

Abstract
Background
Glioblastoma multiforme (GBM), a highly invasive and vascular cancer, responds poorly to conventional cytotoxic therapy. Integrins, widely expressed in GBM and tumor vasculature, mediate cell survival, migration and angiogenesis. Cilengitide is a potent αvβ3 and αvβ5 integrin inhibitor.
Objective
To summarize the preclinical and clinical experience with cilengitide for GBM.
Methods
Preclinical studies and clinical trials evaluating cilengitide for GBM were reviewed.
Results/conclusions
Cilengitide is active and synergizes with external beam radiotherapy in preclinical GBM models. In clinical trials for recurrent GBM, single-agent cilengitide has antitumor benefits and minimal toxicity. Among newly diagnosed GBM patients, single-arm studies incorporating cilengitide into standard external beam radiotherapy/temozolomide have shown encouraging activity with no increased toxicity and have led to a planned randomized Phase III trial.
Keywords: angiogenesis, glioblastoma multiforme, integrins, malignant glioma, vascular endothelial growth factor
1. Introduction
Despite current multimodality therapies integrating surgery, radiation therapy (external beam radiotherapy, XRT) and chemotherapy, outcome for patients with newly diagnosed glioblastoma multiforme (GBM), the most common adult primary CNS tumor, is poor with median progression-free survival (PFS) and overall survival (OS) of only 6.9 and 14.6 months, respectively [1]. Furthermore, following recurrence there is no established therapy. Therefore, better therapies for newly diagnosed GBM patients must be identified, and beneficial salvage therapies following progression are critically needed.
Glioblastoma multiformes universally demonstrate a fecund propensity to invade and migrate extensively within the CNS yet, paradoxically, nearly never metastasize beyond the CNS. They exhibit prolific angiogenesis, driven by markedly upregulated VEGF expression owing to dysregulated growth factor signaling and intra-tumoral hypoxia [2]. Recent results suggest that tumor angiogenesis may be a highly valuable therapeutic target in GBM. Specifically, administration of bevacizumab, a humanized monoclonal antibody against VEGF, in combination with irinotecan, a topoisomerase inhibitor, significantly improves outcome among recurrent patients [3,4]. However, in preclinical GBM models, effective VEGF inhibition can increase tumor cell invasiveness with apparent co-option of normal host vessels [5]. Thus, in addition to VEGF blockade, successful antiangiogenic strategies for GBM may require concomitant targeting of tumor cell migration and invasion.
Integrins, a family of 24 transmembrane receptors, are named for their ability to integrate extracellular and intracellular activities. They are heterodimers composed of paired alpha and beta chains that regulate tumor angiogenesis, invasion and migration by mediating critical cell-to-cell and cell-extracellular matrix interactions [6-8]. Integrins bind specifically, based on respective alpha and beta domain pairings, to several key microenvironment ligands, including vitronectin, fibronectin, laminin, fibroblast-growth factor, MMP-2, thrombospondin, fibrin and fibrinogen. Several integrins including αvβ3 and αvβ5 recognize the arginine-glycine-aspartic acid (RGD) peptide of extracellular ligands. Upon ligand binding, integrins, which lack intrinsic kinase activity, aggregate to form cell membrane focal adhesions. Focal adhesion kinase (FAK) is recruited and autophosphorylates at these sites, in turn activating signaling through phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinase (ERK)/MAPK. Integrins are attractive therapeutic targets owing to increased expression by both GBM cells and tumor vasculature [9-11].
The identification of integrins as key molecular players in multiple tumor cell processes prompted the search for potential inhibitors. Initial αvβ3 and αvβ5 integrin inhibitor candidates were primarily antibodies and cyclic or linear peptides [12]. The peptidomimetics developed included early versions of cilengitide containing the RGD sequence. Cilengitide (EMD 121974, Merck KGaA, Darmstadt, Germany) is a cyclized pentapeptide [Arg-Gly-Asp-DPhe-(NMeVal) ] designed to block integrin-mediated adhesion and migration (Figure 1) [13]. Cilengitide demonstrated selective and potent affinities for isolated immobilized αvβ3 and αvβ5 with an IC50 between 3 and 40 nM and was subsequently demonstrated to directly bind αvβ3 integrin. Cell attachment assays to vitronectin or fibronectin confirmed cellular activity with selective attachment inhibition at 0.4 uM [12,14,15]. Cilengitide's selectivity was documented by minimal inhibition of cell attachment mediated by other integrins. Likewise, binding of platelet glycoprotein IIb/IIIa to fibrinogen was only weakly affected by cilengitide.
Figure 1. Structure of cilengitide highlighting the RGD-binding domain (left) and the structure of integrin αv (red) and β3 (blue).
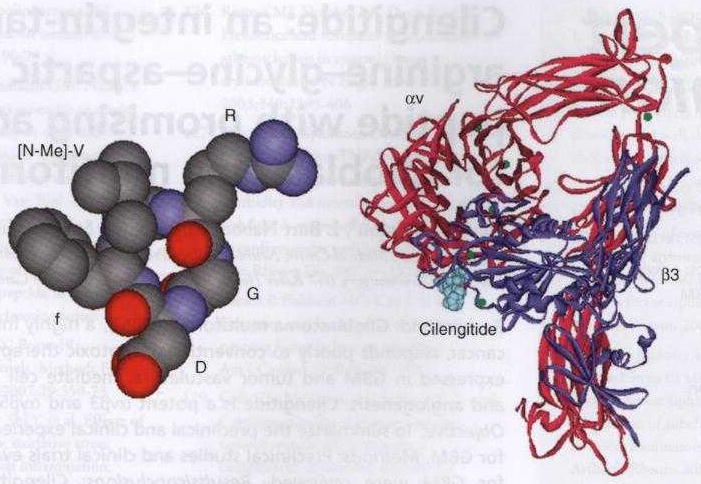
This demonstrates the binding of cilengitide through the RGD domain (right).
