

Abstract
 We report a fundamentally unique approach to the catalytic kinetic resolution of amine-derivatives based on formamide and thioformamide substrates. Readily accessible histidine-containing peptides mediate the kinetic resolutions with as little as 5 mol% of the catalyst. Selectivity factors (krel) were observed as high as 43.7 under simple reaction conditions utilizing Boc2O as the reagent, and when reactions were conducted at room temperature. Mechanistic experiments were conducted that established a higher level of reactivity for thioformamide substrates relative to their formamide analogs. The products of these asymmetric reactions were shown to be readily converted to desirable building blocks such as N-Boc-amines and the parent chiral formamide compounds.
We report a fundamentally unique approach to the catalytic kinetic resolution of amine-derivatives based on formamide and thioformamide substrates. Readily accessible histidine-containing peptides mediate the kinetic resolutions with as little as 5 mol% of the catalyst. Selectivity factors (krel) were observed as high as 43.7 under simple reaction conditions utilizing Boc2O as the reagent, and when reactions were conducted at room temperature. Mechanistic experiments were conducted that established a higher level of reactivity for thioformamide substrates relative to their formamide analogs. The products of these asymmetric reactions were shown to be readily converted to desirable building blocks such as N-Boc-amines and the parent chiral formamide compounds.
The kinetic resolution (KR) of amines is a long-standing problem in the field of asymmetric synthesis (eq 1). Approaches to this problem have included the application of enzymes,1 as well as chiral acylating agents employed in stoichiometric quantities.2 Small-molecule catalytic approaches have also been advanced.3 An elegant strategy based on the use of chiral hydrogen bond donors in combination with achiral nucleophiles as co-catalysts has also appeared recently.4 In each case, important advances have resulted, but significant challenges remain including substrate scope, simplicity of catalysts, and straightforward, room temperature reaction conditions.
 |
(1) |
It is perhaps fair to say that progress in amine KR has trailed the parallel challenge of alcohol KR, wherein a wide variety of catalysts have been introduced that, collectively, address a broad range of substrate types, conceptual strategies and mild reaction conditions.5 Among the challenges associated with amines as substrates is their higher level of reactivity; oftentimes, the background rate of reactions of amines with simple derivatization agents (e.g., anhydrides) is competitive with catalytic rates, leading to low levels of selectivity. Approaches to this problem have included the use of low reactivity amines (e.g., indolines),3a,c or reduced activity electrophiles, such as O-acyl oxazolines, each leading to processes amenable to enantioselective catalysis.3b
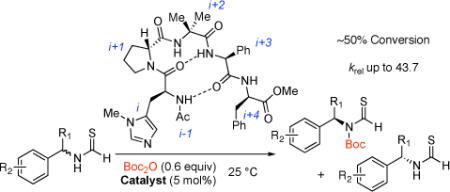
Our approach to the KR of amines and their derivatives has centered on the application of a common catalyst scaffold, based on simple π-methylated histidine derivatives (Pmh-peptides, 1, Scheme 1). The approach has been applied to the selective functionalization of a broad range of alcohols with a variety of electrophiles.6 For amine functionalization, we sought a simple substrate that might exhibit enhanced rates of catalytic acylation, with reduced background rates relative to the reactivity of a free amine. Our hypothesis was that simple formamides (2) would react through a kinetically favorable O-acylation of the formamide oxygen, followed by a rearrangement of the unstable O-acylated intermediate 3 to give the stable imide 4. Our goal was to capitalize on chiral frameworks previously derived from 1 for enantioselectivity. Furthermore, if the amine derivatization reagent were established as the ubiquitous di-t-butyldicarbonate (Boc2O), products such as 4 could then be converted to practical building blocks such as N-Boc-amines (5), or back into the corresponding formamides (2) under straightforward conditions.
Scheme 1.

