

Abstract
Wnt signaling is involved in developmental processes, cell proliferation, and cell migration. Secreted frizzled-related protein 4 (sFRP4) has been demonstrated to be a Wnt antagonist; however, its effects on endothelial cell migration and angiogenesis have not yet been reported. Using various in vitro assays, we show that sFRP4 inhibits endothelial cell migration and the development of sprouts and pseudopodia as well as disrupts the stability of endothelial rings in addition to inhibiting proliferation. sFRP4 interfered with endothelial cell functions by antagonizing the canonical Wnt/β-catenin signaling pathway and the Wnt/planar cell polarity pathway. Furthermore, sFRP4 blocked the effect of vascular endothelial growth factor on endothelial cells. sFRP4 also selectively induced apoptotic events in endothelial cells by increasing cellular levels of reactive oxygen species. In vivo assays demonstrated a reduction in vascularity after sFRP4 treatment. Most importantly, sFRP4 restricted tumor growth in mice by interfering with endothelial cell function. The data demonstrate sFRP4 to be a potent angiogenesis inhibitor that warrants further investigation as a therapeutic agent in the control of angiogenesis-associated pathology.
Wnt signaling pathways have been implicated in the proliferation, survival, differentiation, and migration of various cell types,1–4 including endothelial cells.5,6 Furthermore, Wnt plays an important role in the development of the vasculature under different conditions, including embryonic angiogenesis,7,8 and is known to exert its effect by modulating both cellular and transcriptional events.9,10
The Wnt proteins are a diverse family of secreted glycoproteins that transduce cellular signals by binding to two coreceptor molecules: the transmembrane frizzled receptors and lipoprotein receptor-related proteins 5 or 6.7 There are three Wnt signaling pathways: (i) the canonical, or Wnt-β–catenin pathway, which targets a key cellular regulatory molecule β-catenin; (ii) the Wnt-Calcium- mediated pathway, which mobilizes intracellular calcium to activate calcium/calmodulin-dependant protein kinase II and protein kinase C; and (iii) the Wnt-planar cell polarity pathway, which signals through Rho-associated kinase and c-Jun–N-terminal kinase.11
Secreted frizzled-related protein 4 (sFRP4) is a member of the secreted frizzled-related protein family of Wnt inhibitors that bind directly to Wnt and antagonize both canonical and noncanonical Wnt pathways.12 We have previously shown antiproliferative and proapoptotic roles for sFRP4 during normal homeostasis in tissues such as ovary, corpus luteum, placenta, and mammary gland13–16 but, surprisingly, in pathological states such as mesothelioma17 and colorectal carcinoma,18 sFRP4 did not induce apoptosis of tumor cells.
The exact mechanisms by which Wnt affects angiogenesi remains poorly understood; however, Wnt signaling and the requirements of the canonical Wnt pathway appear to be essential in endothelial cell (EC) commitment developing from embryonic stem cells.19 Furthermore, coexpression of Wnt proteins and Wnt pathway inhibitors by endothelial cells is implicated in the regulation of angiogenesis.20,21
To date there are no published data demonstrating the involvement of sFRP4 in angiogenesis. We report our investigations on the role of sFRP4 on EC physiology using a variety of in vitro assays and its effect on physiological and tumor-associated angiogenesis using in vivo models.
Materials and Methods
Cell Lines and Culture Media
ECV-304 cells were a donation from V. Shah, GI Research Unit, Mayo Clinic, Rochester, MN; Li EA.hy926 cells were donated by C. Edgel, Tissue Culture Facility, UNC Lineburger Comprehensive Cancer Center, University of North Carolina, Chapel Hill; porcine aortic endothelial cells (PAECs) were donated by R. Rieben, University of Bern, Switzerland. Human umbilical vein endothelial cells (HUVECs) were a donation from Dr Chooi-May Lai, Lions Eye Institute, The University of Western Australia. All experiments were performed using EA.hy926 cells, but crucial experiments were repeated in HUVECs and representative experiments are shown whenever possible. However, results were comparable with all cell lines used. Dulbecco's modified Eagle's medium (DMEM) was purchased from Hi-Media, Mumbai, India. Fetal bovine serum was supplied by Invitrogen Life technologies. Anti– β-catenin antibodies were purchased from Millipore. Matrigel was obtained from BD Biosciences, San Jose, CA. Polyether-polyurethane foam sponge was obtained from Amersham Biosciences (SF) Corp., Piscataway, NJ. All other chemicals were of reagent grade and were obtained commercially. For cell culture, cells were maintained in DMEM supplemented with 10% FBS (v/v), 1% penicillin (w/v) and streptomycin (w/v) at 37°C/5% CO2. sFRP4 was supplied by Upstate (Lake Placid, NY).
Animals
Experiments using Wistar rats were conducted with the approval of the Animal Ethics Committee of Anna University, Chennai, India. Homozygous female BALB/c nu/nu athymic mice experiments were conducted with approval from The University of Western Australia Animal Ethics Committee.
In Vitro Studies
Endothelial Wound Scratch Assay
Primary PAECs and 2 immortalized cell lines (ECV-304 and EA.hy926) were cultured in 24-well plates coated in collagen type 2 at 2 × 105 cells per well to produce confluent monolayers. The monolayers were wounded in a line using a standard 100-μl pipette tip and washed with PBS (pH 7.4) to remove cell debris before incubation with different concentrations of sFRP4 (125 and 250 pg/ml). The area of the cell-free wound at selected time points (0 and 8 hours) was recorded using a Nikon digital camera and analyzed using Image J image analysis software (Release α 4.0 3.2). The wound healing effect was calculated as the percentage of remaining cell-free area (at 8 hours) compared with the initial wound area. The reversibility of the sFRP4-mediated effect was studied by washing the cells with PBS 24 hours post treatment.
Endothelial Cell Chemotactic Assay
Migration in endothelial cells was examined using a Boyden chamber migration assay. Collagen-treated polycarbonate membranes (pore size 8 μm) were used for the assay. HUVECs were pre-treated with different concentrations of sFRP4 (0 and 125 pg/ml) for 2 hours and trypsinized; 1 × 105 cells were loaded in the upper chamber of the Boyden apparatus with or without sFRP4. After 4 hours of incubation at 37°C and 5%CO2, the nonmigrated cells of the upper chamber were removed using a cotton swab. The migrated cells on the underside were fixed with 4% paraformaldehyde in PBS (pH 7.4) and stained with propidium iodide (1 μg/ml). The migrated cells were counted at ×20 magnification using a fluorescent microscope (Olympus IX71).
Endothelial Ring Formation
HUVECs were seeded on collagen-coated 12-well plates (1 × 105 cells per well). After 4 hours of incubation at 37°C/5%CO2, the media was changed to medium supplemented with sFRP4 (0 or 125 pg/ml). The cells were incubated with sFRP4 overnight (12 hours) at 37°C/5%CO2. The endothelial cells formed ring-like structures on overnight incubation. The number of rings formed was counted at ×20 magnification using a brightfield phase contrast microscope.
Stability Studies of Ring Structures
HUVECs were seeded as above (1 × 105 cell per well), but on coverslips coated with collagen type 2 in 12-well plates and incubated for 12 hours at 37°C/5%CO2. The endothelial cells formed ring-like structures on overnight incubation. Each coverslip was mounted on a customized live cell chamber and placed on the microscope stage. Single ring structures were identified and the microscope stage was fixed in place, after which the cells were monitored for up to 15 minutes with or without sFRP4 treatment (125 pg/ml). Images were taken at various time points.
Effect of Vascular Endothelial Growth Factor on sFRP4 Action
EA.hy926 cells (which express the vascular endothelial growth factor [VEGF] receptor) were grown to confluence as per the wound scratch protocol. After wounding, cells were incubated with either VEGF (20 nmol/L), Avastin (125 pg/ml), sFRP4 (125 pg/ml), a combination of VEGF and Avastin, or VEGF and sFRP4 for 12 hours. The amount of wound healing was recorded as previously described.
Functional Analysis of the Mechanism of sFRP4 Action in Vitro
Wound Healing under the Influence of LiCl
HUVECs were grown to confluence as per wound scratch assay. After wounding, they were washed with PBS (pH 7.4) to remove cell debris before incubation using the GSK-3β inhibitor, LiCl (500 μmol/L; Sigma, St Louis, MO), alone or in combination with sFRP4 (125 pg/ml) for 4 hours. The amount of cell migration was recorded as previously described.