In this review we will summarize the rationale and current experience of cilengitide in the treatment of GBM. Preclinical studies confirmed both the importance of integrin signaling in GBM as well as the antitumor benefit of integrin antagonists in GBM tumors. The clinical evaluation of cilengitide has followed a well-planned and methodical path initially in recurrent GBM patients and subsequently in newly diagnosed patients (Table 1). Consistent evidence of antitumor benefit with cilengitide across studies highlights the promise of integrin antagonists in the treatment of GBM.
Table 1. Clinical trials evaluating cilengitide in adults with malignant glioma.
| Study | No. of patients | Trial phase | Trial design | Study population | Dose | Ref. |
|---|---|---|---|---|---|---|
| NABTT 9911 | 51 | I | Dose escalation | Recurrent MG | 200 – 2400 mg/m2 | [43] |
| EMD 009 | 81 | II | Randomized | Recurrent GBM | 500 – 2000 mg | [45] |
| NABTC 0302 | 30 | II (peri-operative) | Randomized | Recurrent GBM | 500 versus 2000 mg | [46] |
| EMD 010 | 52 | II | Single-arm | Newly diagnosed GBM | 500 mg | [47] |
| NABTT 0306 | 107 | I/II | Randomized | Newly diagnosed GBM | 500 versus 2000 mg | NA |
EMD: EMD Serono, Inc. (US affiliate of Merck KGaA, Darmstadt, Germany); GBM: Glioblastoma multiforme; MG: Malignant glioma; NA: Not available; NABTC: North American Brain Tumor Consortium; NABTT: New Agents Brain Tumor Therapy Group.
2. Preclinical studies
Integrins, especially αvβ3, regulate cellular properties for both gliomas and endothelial cells, and support their growth-factor-mediated survival [16]. They can act in synchrony with growth factor receptors to coordinate cell proliferation and promote survival. Ligated αvβ3 specifically cooperates with activated growth factor receptors to maintain cell cycle progression and suppress apoptosis [17,18]. Endothelial cell apoptosis is also inhibited by αvβ3 and αvβ5 [19-21].
Blocking αvβ3–ligand interactions disrupts cell attachment, migration and differentiation in response to growth factors in vitro [22-24], inhibits blood vessel formation in vivo [25-27] and retards tumor growth without detectably influencing pre-existing blood vessels [27]. In addition, recent evidence suggests that the antiangiogenic activity of cilengitide also involves inhibiting proliferation and differentiation of human endothelial cell precursor cells [28].
The upregulation of many integrins has been found in glioma cells as compared with normal brain and/or normal astrocytes. Principal roles in tumor astrocyte migration are played by α3β1, αvβ1, αvβ3 and αvβ5 integrins [23,29-31]. Inhibition of the αv integrins with a cyclic RGD peptide impairs angiogenesis, growth and metastasis of solid tumors in vivo [32]. Cilengitide induces apoptosis in U87 glioma cells by preventing adherence to vitronectin and tenascin, matrix protein mediators of brain tumor invasion and growth [33]. Single agent cilengitide has been used in animal models of human glioma, with promising efficacy [34,35].
As cilengitide acts primarily to block survival pathways, enhanced antitumor activity may occur in combination with conventional cytotoxic or pro-apoptotic therapies, including radiation. In fact, cilengitide targeting of αvβ3 integrins synergizes with radio-immunotherapy to increase efficacy and apoptosis in breast cancer [36] and NSCLC xenograft models [37]. In glioma, sublethal radiation may actually promote glioma cell migration and increase αvβ3 expression [38,39]. Most recently, Mikkelsen demonstrated dramatically enhanced antitumor activity of radiotherapy by cilengitide in two-week-old intra-cranial U251 gliomas in nude rats but only when cilengitide was given 4 – 8 h before radiation [40].
3. Clinical studies: recurrent GBM
In an initial Phase I study, 37 patients with advanced solid tumors received a 1 h cilengitide infusion twice-weekly [41]. Successive cohorts received 30 to 1600 mg/m2 without dose-limiting toxicities (DLT) or definition of a maximum-tolerated dose (MTD). Pharmacokinetics were dose-independent and AUC and Cmax increased in a dose proportional manner. The systemic exposure at 120 mg/m2 were within the range of tumor inhibition in preclinical models. Pharmacodynamic studies of serum angiogenic factors did not correlate with activity. Three patients achieved stable disease.
A second Phase I study in advanced solid tumor patients focused on new end points utilizing correlative biology assessments to determine biological activity [42]. Cilengitide was administered twice-weekly at either 600 or 1200 mg/m2 without DLTs or identification of MTD. Among evaluated biological measures, including endothelial cell apoptosis, gene expression profiles, systemic angiogenic factor measurements and tumor tissue mean vessel density, none reliably predicted antitumor activity.
A Phase I trial for recurrent malignant glioma (MG) enrolled 51 patients and escalated from 120 to 2400 mg/m2. Again no consistent or dose-dependent toxicities were observed. Some potentially treatment-related severe toxicities were reported at various dose levels; grade 3 thrombosis, grade 4 myalgia/arthralgia, grade 3 anorexia, grade 3 electrolyte disturbance and grade 3 thrombocytopenia. Also no patient developed intracranial hemorrhage. Two and three patients achieved complete and partial responses, respectively, and six patients achieved stable disease for over 6 months. Extensive pharmacokinetic (PK) analysis concluded that among MG patients: i) cilengitide kinetics were linear; ii) dosing per body surface area did not affect clearance suggesting flat dosing was feasible; and iii) PK parameters were not affected by concurrent use of enzyme inducing anticonvulsants. Correlative studies evaluated perfusion MRI and plasma angiogenic growth factors and showed that decreased tumor perfusion occurred more commonly with higher cilengitide doses (Figure 2) [43].
