

Abstract
 A new mechanistic class of BoNT/A zinc metalloprotease inhibitors, from Echinacea, exemplified by the natural product Dchicoric acid (I1) is disclosed. A detailed evaluation of chicoric acid's mechanism of inhibition reveals that the inhibitor binds to an exosite, displays noncompetitive partial inhibition, and is synergistic with competitive active site inhibitor when used in combination. Other components found in Echinacea, I3 and I4, were also inhibitors of the protease.
A new mechanistic class of BoNT/A zinc metalloprotease inhibitors, from Echinacea, exemplified by the natural product Dchicoric acid (I1) is disclosed. A detailed evaluation of chicoric acid's mechanism of inhibition reveals that the inhibitor binds to an exosite, displays noncompetitive partial inhibition, and is synergistic with competitive active site inhibitor when used in combination. Other components found in Echinacea, I3 and I4, were also inhibitors of the protease.
Neurotoxins of the anaerobic spore forming bacterium Clostridium botulinum are the most lethal human poison. 1 Serotype A (BoNT/A) is the most potent of the several serotypes with an LD50 for a 70 kg human of 0.8 μg. Upon cellular internalization of the holotoxin a light chain (LC) 50 kDa zinc metalloprotease is released. Toxicity results from the metalloprotease's site-specific cleavage of the synaptosomal-associated protein preventing acetylcholine containing vesicles from fusing with the presynaptic neuromuscular junction.2
Currently, there are no approved pharmacological treatments for BoNT intoxication. Although an effective vaccine is available for immuno-prophylaxis,3 vaccine approaches cannot reverse the effects after the toxin has reached its target inside the cell. A small molecule pharmacological intervention, especially one that would be effective against the etiological agent responsible for BoNT intoxication, the light chain protease would be highly desirable and obviate vaccine deficiencies.
The substrate for BoNT/A is SNAP-25, (synaptosomal-associated protein, 25 kDa). The Michaelis complex involves an extensive network of binding interactions ranging from the active site to the opposite surface of the BoNT/A. In the complex, the N-terminal residues of SNAP-25 (147-167) form an α-helix, imbedded in the rear surface of BoNT/A while the C-terminal residues (201-204) form a distorted β-strand, and the spanning residues are mostly extended.4 Both mutagenesis and kinetics have conclusively shown that the N-terminal α-helix and the C-terminal β-sheet are critical for an efficient substrate binding and cleavage, and have been termed α-and β-exosites, respectively.5 Also, substrate truncation experiments reveal that BoNT/A protease requires a long stretch of SNAP-25, (66-amino acids) to have optimal catalytic activity. Likely, it is the extensive enzyme-substrate binding interactions that make the proteases of BoNTs among the most selective known. This multi-site binding strategy incorporating an exceptionally large substrate–enzyme interface area4 probably accounts for the extreme difficulty in producing potent small molecule inhibitors of the enzyme. In effect, the small molecule must be capable of disrupting these protein–protein interactions.6 While considerable efforts have gone into identifying active site inhibitors of BoNT/A, no report of a small molecule exosite inhibitor has been communicated.7 Herein, we provide strong evidence demonstrating that components from the plant Echinacea are potent exosite inhibitor with unexpected synergistic effect when combined with an active site inhibitor.
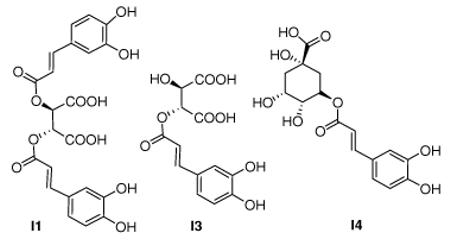
One of the most popular herbs in the US today is the Native American medicinal plant called Echinacea. It has been used for over 400 years to treat infections and wounds and as a general “cure-all”. Main components of Echinacea showing biological and pharmacological activity are the phenolic caffeoyl derivatives8 including I1, I3, and I4, Figure 1. We were intrigued by the structural similarities between the above phenolic caffeoyl derivatives and several known active site inhibitors of BoNT/A, (Fig. 1); in particular the similarity between I2, identified from a high throughput screen9 and D-chicoric acid I1. Interestingly, the unnatural isomer L-chicoric acid (I1′), is a potent inhibitor of the HIV-1 integrase, a metalloenzyme.10 Consequently we tested these Echinacea components for their inhibition of BoNT/A protease.
Figure 1.