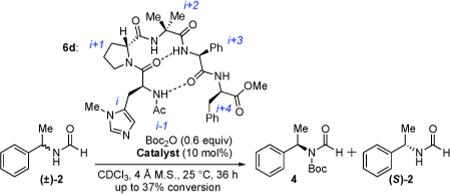
In initial experiments, we were pleased to show that formamides were inert to reactions with Boc2O alone, but subject to catalytic functionalization with Pmh-containing peptides (eq 2). Moreover, catalysts in a β–turn series (6a-e) were found to exhibit enantioselectivity (Table 1). For example, catalyst 6a led to 4 with 79.5:20.5 e.r., at 22% conversion (krel = s-factor7a = 4.6, entry 1).
 |
(2) |
Table 1.
Catalyst Screen and Optimization Results with Formamide Substrate 2.
| Entry | Catalyst | i−1 | i+1 | i+2 | i+3 | i+4 | s-factora |
|---|---|---|---|---|---|---|---|
| 1 | 6a | Boc | d-Pro | Sp6b | l-Val | d-Phe-OMe | 4.6 |
| 2 | 6b | Boc | d-Pro | Aibc | l-Phgd | d-Phe-Ome | 7.8 |
| 3 | 6c | Boc | d-Pipe | Aibc | l-Phgd | d-Phe-Ome | 5.3 |
| 4 | 6d | Ac | d-Pro | Aibc | l-Phgd | d-Phe-Ome | 9.6 |
| 5 | 6e | Boc | d-Pro | Aibc | l-Phgd | −NMe2 | 3.2 |
Calculated using the method of Kagan.7a
1-Aminocyclohexanecarboxylic acid.
1-Aminoisobutyric acid.
Phenylglycine.
Pipecolinic acid.
Optimization of the catalyst structure led to the identification of critical sectors. Catalyst 6b, with alteration of the i+3 residue (L-valine to L-phenylglycine), is incrementally more selective than 6a (s = 7.8, entry 2). Catalyst 6c, with D-pipecolinic acid in place of D-proline, is less selective (s = 5.3, entry 3), implying that secondary structure is important.8 Moreover, modification at the N- and C-termini (6d and 6e, respectively) proved important, revealing that if the N-terminal substituent is exchanged from Boc to acetyl, selectivity is enhanced (s = 9.6, entry 4). This change may be due to the heightened integrity of the β-hairpin structure, associated with a stronger amide-to-amide hydrogen bond.9 In addition, if the catalyst is truncated at the C-terminal side (6e, replacement of the i+4 residue with NMe2, entry 5), the s-factor drops to 3.2. Thus, we established catalyst 6d as our lead catalyst.
While Table 1 revealed an ability to modulate selectivity, reactivity of formamides was modest. Reactions typically proceeded to only ~37% conversion after 36 h at 25 °C. Furthermore, to reach the ideal extent of conversion (50%), 20 mol% catalyst and an excess (1.2 equiv) of Boc2O were needed. Inspired by classical literature regarding the reactivity of thiocarbonyl compounds,10 we hypothesized that thio-formamides would be more reactive. Furthermore, it had been shown that thioformamides react with acyl chlorides via S-acylation followed by intramolecular S-to-N acyl transfer.11 Indeed, a competition experiment involving formamide 2 and the corresponding thioformamide 7 (eq 3) confirmed our hypothesis. Upon exposure to 1 equiv of Boc2O, a 1:1 mixture of 2 and 7 is converted almost exclusively to 8. Moreover, near total conversion is observed within 12 h, with only 5 mol% catalyst, at 25 °C.
 |
(3) |
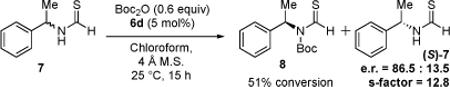
Under conditions of KR, compound 7 reaches the 50% conversion point with only 5 mol% 6d and only 0.6 equiv Boc2O within 15 hours (eq 4). Importantly, substrate 7 undergoes reaction with a nominal s-factor = 12.8 under these conditions.12,13,14 Moreover, thioformamides exhibit the same selectivity trends as their formamide analogs when evaluated with other catalysts.15 In addition, substrates in both formamide and thioformamide series undergo KR with excellent reproducibilty on scales of up to 0.5 mmol.