Figure 2. The perfusion MRI method of dynamic contrast susceptibility was used to determine tumor blood volume and flow and follow changes to these values during treatment with cilengitide.
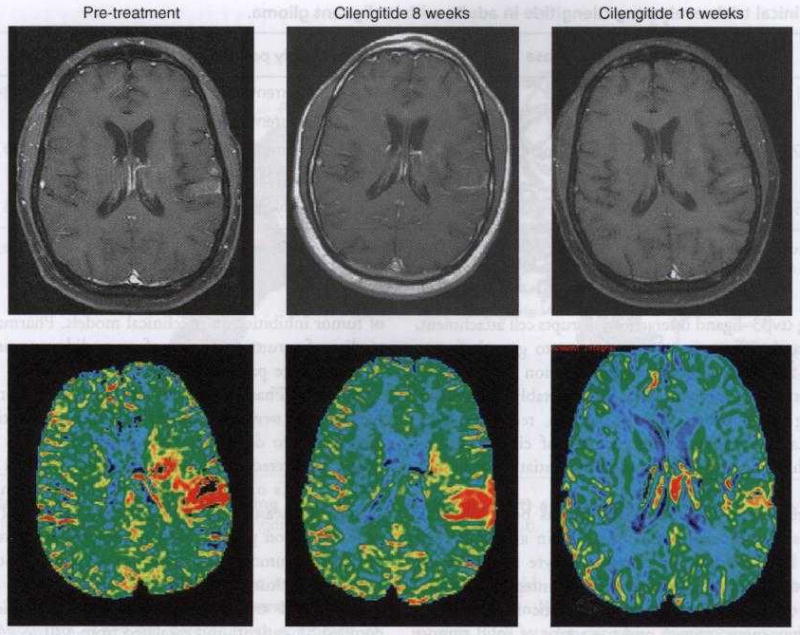
This example demonstrates a glioblastoma multiforme with enhancement on pre-treatment baseline noted on the T1 post gadolinium scan (upper panel) involving the medial and lateral left parietal lobe. The corresponding color map of the cerebral blood volume (bottom panel) reveals substantial signal (red) corresponding to areas of enhancement on the pre-treatment baseline consistent with robust angiogenesis. Of note is the fact that both the gadolinium enhancement and perfusion signals diminish sequentially following 8 and 16 weeks of cilengitide therapy, respectively.
A similar Phase I study enrolled 31 pediatric recurrent MG patients and again there were no DLTs and an MTD was not defined. However, 3 of 13 patients at the highest dose level (2400 mg/m2) experienced self-limiting grade 3 or 4 intracranial hemorrhage (two of these events were asymptomatic). Whereas one patient with a recurrent GBM experienced a complete response, two had stable disease [44].
Based on the encouraging adult MG Phase I experience, two trials were planned to address questions integral to further development of cilengitide for adult GBM. In these studies, the twice-weekly i.v. regimen was retained, however the dosing per square meter was replaced by a flat dosing scheme. First, a randomized Phase II study in recurrent GBM was developed to help determine the optimal dose of cilengitide [45]. In this study, two cilengitide dose levels were evaluated, including an intermediate low (500-mg) dose and an intermediate high (2000-mg) dose because neither MTD nor a clearcut dose-response was determined in the previous Phase I [43]. The primary end point was percentage of patients alive and progression-free at 6 months (6-PFS); the study was not powered to directly compare the two dose-level strata. Key eligibility criteria included: histologically confirmed GBM that recurred following surgery, radiotherapy and no more than one chemotherapy regimen; age ≥ 18 years; measurable, contrast-enhancing tumor; KPS ≥ 70%; and adequate bone marrow, hepatic and renal function. Eligible patients were randomized to receive single-agent cilengitide at a flat dose of either 500 or 2000 mg over 1 h twice-weekly with at least 72 h between infusions. Four-week treatment cycles were repeated until unacceptable toxicity, progressive disease (PD) or consent withdrawal. Stratification factors for randomization included surgery immediately before enrollment (none versus biopsy/subtotal resection) and KPS (70 – 80 versus 90 – 100). A total of 81 patients enrolled, including 41 on the 500-mg arm and 40 on the 2000-mg arm. No significant reproducible toxicities were observed on either arm confirming the excellent safety profile of cilengitide (Table 2). One intracranial hemorrhage occurred but developed at tumor progression, and thus may have been unrelated to cilengitide. Antitumor activity was achieved for both treatment cohorts, however, all outcome measures trended more favorably among patients who received 2000 mg/dose, and none favored the 500-mg cohort. Specifically, the radiographic response (Figure 3) rate, the 6-PFS and the number of patients completing ≥ 12 cycles of therapy were all higher for the 2000-mg cohort (Table 3). Given that effective delivery through the blood–brain barrier poses a challenge for brain tumor patients, a second study was designed to determine whether cilengitide effectively penetrates into GBMs in human patients. In this North American Brain Tumor Consortium (NABTC) study, GBM patients with no more than two previous progressions were randomized to receive three cilengitide doses at either 500 or 2000 mg per dose on days 8, 4 and 1 before planned surgical debulking [46]. Postsurgery, patients received twice-weekly cilengitide at 2000 mg/dose. Among 30 accrued patients, cilengitide was well tolerated with no significant toxicity including no peri-operative hemorrhage or wound healing complications. Determination of intratumoral cilengitide levels 24 h after the last cilengitide dose revealed mean tumor concentrations of 919 and 2561 ng/g of tissue for the 500 and 2000 mg dosing cohorts, respectively, yielding tissue to plasma ratios of 1.83 for the 500-mg dose and 4.17 for the 2000-mg dose. These PK findings confirm that cilengitide is effectively delivered into primary human GBM tumors with good retention for at least 24 h.