Natural products D-Chicoric Acid (I1), Caftaric Acid (I3), Chlorogenic Acid (I4), synthetic hydroxamates I2 and I5.
Thus, I1 was evaluated over an extended concentration range with substrate present at KM (10 μM).11 Surprisingly, partial inhibition was observed. To evaluate this unexpected kinetic inhibition mechanism, concentrations of I1 and the substrate (SNAP-25, amino acids 141-206) were varied.11 A noncompetitive partial inhibition mechanism depicted in Scheme 1 was most consistent with the results. Equation 1 is the rate equation derived from Scheme 1 (Supp. Inf.) where δ is the fractional VMAX at saturating [I1], while KU and KC are the uncompetitive and competitive inhibition constants respectively. Figure 2 presents a global fit of I1 to a matrix of [I1] × [S] from which δ = 0.42 ± 0.04, KU = 1.6 ± 0.3 M, and KC = 0.7 ± 0.1 μM. A submicromolar competitive inhibition constant makes I1 one of the tightest binding small molecules yet discovered for BoNT/A. Intriguingly, at saturation, I1 will only produce 60% inhibition. Consistent with I1, the L-chicoric acid I1′, I3 and I4 were examined in a similar manner and found to exert the same inhibition mechanism. Interestingly, I1′ has virtually the same inhibition potency as I1, although they are enantiomers; while I3 and I4 are about one order of magnitude less potent (see Supp. Inf., Table S1).
Scheme 1.

Chicoric Acid Mechanism of Inhibition and Equation 1
Figure 2.

BoNT/A LC catalysis at varied concentrations of substrate and D-chicoric acid. The substrate is an optimized 66 amino acid sequence of the SNAP 25 bracketing the enzyme's active site.
Partial inhibition is inconsistent with an inhibitor occupying an enzyme's active site since active site residence of either a substrate or an inhibitor physically precludes occupation by the opposite agent. In other words, if an inhibitor binds within the active site, then at saturating inhibitor, the substrate is prevented from binding and catalytic activity falls to zero (producing complete rather than partial inhibition). Therefore, I1, I3, and I4 must associate in an exosite some distance from the active site. Such inhibition has been reported for a number of proteases, but not for the BoNTs.12 Quite likely, the exosite overlaps with a portion of the 66-mer substrate's extended binding region interfering with, but not totally preventing, substrate binding. Simple inhibition experiments do not identify binding site locations. On the other hand, our hypothesized non-occupation of the enzyme active site by phenolic caffeoyl derivatives may be supported by an inhibitor combination study.13 Two inhibitors that bind within the active site of an enzyme will, by definition, be mutually exclusive. In a mutually exclusive inhibitor combination, a plot of 1/Vobserved versus the concentration of one inhibitor at varied but fixed concentrations of the second inhibitor will produce a family of parallel lines as a diagnostic pattern. In contrast, if the inhibitors used in combination are mutually nonexclusive the same plot will produce a family of intersecting lines as a diagnostic pattern. Additionally, the magnitude of the increasing slope with increasing 2nd inhibitor concentration reflects the degree of synergistic binding between the inhibitors. An inhibitor combination study involving chicoric acid is complicated by its partial inhibition as such, the full rate equation was derived (Supp. Inf.) and used in the global fit for the combination study.
To confirm our hypothesis of I1 being an exosite inhibitor, we examined a combination of I2 and I5 (Fig. 1). Both compounds are optimized hydroxamate inhibitors and have been confirmed by kinetics and crystallographic analysis to bind within the metalloprotease's active site through coordination with the catalytic zinc.8,10,14 A global fit to the I2/I5 inhibitor combination experiment was most consistent with the mutually exclusive binding model and clearly visible in the parallel lines of Figure 3 panel A. In contrast, utilizing the combination of I2/I1 produced a pattern of intersecting lines demonstrating non-mutually exclusive binding. Interestingly, a global fit of Equation 3 (Supp. Inf.) to the data produced a synergistic or enhancement factor (α) of 1.7 ± 0.3.
Figure 3.