 |
(4) |
Having established a catalyst that operates at 5 mol% loading, a substrate that exhibits enhanced reactivity and appreciable selectivity at 25 °C, we sought to assess substrate scope (Table 2). In addition to the parent sec-phenethylamine derivative (entry 1), a variety of arylethylamines are excellent substrates for catalyst 6d. In fact, the data presented below represent, in most of the cases, the highest s-factors observed for non-enzymatic KRs of the corresponding amines or their simple amine derivatives, in particular when these reactions are carried out at 25 °C. meta-Methoxy-substituted formamide 9 undergoes the KR exhibiting a nominal s-factor of 32.5 (entry 2). The corresponding meta-phenoxy-substituted compound 10 is also an excellent substrate (s = 25.2, entry 3), while the analogous meta-bromo-substituted compound 11 is somewhat less selective (s = 12.5, entry 4). 3′,5′-Dimethoxy-substituted derivative 12 yields the highest s-factor we have observed to date (s = 43.7, entry 5). para-Substitution is also tolerated. para-Methoxy- and para-phenyl-substituted compounds 13 and 14 exhibited s-factors of 12.2 and 10.2, respectively (entries 6 and 7). 2′-Naphthyl-substituted compound 15 reveals an s-factor of 13.8 (entry 8), and arylpropylamine 16 exhibits an s-factor of 9.4 (entry 9). Even 17, presenting a heteroaryl and a phenyl ring within the substrate, reveals an s-factor of 5.6 (entry 10), suggesting potential for KR of amine derivatives of unusual functionality.
Table 2.
Substrate Scope for the KR of Thioformamides with Peptide 6da
| Entry | Substrate | % Conversionb | e.r.c | Nominal s-factord |
|---|---|---|---|---|
| 1 | 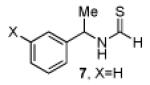 |
51 | 86.5 : 13.5 | 12.8 |
| 2 | 9, X = OMe | 52 | 95.5 : 4.5 | 32.5 |
| 3 | 10, X = OPh | 58 | 97.5 : 2.5 | 25.2 |
| 4 | 11, X = Br | 52 | 88 : 12 | 12.5 |
| 5 | 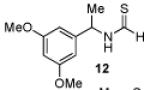 |
52 | 96.5 : 3.5 | 43.7 |
| 6 | 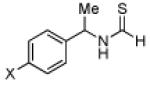 |
52 | 88.2 : 11.8 | 12.2 |
| 7 |
13, X = OMe 14, X = Ph |
53 | 86.5 : 13.5 | 10.2 |
| 8 | 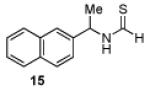 |
54 | 90.8 : 9.2 | 13.8 |
| 9 | 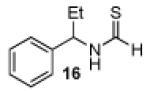 |
52 | 85 : 15 | 9.4 |
| 10 | 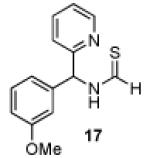 |
53 | 79 : 21 | 5.6 |
Reactions were carried out at 25 °C for 24 h in CHCl3 (0.30 M) with 0.6 equiv Boc2O, 5 mol% of 6d, and in the presence of (TMS)2O as an internal standard and 4 Å mol. sieves. (Entries 3 and 10 employed 0.65 equiv Boc2O.)
Based on substrate relative to internal standard.
Reported for recovered starting material after conversion to the formamide; chiral HPLC analysis. In the case of entry 1, the fast reacting enantiomer was found to be (R)-7, providing(S)-7 as the recovered starting material. The other cases were not explicitly established.
See footnote 12.
The protocol we have established also allows access to amine derivatives of practical utility. For example, the conversion of 8 to carbamate 5 is possible employing oxidative hydrolysis (Scheme 2). Conversely, 8 is converted to formamide 2 by acidic cleavage of the Boc-group and S-to-O exchange.
Scheme 2.

In summary, we have developed a new strategy for the KR of formamides and thioformamides based on catalyst 6d. This work expands the generality of Pmh-based catalysts to include a novel class of amine-based substrates.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health Foundation (GM-068649) for financial support.