Table 2. Toxicities observed in 5% or more patients in Phase II studies with cilengitide.
| Toxicity | Grade | Recurrent GBM (in %): n = 81 (EMD 009 Study) [40] | Newly diagnosed GBM (in %): n = 52 (EMD 010 Study) [42] |
|---|---|---|---|
| Hematologic events | |||
| Anemia | 1 or 2 | 60 | NR |
| 3 or 4 | 0 | 0 | |
| Neutropenia | 1 or 2 | 1 | NR |
| 3 or 4 | 0 | 10 | |
| Thrombocytopenia | 1 or 2 | 0 | NR |
| 3 or 4 | 0 | 13 | |
| Non-hematologic events | |||
| Anorexia | 1 or 2 | <5 | 10 |
| 3 or 4 | 0 | 0 | |
| Diarrhea | 1 or 2 | 6 | <5 |
| 3 or 4 | 0 | 0 | |
| Fatigue | 1 or 2 | 11 | 14 |
| 3 or 4 | 0 | 0 | |
| Headache | 1 or 2 | 5 | < 5 |
| 3 or 4 | 0 | 0 | |
| Nausea | 1 or 2 | 5 | 17 |
| 3 or 4 | 0 | 0 |
EMD: EMD Serono, Inc. (US affiliate of MerckKGaA, Darmstadt, Germany); GBM: Glioblastoma multiforme; NR: Not reported.
Figure 3. Contrast-enhanced magnetic resonance images of representative responses to cilengitide.
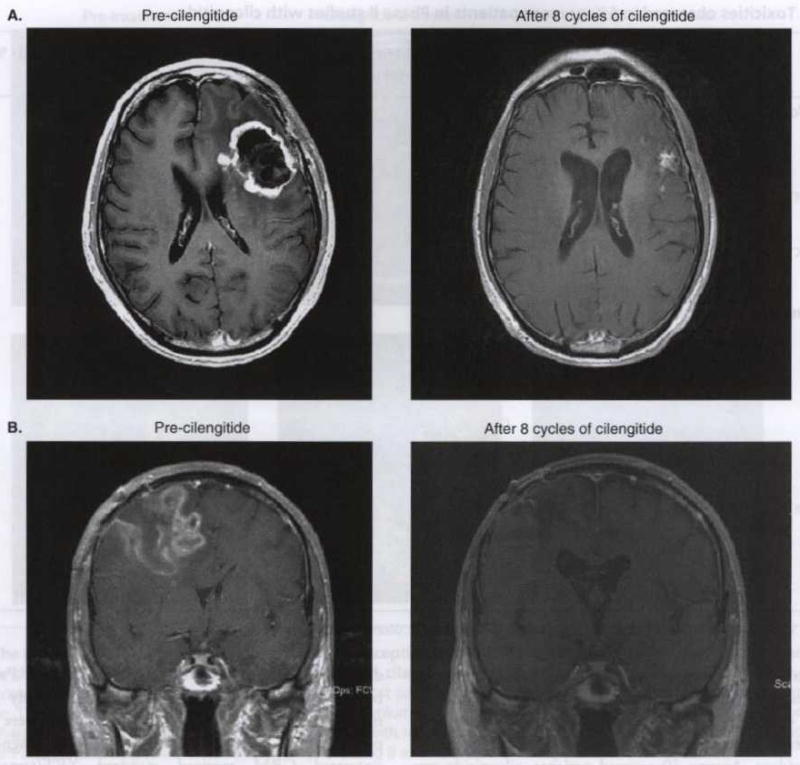
These include a patient who achieved a partial response (A) and a patient who achieved a complete response (B).
Table 3. Efficacy of single-agent cilengitide among recurrent GBM patients at first recurrence by dose level [40].
| Outcome or response | Stratum A (n = 41) (500 mg/day) | Stratum B (n = 40) (2000 mg/day) |
|---|---|---|
| Number of patients with a radiographic response | 2 (5) | 5 (13) |
| Median time to progression (weeks) | 7.9 | 8.1 |
| 95% Cl | 7.7, 15.6 | 7.9, 15.0 |
| 6-month PFS (%) | 10 | 15 |
| 95% Cl | 2.8, 23.7 | 5.7, 29.8 |
| Overall survival (months) | 6.5 | 9.9 |
| 95% Cl | 5.2, 9.3 | 6.4, 15.7 |
| HR | 0.70 [0.43, 1.14], p = 0.15 | |
| Number of patients completing ≥ 12 cycles | 3 (7) | 5 (13) |
| Number of patients completing ≥ 24 cycles | 2 (5) | 2 (5) |
Numbers in parentheses denote percentage unless otherwise indicated.
GBM: Glioblastoma multiforme; HR: Hazard ratio; PFS: Progression-free survival.
The collective cilengitide studies for recurrent MG so far confirm that cilengitide monotherapy is well tolerated, effectively penetrates into GBM tumors and is clinically active.