Proliferation Assay
The proliferation assay was performed as previously reported.22 In summary, HUVECs were seeded at a density of 3000 cells per well in 96 wells and incubated overnight at 37°C/5% CO2, and the following day the cells were treated with sFRP4 (125 pg/ml) in the presence and absence of LiCl (500 μmol/L). The cells were further incubated for 48 hours at 37°C/5%CO2. After incubation the cells were trypsinized and counted in a hemocytometer (Nebauer Improved, Crown Scientific).
Determination of Intracellular Calcium
Calcium levels were determined as previously reported.23 Briefly, intracellular calcium was monitored in HUVECs using the fluorescent indicator Fura-2 acetoxymethyl ester (Fura-2, 1 μmol/L, Molecular Probes). Fluorescence at 340/380 nm excitation and 510 nm emission wavelengths were measured at 1-minute intervals with an exposure of 50 milliseconds on a Hamamatsu Orca ER digital camera attached to an inverted Nikon TE2000-U microscope. Ratiometric 340/380-nm signal of individual HUVECs was quantified using Metamorph 6.3 to measure signal intensity of manually traced cell regions. An equivalent region not containing cells was used for background and was subtracted. Ratiometric 340/380-nm fluorescence was plotted relative to the pretreatment fluorescence and assigned a value of 1.0. Fluorescent ratios recorded for 3 minutes at 37°C just before and the last 3 minutes of a 10-minute exposure to 125 pg/ml sFRP4 were averaged, and increases in fluorescent ratios were reported as a percentage increase from the baseline average. After 10 minutes exposure to 125 pg/ml, a further 125 pg/ml of sFRP4 was added to create a final dose of 250 pg/ml, and the change in fluorescence was monitored as per the 125 pg/ml dose.
Immunoblotting
Whole cell lysates and Western blot analysis were performed as previously described with minor modifications.14 Briefly, HUVECs at 70% confluence were stimulated with LiCl (500 μmol/L; Sigma, St Louis, MO) in DMEM for 30 minutes at 37°C and then sFRP4 (125 pg/ml) or PBS was added and cells incubated for a further 4 hours at 37°C before being harvested for protein. Lysates were prepared by scraping cells in radioimmunoprecipitation assay buffer (150 mmol/L NaCl, 50 mmol/L Tris-HCl pH7.5, 1% triton X100, 0.5% sodium deoxycholate, 0.1% SDS and 0.1 mmol/L PMSF) on ice. The protein content of the lysates was normalized after quantification by Bradford protein estimation.24 The lysates were boiled in Laemmli buffer for 5 minutes at 90°C before loading onto 10% SDS-PAGE gel. After electrophoresis the proteins were transferred onto a nitrocellulose membrane using wet blotting apparatus (Biorad). The membranes were blocked using 5% nonfat milk in Tris-Buffered Saline Tween-20 (TBST) and probed for β-catenin using anti–β-catenin antibody at 1:1000 dilution in TBST (Cell Signaling) and horse radish peroxidase (HRP)-labeled secondary antibody at a dilution of 1:10,000 in TBST (Pierce). The blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce). β-actin antibody was used at a concentration of 1:5000 (Sigma). The secondary antibody was anti-mouse HRP used at 1:10,000 dilution (Pierce).
Isolation of nuclear fragments was performed as previously reported.25 HUVECs were harvested in incubation medium and spun down at 500g for 10 minutes at 4°C, resuspended in PBS, and spun down again using the same conditions. For isolation of nuclei, cell pellets were resuspended in 500 μl of nuclei isolation buffer (10 mmol/L PIPES, 10 mmol/L KCl, 2 mmol/L MgCl2, 1 mmol/L DTT, 0.1% protease-inhibitor cocktail, pH 7.4). Cells were homogenized using a glass potter, layered over 500 μl of 30% sucrose in nuclei isolation buffer, and centrifuged for 10 minutes at 800g and 4°C. The pellet was washed with nuclei isolation buffer and nuclei were lysed with nuclei isolation buffer containing 0.1% SDS for 30 minutes on ice. Cellular debris was spun down and the supernatant stored at −80°C. C-Jun antibody was used at a dilution of 1:1000 in TBST (Cell Signaling) and HRP-labeled secondary antibody at a dilution of 1:10000 in TBST (Pierce). The blots were developed as above, and protein content was quantified using the Bradford assay.
Determination of Levels of H2O2 in EA.hy926
EA.hy926 cells were grown overnight (12 hours) in tissue culture dishes to attain 60% confluence, then washed once with PBS and media changed to that supplemented with different concentrations of sFRP4 (0 or 125 pg/ml). The cells were incubated for 4 hours at 37°C/5%CO2, then the medium was aspirated and cells were washed with PBS (2 times). Cells were incubated in PBS supplemented with 10 μmol/L Amplex Red in the presence of 0.5U HRP for 15 minutes at 37°C/5%CO2. The cells were scraped and centrifuged at 2000g at 4°C and the supernatant was collected. Fluorometric readings using a Cary eclipse fluorometer (Varian, CA) were recorded at 563-nm excitation and 587-nm emission wavelength; the slit width was kept at 5 nm.
Determination of Levels of Superoxides in EA.hy926 Cells
EA.hy926 cells were grown overnight (12 hours) in 12-well plates coated in collagen type 2 (1 × 106 cells per well). The next morning cells were washed with PBS and the media changed to medium supplemented with sFRP4 (0 or 125 pg/ml). Cells were incubated for 4 hours at 37°C/5%CO2, following which 1 mg/ml nitroblue tetrazolium was added to the media and cells incubated for a further 2 hours. After incubation the medium was aspirated and cells were washed with PBS (2 times). Formazan crystals formed by the action of superoxides on nitroblue tetrazolium were dissolved by adding 200 μl of DMSO and the optical density was measured at 540 nm using a spectrophotometer (Varian Cary 4000 uv-vis photometer, Varian, CA).
Catalase Activity Assay
EA.hy926 cells were grown overnight (12 hours) in tissue culture plates coated in collagen type 2 to attain 80% confluence, and the following morning cells were washed with PBS and the media changed to medium supplemented with sFRP4 (0 or 125 pg/ml). Cells were incubated for 4 hours in sFRP4 supplemented medium at 37°C/5%CO2. After incubation, cells were washed 2 times with PBS, scraped in ice cold PBS (500 μl/35 mm dish), and centrifuged to form a pellet at 2000g/4°C. Supernatant was discarded and the pellet was resuspended in 200 μl 50 mmol/L Tris-HCl buffer (pH 7.4). Cells were homogenized on ice using a Dounce homogenizer at 50 strokes. The sample was prepared by diluting lysate in 2 ml 0.1 mol/L PBS buffer (pH 7.4) and 0.1% H2O2 was added, after which the optical density was measured at 15-second intervals up to 3 minutes. The decrease in optical density corresponds to the breakdown of hydrogen peroxide by catalase activity. The activity of the enzyme was plotted as a function of time and expressed as the amount of H2O2 consumed/min/mg of protein.
Investigation of the Influence of Superoxide Dismutase on sFRP4 Wound Healing Effects
EA.hy926 cells were grown to confluence in 24-well plates as described in the wound healing assay. A wound was created as per the wound healing assay and then incubated with either superoxide dismutase (SOD) or nothing to determine whether SOD per se had any impact on wound healing. Next, cells were again grown to confluence and subjected to wounding followed by incubation with either sFRP4 (125 pg/ml) or sFRP4 (125 pg/ml) in combination with SOD (150 SI units). Wound healing was measured for the next 4 hours by taking images of the wound at 0 and 4 hours of incubation.
In Vivo Studies
Chicken Chorioallantoic Membrane Assay
Fourth day incubated chicken eggs were collected from the Poultry Research Station, Nandanam, Chennai. In a modification of the chicken chorioallantoic membrane assay,26 the eggs were broken and gently plated on a cellophane bed in Petri dishes under sterile conditions. Sterile filter paper disks soaked in sFRP4 (0 or 125 pg/ml) were then placed on the egg yolks and incubated for another 12 hours at 37°C. Images were taken using a Nikon digital camera with a stereo microscope at 0, 6, and 12 hours of incubation. Quantification of angiogenesis was performed by using Image J image analysis software (Release α 4.0 3.2).