Footnotes
SUPPORTING INFORMATION. Experimental procedures and characterization. This information is available free of charge via the internet at http://pubs.acs.org
References
- 1.Gotor-Fernández V, Gotor V. Curr. Opin. Drug Disc. Devel. 2009;12:784. [PubMed] [Google Scholar]
- 2.(a) Arseniyadis S, Subhash PV, Valleix A, Mathew SP, Blackmond DG, Wagner A, Mioskowski C. J. Am. Chem. Soc. 2005;127:6138. doi: 10.1021/ja051302+. [DOI] [PubMed] [Google Scholar]; (b) Arseniyadis S, Valleix A, Wagner A, Mioskowski C. Angew. Chem. Int. Ed. 2004;43:3314. doi: 10.1002/anie.200453956. [DOI] [PubMed] [Google Scholar]; (c) Ie Y, Fu GC. Chem. Commun. 2000:119. [Google Scholar]
- 3.(a) Arp FO, Fu GC. J. Am. Chem. Soc. 2006;128:14264. doi: 10.1021/ja0657859. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Arai S, Bellemin-Laponnaz S, Fu GC. Angew. Chem. Int. Ed. 2001;40:234. [PubMed] [Google Scholar]; (c) Birman VB, Jiang H, Li X, Guo L, Uffman EW. J. Am. Chem. Soc. 2006;128:6536. doi: 10.1021/ja061560m. [DOI] [PubMed] [Google Scholar]; (d) Arnold K, Davies B, Herault D, Whiting A. Angew. Chem. Int. Ed. 2008;47:2673. doi: 10.1002/anie.200705643. [DOI] [PubMed] [Google Scholar]
- 4.De CK, Klauber EG, Seidel D. J. Am. Chem. Soc. 2009;131:17060. doi: 10.1021/ja9079435. [DOI] [PubMed] [Google Scholar]
- 5.Wurz RP. Chem. Rev. 2007;107:5570. doi: 10.1021/cr068370e. and references therein. [DOI] [PubMed] [Google Scholar]
- 6.Fiori KW, Puchlopek ALA, Miller SJ. Nature Chem. 2009;1:630. doi: 10.1038/nchem.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Kagan HB, Fiaud JC. Topics in Stereochem. 1988;18:249. [Google Scholar]; (b) Keith JM, Larrow JF, Jacobsen EN. Adv. Synth. Catal. 2001;1:5. [Google Scholar]
- 8.(a) Chung YJ, Christianson LA, Stanger HE, Powell DR, Gellman SH. J. Am. Chem. Soc. 1998;120:10555. [Google Scholar]; (b) Haque TS, Little JC, Gellman SH. J. Am. Chem. Soc. 1996;118:6975. [Google Scholar]
- 9.Gilli P, Pretto L, Bertolasi V, Gilli G. Acc. Chem. Res. 2009;42:33. doi: 10.1021/ar800001k. [DOI] [PubMed] [Google Scholar]
- 10.Wentland MP, Hansen PE, Schow SR, Daum SJ. Tetrahedron Lett. 1989;30:6619. [Google Scholar]
- 11.Walter W, Saha CR. Phosphorus Sulfur. 1985;25:63. [Google Scholar]
-
12.Thioformamides exhibit high cis-trans rotational barriers (eq 5), leading to the possibility of independent reactivity of the cis- and trans-isomers. Thus, each of the isomers may exhibit different enantioselectivity (i.e., a distinct s-factor). It is unclear which rotational isomer is more reactive at this time. This issue could have a significant effect on the calculation of the s-factor. Nonetheless, in the data we report, we calculate a “nominal s-factor,” treating the rotational isomers of starting material and product in a collective fashion.

(5) - 13.Walter W, Shaumann E, Reubke K-J. Angew. Chem. Int. Ed. 1968;7:467. [Google Scholar]
- 14.The inclusion of 4Å molecular sieves leads to modest rate acceleration.
- 15.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.