliovich H., Agostinis P., Agrawal D. K., Alievm G., Askew D. S., Baba M., Baehrecke E. H., Bahr B. A., Ballabio A., et al. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N., Tavernarakis N. (2009). Autophagy and cell death in model organisms. Cell Death Differ. 16, 21-30 [DOI] [PubMed] [Google Scholar]
- Kroemer G., Galluzzi L., Vandenabeele P., Abrams J., Alnemri E. S., Baehrecke E. H., Blagosklonny M. V., El-Deiry W. S., Golstein P., Green D. R., et al. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T., Ohsumi Y., Tokuhisa T., Mizushima N. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032-1036 [DOI] [PubMed] [Google Scholar]
- Liao X., Tang S., Thrasher J. B., Griebling T. L., Li B. (2005). Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol. Cancer Ther. 4, 505-515 [DOI] [PubMed] [Google Scholar]
- Maiuri M. C., Zalckvar E., Kimchi A., Kroemer G. (2007). Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 8, 741-752 [DOI] [PubMed] [Google Scholar]
- Martinez A., Alonso M., Castro A., Pérez C., Moreno F. J. (2002). First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J. Med. Chem. 45, 1292-1299 [DOI] [PubMed] [Google Scholar]
- Mora R., Régnier-Vigouroux A. (2009). Autophagy-driven cell fate decision maker: activated microglia induce specific death of glioma cells by a blockade of basal autophagic flux and secondary apoptosis/necrosis. Autophagy 5, 419-421 [DOI] [PubMed] [Google Scholar]
- Okada M., Adachi S., Imai T., Watanabe K., Toyokuni S. Y., Ueno M., Zervos A. S., Kroemer G., Nakahata T. (2004). A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood 103, 2299-2307 [DOI] [PubMed] [Google Scholar]
- Pierrat B., Simonen M., Cueto M., Mestan J., Ferrigno P., Heim J. (2001). SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics 71, 222-234 [DOI] [PubMed] [Google Scholar]
- Plotkin B., Kaidanovich O., Talior I., Eldar-Finkelman H. (2003). Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J. Pharmacol. Exp. Ther. 305, 974-980 [DOI] [PubMed] [Google Scholar]
- Raymond M. A., Mollica L., Vigneault N., Désormeaux A., Chan J. S., Filep J. G., Hébert M. J. (2003). Blockade of the apoptotic machinery by cyclosporin A redirects cell death toward necrosis in arterial endothelial cells: regulation by reactive oxygen species and cathepsin D. FASEB J. 17, 515-517 [DOI] [PubMed] [Google Scholar]
- Samara C., Syntichaki P., Tavernarakis N. (2008). Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 15, 105-112 [DOI] [PubMed] [Google Scholar]
- Seglen P. O., Gordon P. B. (1982). 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 79, 1889-1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley S. T., Reers M., Mottola-Hartshorn C., Lin M., Chen A., Smith T. W., Steele G. D., Jr, Chen L. B. (1991). Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. USA 88, 3671-3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A., Shanmugam I., Song J., Terranova P. F., Thrasher J. B., Li B. (2007). Lithium suppresses cell proliferation by interrupting E2F-DNA interaction and subsequently reducing S-phase gene expression in prostate cancer. Prostate 67, 976-988 [DOI] [PubMed] [Google Scholar]
- Takacs-Vellai K., Vellai T., Puoti A., Passannante M., Wicky C., Streit A., Kovacs A. L., Müller F. (2005). Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr. Biol. 15, 1513-1517 [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Karbowski M., Yamaguchi H., Kazi A., Wu J., Sebti S. M., Youle R. J., Wang H. G. (2005). Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol. Cell. Biol. 25, 9369-9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Coppola D., Matsushita N., Cualing H. D., Sun M., Sato Y., Liang C., Jung J. U., Cheng J. Q., Mul J. J., et al. (2007). Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142-1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Meyerkord C. L., Wang H. G. (2009). Bif-1/Endophilin B1: a candidate for crescent driving force in autophagy. Cell Death Differ. 16, 947-955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J., Horita H., Redzic J., Hansen K., Frankel A. E., Thorburn A. (2009). Autophagy regulates selective HMGB-1 release in tumor cells that are destined to die. Cell Death Differ. 16, 175-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman E., Fan Y., Stawowczyk M., Chen H. M., Yue Z., Zong W. X. (2008). Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ. 15, 422-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker N. I., Harmon B. V., Gobé G. C., Kerr J. F. (1988). Patterns of cell death. Methods Achiev. Exp. Pathol. 13, 18-54 [PubMed] [Google Scholar]
- Wang Y., Singh R., Massey A. C., Kane S. S., Kaushik S., Grant T., Xiang Y., Cuervo A. M., Czaja M. J. (2008). Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J. Biol. Chem. 283, 4766-4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H., Woods N. T., Dorsey J. F., Takahashi Y., Gjertsen N. R., Yeatman T., Wu J., Wang H. G. (2008). SRC directly phosphorylates Bif-1 and prevents its interaction with Bax and the initiation of anoikis. J. Biol. Chem. 283, 19112-19118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. W., Shao J., Lin J., Zhang N., Lu B. J., Lin S. C., Dong M. Q., Han J. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332-336 [DOI] [PubMed] [Google Scholar]
- Zhong Y., Wang Q. J., Li X., Yan Y., Backer J. M., Chait B. T., Heintz N., Yue Z. (2009). Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11, 468-476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong W. X., Thompson C. B. (2006). Necrotic death as a cell fate. Genes Dev. 20, 1-15 [DOI] [PubMed] [Google Scholar]
- Zong W. X., Ditsworth D., Bauer D. E., Wang Z. Q., Thompson C. B. (2004). Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 18, 1272-1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}