Cotton Plug Method
Ten to 20 mg of sterile absorbent cotton plugs were implanted in the peritoneal cavity of Wistar rats with or without sFRP4 (125 pg/ml). After 8 days the animals were sacrificed and the cotton plug granulomas were extracted to measure the hemoglobin content.
Matrigel Sponge Assay
A polyether-polyurethane sponge (0.5 cm2) was soaked in Matrigel that had been thawed at 4°C and the sponge was implanted in the peritoneal cavity of Wistar rats with or without sFRP4 (125 pg/ml). The animals were sacrificed after eight days, and the resulting granulomas were removed to measure the hemoglobin levels.
Matrigel Plug Assay
Matrigel (20 mg/ml) was injected into the peritoneal cavity of Wistar rats with or without sFRP4 (125 pg/ml). The animals were sacrificed after 8 days to extract the matrigel pellet. The hemoglobin content of the pellet was measured spectrophotometrically.
Image Analysis of Implants
Images of the implants were converted to gray scale and adjusted to suitable brightness and contrast using Adobe Photoshop 7.0 to perform automated image analysis using AngioQuant.27 Next, the images were processed with AngioQuant software as directed by the software tools. The number of junctions (which reflects the number of vessels formed in the implants) was taken as an index of angiogenesis.
Mouse Tumor Models
BALB/c nude mice were injected subcutaneously with 5 × 106 SKOV-3 cells, a human cell line derived from ovarian serous cystadenocarcinoma. On day 5, when tumors had grown to 70 to 100 mm3, 100 μg of Avastin or sFRP4 in 100 μl PBS, or 100 μl PBS alone were injected intraperitoneally twice weekly for 3 weeks. On day 43 the tumors were excised under general anesthesia and immersed in a solution of 4% paraformaldehyde in phosphate buffer (pH 7.2) and processed for paraffin embedding. Five-μm sections were made for routine hematoxylin and eosin staining followed by microscopic examination and photography.
Immunohistochemistry
Cryostat sections of frozen tissue were cut at 6 μm, placed on Super Frost Plus slides, and stored at −20°C until use. Immediately before commencing immunostaining, the sections were washed in PBS buffer for 2 minutes before incubation with 3% H2O2 (in methanol) for 25 minutes at room temperature to block endogenous peroxide activity before 3% bovine serum albumin in PBS for 60 minutes at room temperature to block nonspecific binding. Sections were stained using Mec 13.3 antibody (rat anti-mouse CD-31) at a 1:100 dilution (BD Pharmingen, San Diego, CA) overnight at 4°C and then washed in PBS buffer (2 × 2 minutes) at room temperature. The secondary biotinylated antibody (anti-rat) was applied at a 1:500 dilution (Universal secondary, Vector Laboratories Inc., Burlingame, CA) for 90 minutes at room temperature, followed by 2 × 2 minute washes in PBS. The ABC kit (Vectastain Elite ABC, Vector Laboratories Inc., Burlingame, CA) was added for 60 minutes, again followed by 2 × 2 minute PBS washes. Diaminobenzidine (Sigma, Australia) was used as a chromogen to visualize the antibody–antigen complex. Sections were counterstained in Harris's hematoxylin for 45 seconds, dehydrated, cleared, and mounted in DPX. Slides were examined using an Olympus BX50 with MicroFire (Optronics) microscope connected to a Micro Brightfield MBF camera and using Stereo Investigator software; endothelial cells were counted at ×40 magnification by random sampling of 8 sites, each site being 150 μm × 150 μm in size. Counts were performed for PBS, sFRP4, and Avastin treatment groups.
Detection of Apoptosis
TUNEL Assay
Cell death was localized in tissue by TUNEL analysis.28 Paraffin sections were cut (4 μm) using a microtome and then dewaxed by two washes in toluene (5 minutes). Sections were hydrated through a graded series of ethanol and PBS and then incubated with proteinase K (20 μg/ml in PBS) for 30 minutes at 37°C. Thereafter an in situ apoptosis kit (Apoptosis Detection kit, Chemicon International, Temecula, CA) was used for nick-end labeling according to the manufacturer's protocol. Nuclei with DNA cleavage were visualized with DAB (3,3′-diaminobenzidine tetrahydrochloride), and sections were counterstained with methyl green. Postweaning mammary gland was used as a positive control. Labeling was visualized indirectly with peroxidase-labeled antidigoxigenin antibody. Sections that were near the maximal diameter of the tumor were selected for investigation. Slides were examined using an Olympus BX50 with MicroFire (Optronics) microscope connected to a Micro Brightfield MBF camera and, using Stereo Investigator software, both normal and apoptotic endothelial cells were counted at ×40 magnification by random sampling of 8 sites, each site being 150 μm × 150 μm in size. Counts were performed for PBS, sFRP4, and Avastin treatment groups.
JC-1 Assay
Initiation of apoptosis was determined using the JC-1 technique.29 At the onset of apoptosis the mitochondrial membrane is rapidly depolarized. When the mitochondrial membrane is polarized the JC-1 dye (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolyl-carbocyanine iodide) aggregates and fluoresces red. On depolarization, JC-1 forms a green fluorescent monomer, so the ratio of aggregated to monomeric JC-1 gives a quantitative representation of the extent of mitochondrial membrane permeability. Apoptotic cells primarily demonstrate green fluorescence, whereas healthy cell fluoresce red/green. Briefly, cells were grown at 1 × 105 c/ml in 96-well plates for 24 hours. After either 1 or 3 hours of 1 μmol/L STS treatment, media was removed and 50 μl of 2.5 mmol/L JC-1 (Molecular Probes T-3168) dye diluted 1:75 in Hams F12-K was added and incubated for 60 minutes. Positive controls were conducted by the addition of 50 μmol/L FCCP (carbonyl cyanide p-[trifluoromethoxy] phenylhydrazone). JC-1 was removed and cells washed with 5% bovine serum albumin in PBS. Plates were analyzed using a FluoStar fluorescent plate reader. The green monomeric form has absorption/emission maxima of 510/527 nm while the aggregate (red) form has absorption/emission maxima of 585/590 nm.30 Raw values at 590 nm were divided by the raw values of the corresponding well at 520 nm.
Statistical Analyses
Data are presented as mean ± SEM. n for each experiment is indicated in figure legends and refers to the number of well replicates per group. Analysis of variance was used to assess variation between control and treatment groups. For comparisons in which F test for the analysis of variance reached statistical significance (P < 0.05), differences were assessed by least significant difference test.
Results
In Vitro Studies
We used a wound scratch assay using two concentrations of sFRP4 (125pg/ml and 250pg/ml) on three different endothelial cell lines; primary PAECs and the immortalized human EC lines, ECV-304 and EA.hy926. In all three cell types, sFRP4 induced a significant reduction (P < 0.05) in EC migration (Figure 1A). Treatment with 250 pg/ml and 125 pg/ml were equally effective in inhibiting wound healing (Figure 1A), thus the 125 pg/ml dose was used in subsequent studies. Inhibitory effects of sFRP4 on PAEC migration were maintained up to 24 hours after treatment as indicated in the time course results (Figure 1B). However, this inhibition was abolished within three hours of washing for the 125 pg/ml dose. The duration of these inhibitory effects on EC migration healing suggest that sFRP4 exerts its effect by inhibiting both proliferation and migration of endothelial cells.
Figure 1.