4. Clinical studies: newly diagnosed GBM
The low toxicity and encouraging activity observed among recurrent MG patients [43,45], as well as synergistic interaction of cilengitide with radiation therapy in preclinical GBM models [40], compelled the evaluation of cilengitide for newly diagnosed GBM patients in two initial studies. Preliminary results of the first of these trials (EMD study #010), a multi-center, single-arm, Phase II study, were recently presented [47]. Fifty-two adults with newly diagnosed and untreated GBM received standard XRT/temozolomide (TMZ) followed by six TMZ cycles as per the recent EORTG/NCIC study [1]. All patients also received 500 mg of cilengitide administered twice-weekly beginning one week before XRT/TMZ and continued through six post-XRT, TMZ cycles. Cilengitide maintenance after the end of adjuvant TMZ was optional and seven patients continued cilengitide for up to 2 years. Toxicity was comparable to that observed with XRT/TMZ alone. Specifically, hematologic toxicity was modest and significant non-hematologic toxicities were uncommon (Table 2). Compliance was excellent with 96, 83 and 88% of patients completing planned XRT, planned TMZ during XRT and planned cilengitide during XRT, respectively. With a median follow-up of 18 months, a modest improvement in PFS and OS was noted compared to that of standard TMZ chemoradiation (Table 4) [1]. Of note, patients with lowered methylguanine methyltransferase (MGMT) expression based on gene promoter methylation [48] appeared to benefit more with the addition of cilengitide therapy, whereas patients with unmethylated MGMT had no apparent benefit with cilengitide. Further assessment of this association, as well as elucidation of potential mechanisms of action, is required in future studies.
Table 4. Summary of Cilengitide dose levels and activity in malignant glioma clinical trials compared with historical controls.
| Study | Phase | No. of patients | Population | Cilengitide dose | ORR | PFS | Median OS (months) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Recurrent patients | ||||||||
| NABTT 9911 | I | 51 | MG | 120 – 2400 mg/m2 | 10% | NA | 5.6 | [38] |
| EMD 009 | II | 41 | GBM | 500 mg | 5% | 10% | 6.5 | [40] |
| 40 | GBM | 2000 mg | 15% | 15% | 9.9 | |||
| NABTC 03-02 | II | 8 | GBM | 500 mg | TE | TE | TE | [41] |
| 11 | GBM | 2000 mg | TE | TE | TE | |||
| Historical control | II | 112 | GBM | NA | 5% | 21% | 7.6 | [44] |
| Newly diagnosed patients | ||||||||
| EMD 010 | II | 22 | GBM; MGMT+ | 500 mg | NA | 3.4 months | 13.1 | [42] |
| 23 | GBM; MGMT− | 500 mg | NA | 13.4 months | NA | |||
| NABTT 0306 | II | −50 | GBM | 500 mg | TE | TE | TE | NA |
| −50 | GBM | 2000 mg | TE | TE | TE | |||
| Historical control | II | 60 | GBM; MGMT+ | NA | NA | 5.3 months | 12.7 | [43] |
| II | 46 | GBM; MGMT− | NA | NA | 10.3 months | 21.7 | ||
EMD: EMD Serono, Inc. (US affiliate of MerckKGaA, Darmstadt, Germany); GBM: Glioblastoma multiforme, MG: Malignant glioma; MGMT−: Methylguanine methyltransferase gene promoter methylated; MGMT+: Methylguanine methyltransferase gene promoter not methylated; NA: Not available; NABTC: North American Brain Tumor Consortium; NABTT: New Agents Brain Tumor Therapy Group; ORR: Overall response rate; OS: Overall survival; PFS: Progression-free survival; TE: Too early.
The second study evaluating cilengitide for newly diagnosed GBM patients was done by the NABTT. In this study, cilengitide was also administered twice-weekly throughout standard XRT/TMZ, during six monthly cycles of post-XRT TMZ and continued thereafter until disease progression. After a Phase I, safety lead-in with three, six-patient cohorts at cilengitide doses of 500, 1000 and 2000 mg, respectively, the protocol evaluated lower and higher doses by randomizing patients to receive either 500 or 2000 mg of cilengitide per dose with standard XRT/TMZ. MGMT will be assessed from archival tumor tissue and correlated with outcome. This study recently completed accrual and results are forthcoming.
Based on the encouraging Phase I and II trial results for recurrent GBM, and the recently reported newly diagnosed GBM study, a large, international, randomized Phase III study evaluating the addition of cilengitide to standard TMZ chemoradiation compared to standard TMZ chemoradiation alone for newly diagnosed GBM patients with methylated MGMT tumors will start in 2008.
5. Conclusion
Conventional cyototoxic agents have nominally improved outcome for GBM patients. Inhibiting key cellular processes such as angiogenesis and invasion represent alternative therapeutic strategies with promising potential for this challenging patient population. Integrins critically regulate many tumor cell processes including invasion, migration, angiogenesis, proliferation and survival. Cilengitide, a selective inhibitor of αvβ3 and αvβ5 integrins, has activity in preclinical GBM models and has shown promising activity with minimal toxicity as single-agent therapy in recurrent GBM patients. Two recent studies have explored the impact of cilengitide integration into standard TMZ chemoradiation for newly diagnosed GBM patients. Preliminary results suggest that cilengitide is well tolerated and may improve outcome, particularly for patients with low MGMT rumors. The overall results so far suggest that further evaluation of cilengitide and other integrin antagonists for GBM is warranted.
6. Key issues
Glioblastoma multiforme, the most common primary CNS tumor, has a dismal outcome. Current ‘state-of-the-art’ therapy, including surgery and chemoradiation, achieves a median 7 month PFS and only 15 month median OS survival. At 2 years, only 25 – 30% of patients are alive.
Salvage therapies at recurrence are ineffective; thus recurrent MG patients remain a principal unmet oncologic need.
Malignant gliomas are highly invasive and angiogenic neoplasms. Integrins are key mediators of GBM cell and tumor endothelial cell motility, and thus critically regulate invasion and angiogenesis. Integrins also activate intracellular signaling pathways that contribute to tumor cell proliferation and survival.