Panel A: I2 in combination with I5 displaying mutually exclusive inhibition. Panel B: I2 in combination with I1 displaying synergistic inhibition.
We have disclosed a new mechanistic class of BoNT/A zinc metalloprotease inhibitors exemplified by the natural product chicoric acid. A detailed evaluation of chicoric acid's inhibition mechanism reveals that the inhibitor binds to an exosite, displays noncompetitive partial inhibition, and is synergistic with competitive inhibitor (I2) when used in combination. The ability to inhibit an exosite by a small molecule is no simple feat as this requires the disruption of protein–protein interactions.6 Our work also highlights how natural products could provide a rewarding frontier for the BoNT drug discovery and development. Future research along these lines will be reported in due course.
Supplementary Material
Acknowledgment
The authors gratefully acknowledge support of this project by the National Institute of Allergy and Infectious Diseases, National Institute of Health and the Department of Health and Human Services under contract numbers N01-AI30050 and AI080671.
Footnotes
Supporting Information Available: Full experimental procedures, derivations and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. Nature Rev. Drug Disc. 2005;4:281–297. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simpson LL. Annu. Rev. Pharmacol. Toxicol. 2004;44:167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 3.Willis B, Eubanks LM, Dickerson TJ, Janda KD. Angew. Chem. Int. Ed. 2008;47:8360–8379. doi: 10.1002/anie.200705531. references cited therein. [DOI] [PubMed] [Google Scholar]
- 4.Breidenbach MA, Brunger AT. Nature. 2004;432:925–929. doi: 10.1038/nature03123. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Barbieri JT. J. Biol. Chem. 2006;281:10906–10911. doi: 10.1074/jbc.M513032200. [DOI] [PubMed] [Google Scholar]
- 6.(a) Arkin MR, Wells JA. Nat. Rev. Drug Dis. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]; (b) Toogood PL. J. Med. Chem. 2002;45:1543–1558. doi: 10.1021/jm010468s. [DOI] [PubMed] [Google Scholar]
- 7.BoNT/A noncompetitive inhibition has been reported, however, these studies have utilized the small peptide substrate SNAPtide™, which is known to produce an inner filter effect confounding mechanistic interpretations. Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, Janda KD. J. Comb. Chem. 2006;8:513–521. doi: 10.1021/cc060010h.Cai S, Lindo P, Park J-B, Vasa K, Singh BR. Toxicon. doi: 10.1016/j.toxicon.2009.11.017. in press.Lai H, Feng M, Roxas-Duncan V, Dakshanamurthy S, Smith LA, Yang DCH. Arch. Biochem. Biophys. 2009;491:75–84. doi: 10.1016/j.abb.2009.09.008.
- 8.Cheminat A, Zawatzky R, Becker H, Brouillard R. Phytochemistry. 1988;27:2787–2794. [Google Scholar]
- 9.Boldt GE, Kennedy JP, Janda KD. Organic Letters. 2006;8:1729–1732. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reinke RA, Lee DJ, McDougall BR, King PJ, Victoria J, Mao Y, Lei X, Reinecke MG, Robinson WE., Jr. Virology. 2004;326:203–219. doi: 10.1016/j.virol.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Čapková K, Hixon MS, McAllister LA, Janda KD. Chem. Commun. 2008;14:3525–3527. doi: 10.1039/b808305c. [DOI] [PubMed] [Google Scholar]
- 12.Bock PE, Panizzi P, Verhamme IMA. J Thromb. Haemost. 2007;5:81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley & Sons; Hoboken, NJ: 1975. [Google Scholar]
- 14.Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. Chem Biol. 2007;14:533–542. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.