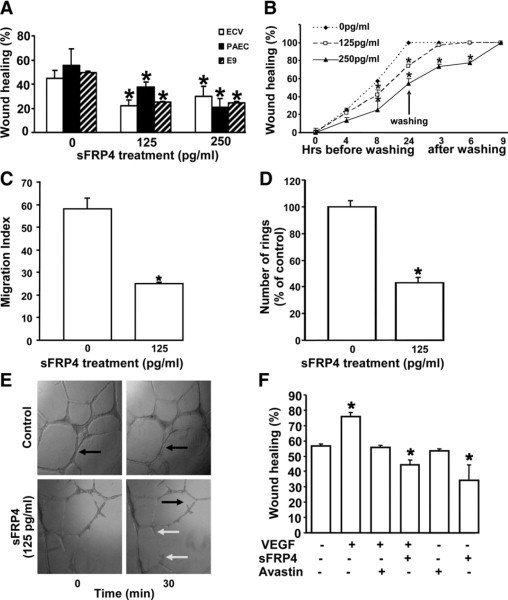
Effect of sFRP4 on wound scratch assay. A: A slower movement of ECs in comparison with that of control was recorded under both 125 and 250 pg/ml sFRP4 treatments (*125 pg/ml P < 0.05; *250 pg/ml P < 0.05, n = 3). B: Withdrawing sFRP4 treatment of PAECs by washing recovered wound healing effects (*125 pg/ml P < 0.05, n = 3) after 6 hours. C: sFRP4 blocked migration of HUVECs in Boyden chamber experiment (*125 pg/ml P < 0.05, n = 8). D: sFRP4 inhibited ring formation by HUVECs. Fewer rings were counted in sFRP4 sets than that of control (*125 pg/ml P < 0.05). E: Time-dependent sFRP4 treatment destabilized the ring structures in the endothelial layer. The black arrows in the control group indicate rings. In the sFRP4 treated group, the black arrow indicates the breakdown of a ring structure, whereas the white arrows show loss of sprouts and pseudopodia. F: Comparative analysis of sFRP4 (125 pg/ml) and Avastin (125 pg/ml) actions on wound healing with or without VEGF (20 nmol/L). Both sFRP4 and Avastin blocked VEGF-dependent wound healing, whereas sFRP4 is more efficient than Avastin in blocking the VEGF-dependent wound healing *P < 0.01, n = 6.
Chemotactic migration of endothelial cells is a key event in angiogenesis, and we investigated the effect of sFRP4 on EC migration using a Boyden's chamber migration assay and HUVECs. EC migration was significantly reduced (P < 0.01) after sFRP4 treatment of 125 pg/ml (Figure 1C).
Endothelial cell ring formation and ring stability also were examined using HUVECs. sFRP4 administration induced a significant reduction in numbers of endothelial rings formed (P < 0.01; Figure 1D) and a reduction of pseudopodia, together with a loss of stability of ring structures within 30 minutes of sFRP4 treatment (Figure 1E). Together these results demonstrate inhibition of endothelial cell spreading, development of pseudopodia, and sprout formation, all of which are important in the initiation and development of angiogenesis.
The interaction between sFRP4 and VEGF was also examined using the wound scratch assay. Addition of VEGF to EA.hy926 cells (which express the VEGF receptor) predictably increased the percentage of wound healing (P < 0.01), whereas addition of the VEGF receptor blocker Avastin restored wound healing activity to control levels. sFRP4, both in the presence or absence of VEGF, significantly reduced (P < 0.01) wound healing (Figure 1F) to a greater extent than observed for Avastin (P < 0.01). Thus, exogenous VEGF is unable to block the effects of sFRP4 on cell migration.
Functional Analysis of sFRP4-Wnt Signaling Pathway Interaction in Vitro
Our in vitro data demonstrated a significant effect of sFRP4 on migration; therefore, we examined the expression of nuclear β-catenin in HUVECs treated with sFRP4 (125 and 250 pg/ml). There were negligible levels of nuclear β-catenin protein detectable by Western blot analysis after treatment with sFRP4 (see Supplemental Figure S1 at http://ajp.amjpathol.org). We then examined the effect of sFRP4 treatment on the Wnt signaling pathway using HUVECs.
Wnt/β-Catenin Pathway
The effect of sFRP4 on HUVECs after activation of the Wnt/β-catenin pathway was examined. LiCl is known to block glycogen synthase kinase-3 (GSK-3β), a downstream target for Wnt signaling, thereby increasing cellular levels of β-catenin.31 We found the addition of LiCl (20 mmol/L) had no effect on HUVEC migration (Figure 2A), whereas sFRP4 produced a significant reduction in cell migration (P < 0.05). However, the addition of LiCl promoted cell proliferation, but this effect was significantly reduced after administration of sFRP4 (P < 0.05; Figure 2B). The data demonstrate that sFRP4 is able to block Wnt signaling via the canonical β-catenin pathway. We also investigated whether sFRP4 was able to antagonize one or more other Wnt signaling pathways to exert its inhibitory effects.
Figure 2.
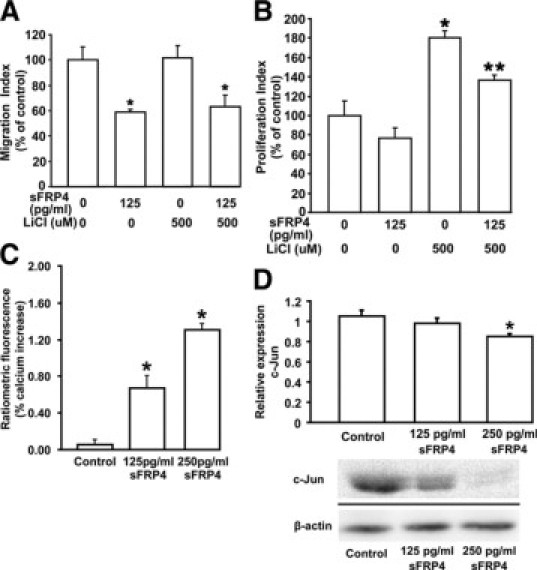
Analysis of sFRP4-Wnt signaling pathway interaction in vitro. A: sFRP4 blocked endothelial cell migration. HUVECs treated with LiCl had no effect on migration, whereas there was a significant reduction in migration after sFRP4 treatment, even in the presence of LiCl (*P < 0.05, n = 3). B: HUVECs treated with a combination of LiCl and sFRP4 showed a lower cell proliferation index than that of LiCl-treated cells (*LiCl P < 0.05; **sFRP4+LiCl P < 0.05, n = 3). C: Exposure of HUVECs to sFRP4 is associated with a significant increase in intracellular Ca2+ (*125 pg/ml P < 0.05; *250 pg/ml P < 0.05. Control n = 4, sFRP4 n = 7). D: Exposure of HUVECs to sFRP4 resulted in a decrease in nuclear c-Jun levels. β-actin was used as loading control. Results of Western blot analysis for c-Jun (dissected from the gel) are shown beneath the graph (125 pg/ml P = 0.1; *250 pg/ml P < 0.01, n = 3).
Wnt/Calcium Pathway
The impact of sFRP4 on the Wnt/calcium pathway was examined using HUVECs. After administration of sFRP4 there was a steady time-dependent increase in intracellular calcium levels. The resultant significant increase (P < 0.05) in intracellular calcium levels once steady state was reached is shown in Figure 2C. Our findings indicate that sFRP4 not only blocks the Wnt/β-catenin pathway but it also activates the Wnt/Calcium pathway.
Wnt/Planar Cell Polarity Pathway
Involvement of sFRP4 in the planar cell polarity/c-Jun NH2-terminal kinase (JNK) pathway was also examined using HUVECs. Western blot analysis demonstrated a reduction in nuclear c-Jun protein expression after treatment with 125 and 250 pg/ml sFRP4 (Figure 2D). Therefore, our data demonstrate a regulatory role of sFRP4 in all three Wnt signaling pathways.
Involvement of sFRP4 in Redox Activation
Reactive oxygen species (ROS), particularly H2O2, promote cell death in tumor models via a stress-induced apoptotic pathway,32 whereas FoxO (a Forkhead transcription factors class) antagonizes ROS effects.33 ROS may perform a role in the antiangiogenic effects of sFRP4 through the β-catenin–T-cell factor (Tcf) axis because FoxO-mediated transcription requires binding of β-catenin.34 β-catenin is required for the transcriptional activity of the Tcf family of transcription factors, which are the downstream effectors of the Wnt/β-catenin pathway.9,35
The involvement of ROS as a possible effector pathway for sFRP4 was investigated in vitro with EA.hy926 cells. Incubation with sFRP4 increased levels of both superoxide and H2O2 (Figure 3, A and B), a consequence of reduced levels of cellular catalase (Figure 3C), a free radical scavenger essential for maintenance of normal cell function. Administration of SOD, which can counteract the activation of the ROS pathway, resulted in restoration of the inhibitory effect of sFRP4 on wound healing (Figure 3D). These results indicate that sFRP4 is able to activate ROS, most likely because of its ability to block Wnt/β-catenin interaction, thereby interfering with FoxO-mediated transcription and, subsequently, Tcf activity.