Cilengitide, a cyclized RGD-containing pentapeptide that potently and selectively inhibits αvβ3 and αvβ3 integrins, suppresses tumor cell migration and angiogenesis in vitro and exhibits in vivo antitumor activity in orthotopic GBM models including synergy with XRT.
Preliminary encouraging antitumor activity has been observed among recurrent GBM patients treated with single-agent cilengitide, with minimal toxicity including no increased risk of intracranial hemorrhage. Following presurgery administration in GBM patients, cilengitide is effectively delivered to achieve adequate intratumoral concentrations that persist for at least 24 h.
One study incorporating cilengitide into standard TMZ chemoradiation for newly diagnosed GBM patients has been reported so far and describes encouraging modest benefit compared to historical controls with no increase in toxicity. A second study has recently been completed and a randomized Phase III study is planned.
Future efforts will also explore further combinatorial regimens and attempt to identify more reliable biomarkers to predict response.
7. Expert opinion
Improvement in outcome for GBM, the most common primary brain tumor in adults, remains a principal challenge in oncology. Outcome with conventional cytotoxic therapy, the current standard of care for newly diagnosed patients, is poor and no effective salvage therapy is established for recurrent tumors. New therapeutics targeting activated signaling mechanisms critical to GBM cell growth, survival, invasion and angiogenesis represent promising alternative treatment approaches that may provide more specific yet less toxic therapeutic benefit.
However, development of a targeting therapeutic for cancer population must satisfy several incremental criteria. First, the therapeutic target must be present and biologically significant. The αvβ3 and αvβ5 integrins, specific targets of cilengitide, are key cell adhesion molecules expressed by both GBM cells and tumor vasculature that actively mediate tumor invasion, angiogenesis, survival and proliferation. Second, a potential therapeutic must successfully inhibit its intended target and in so doing, achieve a meaningful antitumor effect. In preclinical studies, cilengitide potently suppressed integrin-mediated tumor cell invasion and angiogenesis in vitro, whereas in vivo studies confirm antitumor benefit in orthotopic GBM models as well as a synergistic interaction with XRT. Third, a potential therapeutic must exhibit a favorable toxicity profile in the intended patient population. CNS tumor patients are deliberately excluded in the development of many therapeutics owing to concerns about neurologic toxicity or intracranial hemorrhage. Across several Phase I and II studies, cilengitide demonstrates a remarkably favorable safety profile among adults with MG. In addition, cilengitide does not appear to increase the risks of intracranial hemorrhage or wound healing complications. Furthermore, the minimal frequency and severity of adverse events from cilengitide suggest that it may be safely combined with other therapeutics such as cytotoxics. Results of a recent Phase II study for newly diagnosed GBM patients support this notion in that the addition of cilengitide did not appear to increase toxicity of TMZ chemoradiation.
Fourth, a potential therapeutic must reach and inhibit its intended target. Delivery into CNS tumors is particularly challenging owing to limitations posed by the blood–brain barrier. Nonetheless, preliminary results of a recently done clinical trial confirm that elevated intratumoral concentrations of cilengitide are achieved and maintained for at least 24 h, with higher levels achieved with 2000 mg dosing compared to 500 mg/dose.
Finally, the ‘bottom-line’ for any potential therapeutic is meaningful improvement in outcome. The available cumulative experience with cilengitide among GBM patients including monotherapy Phase I and II studies in the recurrent setting as well as single-arm data evaluating the addition of cilengitide to standard TMZ chemoradiation for newly diagnosed GBM patients suggests that cilengitide has activity for this disease. Among recurrent GBM patients, single-agent cilengitide achieved a 10% objective radiographic response (ORR) rate that compares favorably to 5% ORR rate for TMZ [49]. Furthermore, recurrent patients who achieved an ORR remained progression-free for 10.8 – 36 months (median 17 months). Although antitumor benefit was noted among both the 500 and 2000 mg/dose cohorts treated on the recurrent Phase II study, all outcome parameters, including ORR, PFS, 6-PFS and OS, trended more favorably among patients who received 2000 mg/dose. As no difference in toxicity was noted between the two dosing cohorts, future clinical trials will likely incorporate the 2000 mg/dose schedule.
Among newly diagnosed GBM patients, the administration of cilengitide at 500 mg/dose with standard TMZ chemoradiation appears to improve outcome compared to historical controls. Interestingly, improvement in outcome was notable only among patients with MGMT promoter methylation. The mechanism underlying improved outcome among patients with MGMT methylated tumors is not yet clear and requires further clarification. A second recently completed study will further address the antitumor benefit of cilengitide administered at either 500 or 2000 mg/dose for newly diagnosed GBM patients treated with TMZ chemoradiation. Finally a Phase III study randomizing newly diagnosed GBM patients with MGMT methylated tumors to either standard TMZ chemoradiation or standard TMZ chemoradiation plus cilengitide administered at 2000 mg/dose is expected to initiate accrual in mid-2008.
Thus, the development of cilengitide for GBM has followed a well-planned, methodical path with positive results at each assessment point supporting further incremental evaluation. Future considerations include additional combinatorial partners such as VEGF-inhibiting agents, alternative targeting agents and additional cytotoxics. Additionally, biomarkers beyond MGMT will also be critical to most effectively incorporate this promising agent into a multi-agent treatment regimen for patients with a high likelihood of response while permitting the consideration of alternative agents among patients with less predicted benefit.
Acknowledgments
This study was supported by National Institutes of Health grant nos. 1-P50-CA108786-01, NS20023, and CA11898 and by grant no. MO1 RR 30 through the General Clinical Research Centers Program, National Center for Research Resources, National Institutes of Health.