Figure 3.
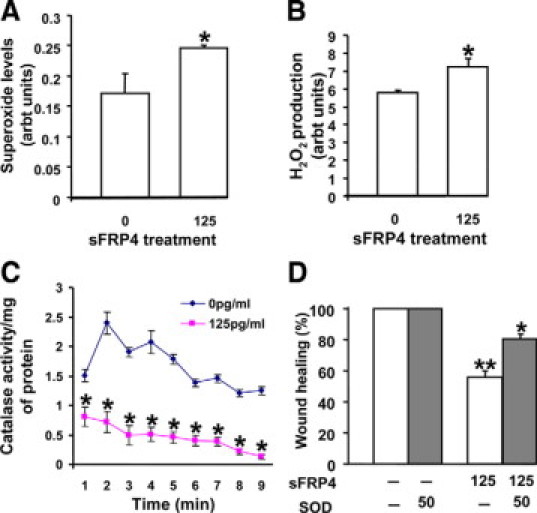
Involvement of sFRP4 in redox activation. A: sFRP4 treatment enhanced superoxide generation in sFRP4-treated cells (*125 pg/ml P < 0.05, n = 3). B: sFRP4 increased hydrogen peroxide formation. The level of hydrogen peroxide in 125 pg/ml sFRP4-treated cells was increased compared with control (*125 pg/ml P < 0.05, n = 3). C: The catalase activity decreased in sFRP4-treated cells in a time-dependent manner (*125 pg/ml P < 0.05, n = 3). D: The addition of superoxide dismutase (SOD; 150 SI units) was able to restore the effect of sFRP4 on wound healing (*125 pg/ml P < 0.01, n = 3). EA.hy926 cells were used for these studies.
Thus, sFRP4 clearly demonstrated antiangiogenic effects on endothelial cells in vitro, and subsequent investigations examined its potential as an angiogenesis inhibitor in vivo.
Investigation of sFRP4 Effects in an in Vivo Environment
We examined the effect of recombinant sFRP4 on the whole vascular bed of the developing embryo (day 3) using a modified chicken chorioallantoic membrane assay.26 A significant reduction in vascularity was observed in the area of sFRP4 application. This reduction of vascularity is illustrated in Figure 4A, which comprises control and sFRP4 treated groups at 0, 6, and 12 hours posttreatment, together with their corresponding mean vessel lengths.
Figure 4.
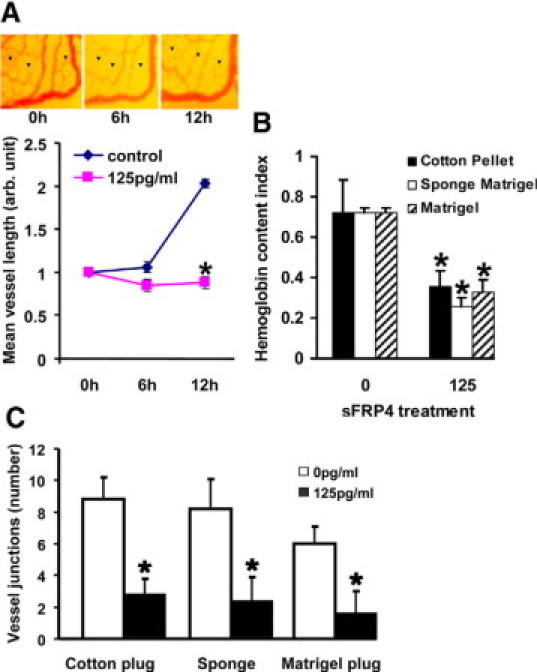
Effect of sFRP4 on ex vivo and in vivo angiogenesis. A: It is evident from the representative images and graph that angiogenesis was blocked by sFRP4 treatment. (*125 pg/ml P < 0.05, n = 3). B: sFRP4 restricts in vivo angiogenesis. sFRP4 treated implants contained less hemoglobin than control implants (*125 pg/ml P < 0.05, n = 3), indicating a reduction in numbers of angiogenic vessels. C: Quantitation of vessel junctions for each implant demonstrating significantly fewer vessels (*125 pg/ml P < 0.05, n = 3) after sFRP4 treatment.
Vessel formation and the effect of sFRP4 were examined using three in vivo assays (cotton plug, Matrigel sponge, and Matrigel plug implants). After sFRP4 treatment, all groups showed a significant decrease in the amount of hemoglobin (Figure 4B), indicating a reduction in vascularization from control levels. The number of junctions (branch points) was also quantified, with all treatment groups showing a significant reduction (Figure 4C).
Finally, the effect of sFRP4 on the growth of an established aggressive tumor was then examined using a murine tumor model. SKOV-3 cells, an aggressive human tumor cell line derived from ovarian serous cystadenocarcinoma, were implanted subcutaneously into BALB/c nude mice. Strikingly, over a treatment period of 3 weeks, the efficacy of sFRP4 to inhibit angiogenesis mimicked the effects of the VEGF-blocking antibody Avastin, a clinically approved angiogenesis inhibitor (Figure 5, A and B). Furthermore, endothelial cell numbers were significantly reduced (P < 0.05) in both sFRP4 and Avastin-treated groups compared with the control (Figure 5C). Microscopically, there was no evidence of cellular degeneration among the tumor cells, however the cytoplasm within the SKOV cells in the treated groups (Avastin and sFRP4) was enlarged and resembled cysts (see Supplemental Figure S2 at http://ajp.amjpathol.org). Treatment with sFRP4 or Avastin did not result in any evidence of systemic toxicity or adverse effects as illustrated by unchanged animal behavior, bodyweight, or macroscopic appearance of the liver or kidney at necropsy. Liver and kidney samples examined using light microscopy showed no adverse effects of treatment (see Supplemental Figure S3 at http://ajp.amjpathol.org).
Figure 5.
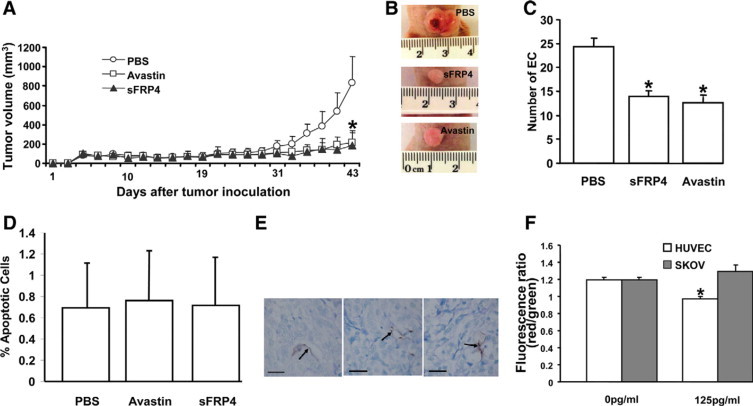
Inhibitory effect of sFRP4 on SKOV tumors. A: Over a treatment period of 3 weeks the efficacy of sFRP4 mimicked the effects of the VEGF-blocking antibody Avastin. B: Photographs of the gross morphology of the tumors highlighting the differences in size. C:In vivo quantification of EC numbers per field of view in tumors from PBS, sFRP4 (100 μg/100 μl), and Avastin (125 μg/100 μl) groups showing a significant reduction in EC numbers (as determined by CD31 staining) in the treated groups (*P < 0.05, n = 6). D: The percentage of apoptotic endothelial cells counted per field of view did not vary between treatment groups (n = 6). E: Microscopy showing results of a TUNEL assay for each treatment group (125pg/ml). Arrows indicate endothelial cells undergoing apoptosis. Scale bar = 50 μm. F:In vitro evaluation of apoptosis using JC-1 assay clearly demonstrating a significant increase (*P < 0.05, n = 3) in apoptosis in HUVECs, whereas SKOV cells remained unaffected.
sFRP4 and Avastin treated tumors showed few apoptotic events that did not differ from control levels, as determined using TUNEL assay (Figure 5D). The few sites of apoptotic activity were localized to the endothelium of sFRP4 and Avastin-treated tumors (Figure 5E). This observation, together with strong in vitro evidence, suggests that sFRP4 acts on tumor blood vessels and thus indirectly restricts tumor growth. To test this hypothesis, HUVECs and SKOV-3 cells were exposed to sFRP4 in vitro for 24 hours and, using a JC-1 assay, we observed that sFRP4 induced apoptosis in HUVECs (Figure 5F). In contrast, SKOV-3 cells exhibited no change in viability, suggesting that sFRP4 indeed predominantly inhibits angiogenesis by selectively targeting EC.
Discussion
Although the sFRPs are normally associated with antagonism of the Wnt signaling pathway, depending on their concentration, they may promote Wnt activity.12 Our data demonstrate a steady increase in intracellular calcium levels after sFRP4 treatment, and it has been shown that such a prolonged but low increase in total intracellular calcium leads to activation of calcineurin.36 Recently, sFRP2 has been shown to stimulate the Wnt/Ca2+ pathway by the dephosphorylation of cytoplasmic nuclear factor associated with T cells (NFAT) by calcineurin, leading to translocation of NFAT into the nucleus where it acts as a transcription factor.37 This may explain the ability of sFRP4 to activate the Wnt/Ca2+ pathway in our studies.
LiCl has been shown to block the activation of GSK3-β, thereby increasing cytoplasmic β-catenin levels and allowing β-catenin translocation to the nucleus. In the nucleus it can bind to transcription factors of the T-cell factor/lymphocyte enhancing factor family (Tcf/Lef) and upregulate genes associated with angiogenesis such as the matrix metalloproteinase 7 and the extracellular matrix component fibronectin; as well as genes associated with migration and proliferation such as cyclin D1, c-myc, cyclooxygenase-2, and VEGF.38 A recent report has demonstrated that, in mice, Wnt7a and Wnt7b are required for normal angiogenesis in the brain and that blockade of Wnt/β-catenin also disrupts angiogenesis in the developing mouse brain.39
We have demonstrated that sFRP4 suppresses the translocation of β-catenin into the nucleus of HUVECs to adversely affect EC functions (see Supplemental Figure S1 at http://ajp.amjpathol.org). Furthermore, the ability of sFRP4 to antagonize the Wnt/Planar Cell Polarity pathway by reducing nuclear c-Jun expression has a synergistic effect on reducing EC functions because JNK is required for β-catenin nuclear localization.40 The ability for sFRP4 to antagonize both the canonical β-catenin and noncanonical c-JNK pathways is in accordance with a previous study, which reported that sFRP4 antagonized Wnt7a signaling via both the canonical β-catenin and noncanonical c-JNK pathways in endometrial cancer cells.41
Therefore, sFRP4 exhibits a multilevel effect on β-catenin. It directly antagonizes the canonical Wnt/β-catenin pathway and also the noncanonical Wnt/planar cell polarity pathway. Additionally, its activation of the Wnt/Ca2+ pathway has an indirect effect in that it can antagonize the Wnt/β-catenin pathway.42–46 Furthermore, reduction of β-catenin correlates with EC apoptosis.47,48 Our data are in accordance with earlier findings, which reported that Wnt/β-catenin signaling is involved in the regulation of EC migration and cell cycle progression.49 Based on our findings we have proposed a model through which sFRP4 interacts with the Wnt signaling pathway (Figure 6).
Figure 6.
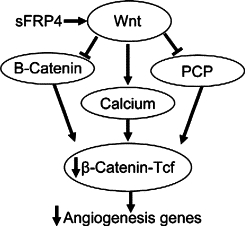
Interaction between sFRP4 and the Wnt signaling pathway. sFRP4 antagonizes the Wnt/β-catenin and Wnt/PCP pathways while activating the Wnt/Ca2+ pathway, the net result being a reduction in translocation of β-catenin to the nucleus. As a consequence, there is diminished β-catenin–Tcf interaction, resulting in a reduction in the transcription of angiogenesis genes.
It has recently been reported that bone morphogenetic protein 2 (BMP-2) can induce angiogenesis in human PAECs by activating both Wnt/β-catenin and Wnt/PCP signaling pathways.22 It is interesting to observe that pro-angiogenic (BMP-2) and antiangiogenic (sFRP4) proteins use the same Wnt signaling pathways to exert their effects.
Our investigations into the effect of sFRP4 on ROS demonstrated a significant increase in the generation of superoxide and H2O2. It has been shown that increased cellular oxidative stress (exemplified by H2O2) relocalizes FOXO to the nucleus where it promotes the association of FOXOs with β-catenin and competes with the binding of β-catenin to Tcf. H2O2 promotes FOXO-mediated transcription at the expense of β-catenin/Tcf–mediated transcription.50,51 Nitric Oxide (NO) synthesized by endothelial NO synthase (eNOS) is essential for EC survival, migration, and postnatal neovascularization. The transcriptional repression of eNOS by FOXOs might also contribute to the antiangiogenic effects of FOXOs on EC. In addition, changes in expression of several extracellular matrix proteins, such as collagen and matrix metalloproteinases, indicate that FOXOs might also be involved in regulating vessel remodeling.52 Our data demonstrated that sFRP4 facilitates the selective apoptosis of endothelial cells in vitro (Figure 5F) and this may, in part, be related to the activation of cellular reactive oxygen species, which then facilitate nuclear entry of FOXO (which preferentially binds to the limited pool of nuclear β-catenin), thus driving the cell toward apoptosis. A proposed model for the interaction between ROS and Wnt (through its action on nuclear β-catenin) is shown in Figure 7.
Figure 7.
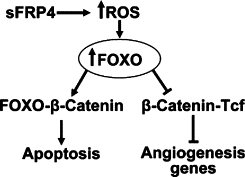
Interaction between sFRP4 and ROS. sFRP4 increases intracellular ROS, which drives FOXO translocation into the nucleus where it preferentially binds to β-catenin, thereby promoting cell apoptosis while simultaneously blocking β-catenin–Tcf interaction, which results in down-regulation of angiogenesis-associated genes.
Our examination of a possible interaction between sFRP4 and VEGF found that the application of exogenous VEGF was unable to block the effects of sFRP4 on cell migration. A relationship between VEGF and the Wnt pathway has been reported previously,53 and the authors reported that VEGF induced EC migration and proliferation through tyrosine phosphorylation of β-catenin that was dependant on protein kinase C. Recently the Wnt/β-catenin pathway has been reported to regulate VEGF-A gene transcription.54 Our in vitro results indicate that sFRP4 inhibits EC migration and proliferation by blocking β-catenin entry into the nucleus, thereby preventing transcription of angiogenesis associated genes (eg, VEGF). The non-canonical Wnt5a signaling pathway has been recently demonstrated to induce proliferation and survival of ECs while up-regulating matrix metalloproteinase-1 expression in ECs.5 The primary step in angiogenesis involves breakdown of the extracellular matrix,55 and our in vitro data demonstrate that sFRP4 prevents formation of pseudopodia, EC ring formation, and disruption of ring structures in EC via its effects on both canonical and noncanonical Wnt signaling pathways by reducing nuclear entry of β-catenin. In addition, the activation of ROS by sFRP4 also reduces the nuclear levels of β-catenin. The net result is that sFRP4 not only reduces migration and proliferation of ECs, but also drives ECs toward apoptosis and, thereby, inhibits angiogenesis.
Examination of the effects of sFRP4 in an in vivo environment demonstrated that sFRP4 can significantly reduce the development of blood vessels in both the physiological setting (chicken chorioallantoic membrane assay) and wound healing response (cotton plug and matrigel implants). Furthermore, we were able to demonstrate that sFRP4 can halt the growth of the aggressive SKOV-3 ovarian tumor via its antiangiogenic properties without affecting tumor cell viability, a finding similar to earlier studies.17,18 VEGF is produced by SKOV-3 cells56 and can increase vessel permeability, thereby facilitating tumor growth by aiding the passage of macromolecules and growth factors directly into the tumor environment. Furthermore, VEGF promotes endothelial cell survival,57 and together these VEGF-induced effects may act to counter sFRP4 inhibition of angiogenesis but are insufficient to support further tumor growth. Thus, sFRP4 treatment resulted in developmental stasis of the tumor. Surprisingly, our in vivo tumor data demonstrated no appreciable difference in numbers of apoptotic ECs among all treatment groups. However, in previous research, TUNEL-positive ECs were detected with very low frequency, which did not correspond with the dramatic loss of capillarization present in the regressing corpus luteum (approximately 1 in 100 apoptotic cells).58 The authors discovered this phenomenon was attributable to a combination of rounding and subsequent detachment of ECs from the basement membrane together with contractive occlusion of blood vessels. Hence, it is not surprising that, because of the speed of the apoptotic process, ECs can be rapidly dispersed into the blood stream in vivo and avoid being detected using apoptosis assays.