Footnotes
Declaration of interest: T Mikkelsen has received research funding from Merck KGaA. LB Nabors has served on the advisory board for Merck, has received honorarium and has contributed to the writing. R Stupp has acted as a consultant for Merck KGaA sponsored trials and has received honoraria.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Fischer I, Gagner JP, Law M, et al. Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol. 2005;15(4):297–310. doi: 10.1111/j.1750-3639.2005.tb00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13(4):1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 4.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25(30):4722–9. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 5.Rubenstein JL, Kim J, Ozawa T, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2(4):306–14. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tucker GC. Integrins: molecular targets in cancer therapy. Curr Oncol Rep. 2006;8(2):96–103. doi: 10.1007/s11912-006-0043-3. [DOI] [PubMed] [Google Scholar]
- 7.Stupack DG. The biology of integrins. Oncology (Williston Park) 2007;21(9 Suppl 3):6–12. [PubMed] [Google Scholar]
- 8.Moschos SJ, Drogowski LM, Reppert SL, Kirkwood JM. Integrins and cancer. Oncology (Williston Park) 2007;21(9 Suppl 3):13–20. [PubMed] [Google Scholar]
- 9.Bello L, Francolini M, Marthyn P, et al. Alpha(v)beta3 and alpha(v)beta5 integrin expression in glioma periphery. Neurosurgery. 2001;49(2):380–9. doi: 10.1097/00006123-200108000-00022. discussion 90. [DOI] [PubMed] [Google Scholar]
- 10.Gingras MC, Roussel E, Bruner JM, et al. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J Neuroimmunol. 1995;57(12):143–53. doi: 10.1016/0165-5728(94)00178-q. [DOI] [PubMed] [Google Scholar]
- 11.Gladson CL. Expression of integrin alpha v beta 3 in small blood vessels of glioblastoma tumors. J Neuropathol Exp Neurol. 1996;55(11):1143–9. doi: 10.1097/00005072-199611000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Goodman SL, Holzemann G, Sulyok GA, Kessler H. Nanomolar small molecule inhibitors for alphav(beta)6, alphav(beta)5, and alphav(beta)3 integrins. J Med Chem. 2002;45(5):1045–51. doi: 10.1021/jm0102598. [DOI] [PubMed] [Google Scholar]
- 13.Dechantsreiter MA, Planker E, Matha B, et al. N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta 3;integrin antagonists. J Med Chem. 1999;42(16):3033–40. doi: 10.1021/jm970832g. [DOI] [PubMed] [Google Scholar]
- 14.Germer M, Kanse SM, Kirkegaard T, et al. Kinetic analysis of integrin-dependent cell adhesion on vitronectin – the inhibitory potential of plasminogen activator inhibitor-1 and RGD peptides. Eur J Biochem FEBS. 1998;253(3):669–74. doi: 10.1046/j.1432-1327.1998.2530669.x. [DOI] [PubMed] [Google Scholar]
- 15.Belvisi L, Riccioni T, Marcellini M, et al. Biological and molecular properties of a new alpha(v)beta3/alpha(v)beta5 integrin antagonist. Mol Cancer Ther. 2005;4(11):1670–80. doi: 10.1158/1535-7163.MCT-05-0120. [DOI] [PubMed] [Google Scholar]
- 16.Rice J, Courter DL, Giachelli CM, Scatena M. Molecular mediators of alphavbeta3-induced endothelial cell survival. J Vasc Res. 2006;43(5):422–36. doi: 10.1159/000094884. [DOI] [PubMed] [Google Scholar]
- 17.Stupack DG, Cho SY, Klemke RL. Molecular signaling mechanisms of cell migration and invasion. Immunol Res. 2000;21(23):83–8. doi: 10.1385/IR:21:2-3:83. [DOI] [PubMed] [Google Scholar]
- 18.Stupack DG, Cheresh DA. Get a ligand, get a life: integrins, signaling and cell survival. J Cell Sci. 2002;115(Pt 19):3729–38. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- 19.Maubant S, Saint-Dizier D, Boutillon M, et al. Blockade of alpha v beta3 and alpha v beta5 integrins by RGD mimetics induces anoikis and not integrin-mediated death in human endothelial cells. Blood. 2006;108(9):3035–44. doi: 10.1182/blood-2006-05-023580. [DOI] [PubMed] [Google Scholar]
- 20.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264(5158):569–71. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 21.Aguzzi MS, Giampietri C, De Marchis F, et al. RGDS peptide induces caspase 8 and caspase 9 activation in human endothelial cells. Blood. 2004;103(11):4180–7. doi: 10.1182/blood-2003-06-2144. [DOI] [PubMed] [Google Scholar]
- 22.Ding Q, Stewart J, Jr, Prince CW, et al. Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain: differences in integrin signaling during cell adhesion to osteopontin versus vitronectin. Cancer Res. 2002;62(18):5336–43. doi: 10.1100/tsw.2002.247. [DOI] [PubMed] [Google Scholar]
- 23.Friedlander DR, Zagzag D, Shiff B, et al. Migration of brain tumor cells on extracellular matrix proteins in vitro correlates with tumor type and grade and involves alphaV and betal integrins. Cancer Res. 1996;56(8):1939–47. [PubMed] [Google Scholar]; • Early report linking integrin expression and glioma migration.