Our in vivo data also highlight the endothelial-specific action of sFRP4 observed as a significant reduction in the number of ECs within the tumor mass. This finding is comparable with the antiangiogenic properties demonstrated by Avastin in selectively reducing the number of ECs.59
In summary, these studies demonstrate a previously unknown role of sFRP4 as an inhibitor of angiogenesis and illustrate sFRP4 inhibition of blood vessel formation both in vitro and in vivo. This unique antiangiogenic effect of sFRP4 affects the Wnt/β-catenin and PCP signaling pathways to inhibit the accumulation of β-catenin in the nucleus of endothelial cells. As a consequence, sFRP4 inhibits the migration and proliferation of endothelial cells, both crucial steps in angiogenesis. In addition, sFRP4 is able to increase levels of ROS in ECs, thereby promoting EC apoptosis. Thus, our data promote sFRP4 as a potent angiogenesis inhibitor, which has therapeutic potential for the control of angiogenesis-dependent pathologies.
Acknowledgements
We thank Vijay Shah (GI Research Unit, Mayo Clinic, Rochester, MN) for the ECV-304 cells, Cora-Jean Edgel (Tissue Culture Facility, UNC Lineburger Comprehensive Cancer Center, University of North Carolina, Chapel Hill) for the Li EA.hy926 cells Robert Rieben (University of Bern, Switzerland) for the PAECs and Dr. Chooi-May Lai (Lions Eye Institute, The University of Western Australia) for the HUVECs. We thank Robert Friis (University of Bern, Switzerland) for his scientific advice. We also thank Mary Lee, Leonie Khoo, Guy Ben-Ary, Maike Bollen, Greg Cozens, Uttara Saran, Simon Mahoney, David Longman, and Bernadette Pedersen (School of Anatomy and Human Biology, The University of Western Australia) for their technical assistance and Simon Handford (Office of Industry and Innovation, The University of Western Australia) for his support and help with the patent application and commercialization of this protein.
Footnotes
Supported by the Cancer Council of Western Australia (A.D.); National Health and Medical Research Council (NHMRC) project grants #458627 and 572578 and Cancer Council WA Fellowship (R.G.); a research grant from K.B. Chandrashekar Research Foundation, India (S.C.); NHMRC project grants #254504 and #404002 and is a recipient of a NHMRC Career Development Award (L.H.) and a National Heart Foundation of Australia/NHMRC Postgraduate Research Scholarship (H.V.).
The authors have filed a patent application on the use of sFRP4 as an angiogenesis inhibitor.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
References
- 1.Nusse R, Varmus HE. Wnt genes. Cell. 1992;69:1073–1087. doi: 10.1016/0092-8674(92)90630-u. [DOI] [PubMed] [Google Scholar]
- 2.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh JC, Rattner A, Smallwood PM, Nathans J. Biochemical characterization of Wnt-frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc Natl Acad Sci USA. 1999;96:3546–3551. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finch PW, He X, Kelley MJ, Uren A, Schaudies RP, Popescu NC, Rudikoff S, Aaronson SA, Varmus HE, Rubin JS. Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci USA. 1997;94:6770–6775. doi: 10.1073/pnas.94.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masckauchan TN, Agalliu D, Vorontchikhina M, Ahn A, Parmalee NL, Li CM, Khoo A, Tycko B, Brown AM, Kitajewski J. Wnt5a signaling induces proliferation and survival of endothelial cells in vitro and expression of MMP-1 and Tie-2. Mol Biol Cell. 2006;17:5163–5172. doi: 10.1091/mbc.E06-04-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodwin AM, Kitajewski J, D'Amore PA. Wnt1 and Wnt5a affect endothelial proliferation and capillary length; Wnt2 does not. Growth Factors. 2007;25:25–32. doi: 10.1080/08977190701272933. [DOI] [PubMed] [Google Scholar]
- 7.Uren A, Reichsman F, Anest V, Taylor WG, Muraiso K, Bottaro DP, Cumberledge S, Rubin JS. Secreted frizzled-related protein-1 binds directly to Wingless and is a biphasic modulator of Wnt signaling. J Biol Chem. 2000;275:4374–4382. doi: 10.1074/jbc.275.6.4374. [DOI] [PubMed] [Google Scholar]
- 8.Bafico A, Gazit A, Pramila T, Finch PW, Yaniv A, Aaronson SA. Interaction of frizzled related protein (FRP) with Wnt ligands and the frizzled receptor suggests alternative mechanisms for FRP inhibition of Wnt signaling. J Biol Chem. 1999;274:16180–16187. doi: 10.1074/jbc.274.23.16180. [DOI] [PubMed] [Google Scholar]
- 9.Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- 10.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 11.Turashvili G, Bouchal J, Burkadze G, Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology. 2006;73:213–223. doi: 10.1159/000098207. [DOI] [PubMed] [Google Scholar]
- 12.Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- 13.White L, Suganthini G, Friis R, Dharmarajan A, Charles A. Expression of secreted frizzled-related protein 4 in the primate placenta. Reprod Biomed Online. 2009;18:104–110. doi: 10.1016/s1472-6483(10)60431-2. [DOI] [PubMed] [Google Scholar]
- 14.Constantinou T, Baumann F, Lacher MD, Saurer S, Friis R, Dharmarajan A. SFRP-4 abrogates Wnt-3a-induced beta-catenin and Akt/PKB signalling and reverses a Wnt-3a-imposed inhibition of in vitro mammary differentiation. J Mol Signal. 2008;3:10–23. doi: 10.1186/1750-2187-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drake JM, Friis RR, Dharmarajan AM. The role of sFRP4, a secreted frizzled-related protein, in ovulation. Apoptosis. 2003;8:389–397. doi: 10.1023/a:1024181203729. [DOI] [PubMed] [Google Scholar]
- 16.Hsieh M, Mulders SM, Friis RR, Dharmarajan A, Richards JS. Expression and localization of secreted frizzled-related protein-4 in the rodent ovary: evidence for selective up-regulation in luteinized granulosa cells. Endocrinology. 2003;144:4597–4606. doi: 10.1210/en.2003-0048. [DOI] [PubMed] [Google Scholar]
- 17.Fox S, Dharmarajan A. WNT signaling in malignant mesothelioma. Front Biosci. 2006;11:2106–2112. doi: 10.2741/1953. [DOI] [PubMed] [Google Scholar]
- 18.Feng Han Q, Zhao W, Bentel J, Shearwood AM, Zeps N, Joseph D, Iacopetta B, Dharmarajan A. Expression of sFRP-4 and beta-catenin in human colorectal carcinoma. Cancer Lett. 2006;231:129–137. doi: 10.1016/j.canlet.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Gilner JB, Bautch VL, Wang DZ, Wainwright BJ, Kirby SL, Patterson C. Wnt2 coordinates the commitment of mesoderm to hematopoietic, endothelial, and cardiac lineages in embryoid bodies. J Biol Chem. 2007;282:782–791. doi: 10.1074/jbc.M606610200. [DOI] [PubMed] [Google Scholar]
- 20.Dufourcq P, Couffinhal T, Ezan J, Barandon L, Moreau C, Daret D, Duplaa C. FrzA, a secreted frizzled related protein, induced angiogenic response. Circulation. 2002;106:3097–3103. doi: 10.1161/01.cir.0000039342.85015.5c. [DOI] [PubMed] [Google Scholar]
- 21.Goodwin AM, Sullivan KM, D'Amore PA. Cultured endothelial cells display endogenous activation of the canonical Wnt signaling pathway and express multiple ligands, receptors, and secreted modulators of Wnt signaling. Dev Dyn. 2006;235:3110–3120. doi: 10.1002/dvdy.20939. [DOI] [PubMed] [Google Scholar]
- 22.de Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, Rabinovitch M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol. 2009;184:83–99. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viola HM, Arthur PG, Hool LC. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ Res. 2007;100:1036–1044. doi: 10.1161/01.RES.0000263010.19273.48. [DOI] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Chopra M, Dharmarajan AM, Meiss G, Schrenk D. Inhibition of UV-C light-induced apoptosis in liver cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2009;111:49–63. doi: 10.1093/toxsci/kfp128. [DOI] [PubMed] [Google Scholar]
- 26.Auerbach R, Lewis R, Shinners B, Kubai L, Akhtar N. Angiogenesis assays: a critical overview. Clin Chem. 2003;49:32–40. doi: 10.1373/49.1.32. [DOI] [PubMed] [Google Scholar]
- 27.Niemisto A, Dunmire V, Yli-Harja O, Zhang W, Shmulevich I. Robust quantification of in vitro angiogenesis through image analysis. IEEE Trans Med Imaging. 2005;24:549–553. doi: 10.1109/tmi.2004.837339. [DOI] [PubMed] [Google Scholar]
- 28.Berg MN, Dharmarajan AM, Waddell BJ. Glucocorticoids and progesterone prevent apoptosis in the lactating rat mammary gland. Endocrinology. 2002;143:222–227. doi: 10.1210/endo.143.1.8584. [DOI] [PubMed] [Google Scholar]
- 29.White L, Dharmarajan A, Charles A. Caspase-14: a new player in cytotrophoblast differentiation. Reprod Biomed Online. 2007;14:300–307. doi: 10.1016/s1472-6483(10)60871-1. [DOI] [PubMed] [Google Scholar]
- 30.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Jr, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clement-Lacroix P, Ai M, Morvan F, Roman-Roman S, Vayssiere B, Belleville C, Estrera K, Warman ML, Baron R, Rawadi G. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci USA. 2005;102:17406–17411. doi: 10.1073/pnas.0505259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banhegyi G, Benedetti A, Csala M, Mandl J. Stress on redox. FEBS Lett. 2007;581:3634–3640. doi: 10.1016/j.febslet.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 33.Burgering BM, Medema RH. Decisions on life and death: fOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukoc Biol. 2003;73:689–701. doi: 10.1189/jlb.1202629. [DOI] [PubMed] [Google Scholar]
- 34.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 35.Staal FJ, Clevers H. Tcf/Lef transcription factors during T-cell development: unique and overlapping functions. Hematol J. 2000;1:3–6. doi: 10.1038/sj.thj.6200001. [DOI] [PubMed] [Google Scholar]
- 36.Pandur P, Maurus D, Kuhl M. Increasingly complex: new players enter the Wnt signaling network. Bioessays. 2002;24:881–884. doi: 10.1002/bies.10164. [DOI] [PubMed] [Google Scholar]
- 37.Courtwright A, Siamakpour-Reihani S, Arbiser JL, Banet N, Hilliard E, Fried L, Livasy C, Ketelsen D, Nepal DB, Perou CM, Patterson C, Klauber-Demore N. Secreted frizzle-related protein 2 stimulates angiogenesis via a calcineurin/NFAT signaling pathway. Cancer Res. 2009;69:4621–4628. doi: 10.1158/0008-5472.CAN-08-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodwin AM, D'Amore PA. Wnt signaling in the vasculature. Angiogenesis. 2002;5:1–9. doi: 10.1023/a:1021563510866. [DOI] [PubMed] [Google Scholar]
- 39.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–634. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carmon KS, Loose DS. Secreted frizzled-related protein 4 regulates two Wnt7a signaling pathways and inhibits proliferation in endometrial cancer cells. Mol Cancer Res. 2008;6:1017–1028. doi: 10.1158/1541-7786.MCR-08-0039. [DOI] [PubMed] [Google Scholar]
- 42.Schneider I, Houston DW, Rebagliati MR, Slusarski DC. Calcium fluxes in dorsal forerunner cells antagonize beta-catenin and alter left-right patterning. Development. 2008;135:75–84. doi: 10.1242/dev.004713. [DOI] [PubMed] [Google Scholar]
- 43.Topol L, Jiang X, Choi H, Garrett-Beal L, Carolan PJ, Yang Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J Cell Biol. 2003;162:899–908. doi: 10.1083/jcb.200303158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishitani T, Kishida S, Hyodo-Miura J, Ueno N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J, Matsumoto K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol Cell Biol. 2003;23:131–139. doi: 10.1128/MCB.23.1.131-139.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westfall TA, Brimeyer R, Twedt J, Gladon J, Olberding A, Furutani-Seiki M, Slusarski DC. Wnt-5/pipetail functions in vertebrate axis formation as a negative regulator of Wnt/beta-catenin activity. J Cell Biol. 2003;162:889–898. doi: 10.1083/jcb.200303107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torres MA, Yang-Snyder JA, Purcell SM, DeMarais AA, McGrew LL, Moon RT. Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early Xenopus development. J Cell Biol. 1996;133:1123–1137. doi: 10.1083/jcb.133.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein D, Demory A, Peyre F, Kroll J, Geraud C, Ohnesorge N, Schledzewski K, Arnold B, Goerdt S. Wnt2 acts as an angiogenic growth factor for non-sinusoidal endothelial cells and inhibits expression of stanniocalcin-1. Angiogenesis. 2009;12:251–265. doi: 10.1007/s10456-009-9145-5. [DOI] [PubMed] [Google Scholar]
- 48.Wu WB, Peng HC, Huang TF. Disintegrin causes proteolysis of beta-catenin and apoptosis of endothelial cells. Involvement of cell-cell and cell-ECM interactions in regulating cell viability. Exp Cell Res. 2003;286:115–127. doi: 10.1016/s0014-4827(03)00105-8. [DOI] [PubMed] [Google Scholar]
- 49.Hanai J, Gloy J, Karumanchi SA, Kale S, Tang J, Hu G, Chan B, Ramchandran R, Jha V, Sukhatme VP, Sokol S. Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol. 2002;158:529–539. doi: 10.1083/jcb.200203064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoogeboom D, Essers MA, Polderman PE, Voets E, Smits LM, Burgering BM. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J Biol Chem. 2008;283:9224–9230. doi: 10.1074/jbc.M706638200. [DOI] [PubMed] [Google Scholar]
- 51.Almeida M, Han L, Martin-Millan M, O'Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem. 2007;282:27298–27305. doi: 10.1074/jbc.M702811200. [DOI] [PubMed] [Google Scholar]
- 52.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen AW, Carbajal JM, Schaeffer RC., Jr VEGF stimulates tyrosine phosphorylation of beta-catenin and small-pore endothelial barrier dysfunction. Am J Physiol. 1999;277:H2038–H2049. doi: 10.1152/ajpheart.1999.277.5.H2038. [DOI] [PubMed] [Google Scholar]
- 54.Zhang X, Gaspard JP, Chung DC. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 2001;61:6050–6054. [PubMed] [Google Scholar]
- 55.Giambernardi TA, Grant GM, Taylor GP, Hay RJ, Maher VM, McCormick JJ, Klebe RJ. Overview of matrix metalloproteinase expression in cultured human cells. Matrix Biol. 1998;16:483–496. doi: 10.1016/s0945-053x(98)90019-1. [DOI] [PubMed] [Google Scholar]
- 56.Mesiano S, Ferrara N, Jaffe RB. Role of vascular endothelial growth factor in ovarian cancer: inhibition of ascites formation by immunoneutralization. Am J Pathol. 1998;153:1249–1256. doi: 10.1016/S0002-9440(10)65669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251–275. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- 58.Modlich U, Kaup FJ, Augustin HG. Cyclic angiogenesis and blood vessel regression in the ovary: blood vessel regression during luteolysis involves endothelial cell detachment and vessel occlusion. Lab Invest. 1996;74:771–780. [PubMed] [Google Scholar]
- 59.Kim TI, Kim SW, Kim S, Kim T, Kim EK. Inhibition of experimental corneal neovascularization by using subconjunctival injection of bevacizumab (Avastin) Cornea. 2008;27:349–352. doi: 10.1097/ICO.0b013e31815cf67d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.