- 24.Nisato RE, Tille JC, Jonczyk A, et al. alpha v beta 3 and alpha v beta 5 integrin antagonists inhibit angiogenesis in vitro. Angiogenesis. 2003;6(2):105–19. doi: 10.1023/B:AGEN.0000011801.98187.f2. [DOI] [PubMed] [Google Scholar]
- 25.Fu Y, Ponce ML, Thill M, et al. Angiogenesis inhibition and choroidal neovascularization suppression by sustained delivery of an integrin antagonist, EMD478761. Invest Ophthalmol Vis Sci. 2007;48(11):5184–90. doi: 10.1167/iovs.07-0469. [DOI] [PubMed] [Google Scholar]
- 26.Friedlander M, Theesfeld CL, Sugita M, et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci USA. 1996;93(18):9764–9. doi: 10.1073/pnas.93.18.9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drake CJ, Cheresh DA, Little CD. An antagonist of integrin alpha v beta 3 prevents maturation of blood vessels during embryonic neovascularization. J Cell Sci. 1995;108(Pt 7):2655–61. doi: 10.1242/jcs.108.7.2655. [DOI] [PubMed] [Google Scholar]
- 28.Loges S, Butzal M, Otten J, et al. Cilengitide inhibits proliferation and differentiation of human endothelial progenitor cells in vitro. Biochem Biophys Res Commun. 2007;357(4):1016–20. doi: 10.1016/j.bbrc.2007.04.060. [DOI] [PubMed] [Google Scholar]
- 29.Uhm JH, Gladson CL, Rao JS. The role of integrins in the malignant phenotype of gliomas. Front Biosci. 1999;4:D188–99. doi: 10.2741/uhm. [DOI] [PubMed] [Google Scholar]
- 30.Rutka JT, Muller M, Hubbard SL, et al. Astrocytoma adhesion to extracellular matrix: functional significance of integrin and focal adhesion kinase expression. J Neuropathol Exp Neurol. 1999;58(2):198–209. doi: 10.1097/00005072-199902000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Paulus W, Baur I, Schuppan D, Roggendorf W. Characterization of integrin receptors in normal and neoplastic human brain. Am J Pathol. 1993;143(1):154–63. [PMC free article] [PubMed] [Google Scholar]
- 32.Buerkle MA, Pahernik SA, Sutter A, et al. Inhibition of the alpha-nu integrins with a cyclic RGD peptide impairs angiogenesis, growth and metastasis of solid tumours in vivo. Br J Cancer. 2002;86(5):788–95. doi: 10.1038/sj.bjc.6600141. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Preclinical activity of cyclic RGD integrin antagonist.
- 33.Taga T, Suzuki A, Gonzalez-Gomez I, et al. Alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer. 2002;98(5):690–7. doi: 10.1002/ijc.10265. [DOI] [PubMed] [Google Scholar]
- 34.MacDonald TJ, Taga T, Shimada H, et al. Preferential susceptibility of brain tumors to the antiangiogenic effects of an alpha(v) integrin antagonist. Neurosurgery. 2001;48(1):151–7. doi: 10.1097/00006123-200101000-00026. [DOI] [PubMed] [Google Scholar]; •• Early preclinical report of integrin antagonist in CNS tumor models.
- 35.Yamada S, Bu XY, Khankaldyyan V, et al. Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice. Neurosurgery. 2006;59(6):1304–12. doi: 10.1227/01.NEU.0000245622.70344.BE. discussion 12. [DOI] [PubMed] [Google Scholar]
- 36.Burke PA, DeNardo SJ, Miers LA, et al. Cilengitide targeting of alpha(v)beta 3;integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res. 2002;62(15):4263–72. [PubMed] [Google Scholar]; •• Preclinical synergy of cilengitide and XRT in breast cancer.
- 37.Albert JM, Cao C, Geng L, et al. Integrin alpha v beta 3 antagonist Cilengitide enhances efficacy of radiotherapy in endothelial cell and non-small-cell lung cancer models. Int J Radiat Oncol Biol Phys. 2006;65(5):1536–43. doi: 10.1016/j.ijrobp.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 38.Abdollahi A, Griggs DW, Zieher H, et al. Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin Cancer Res. 2005;11(17):6270–9. doi: 10.1158/1078-0432.CCR-04-1223. [DOI] [PubMed] [Google Scholar]; •• Synergy of cilengitide and XRT in glioma.
- 39.Wick W, Wick A, Schulz JB, et al. Prevention of irradiation-induced glioma cell invasion by temozolomide involves caspase 3 activity and cleavage of focal adhesion kinase. Cancer Res. 2002;62(6):1915–9. [PubMed] [Google Scholar]
- 40.Mikkelsen T, Nelson K, Brown S, et al. Cilengitide and synergy with radiation. 12th Annual Meeting of the Society of Neuro-Oncology; Dallas, TX. 2007. p. 486. [Google Scholar]
- 41.Eskens FA, Dumez H, Hoekstra R, et al. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer. 2003;39(7):917–26. doi: 10.1016/s0959-8049(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 42.Hariharan S, Gustafson D, Holden S, et al. Assessment of the biological and pharmacological effects of the alpha nu beta3 and alpha nu beta5 integrin receptor antagonist, cilengitide (EMD 121974), in patients with advanced solid tumors. Ann Oncol. 2007;18(8):1400–7. doi: 10.1093/annonc/mdm140. [DOI] [PubMed] [Google Scholar]
- 43.Nabors LB, Mikkelsen T, Rosenfeld SS, et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 2007;25(13):1651–7. doi: 10.1200/JCO.2006.06.6514. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First clinical trial of cilengitide in malignant glioma.
- 44.MacDonald TJ, Stewart CF, Kocak M, et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium Study PBTC-012. J Clin Oncol. 2008;26(6):919–24. doi: 10.1200/JCO.2007.14.1812. [DOI] [PubMed] [Google Scholar]
- 45.Reardon DA, Fink K, Nabors LB, et al. Phase IIa trial of cilengitide (EMD121974) single-agent therapy in patients (pts) with recureetn glioblastoma (GBM): EMD 121974-009. In: Grunberg MDSM, editor. 43rd Annual Meeting of American Society of Clinical Oncology; Chicago, IL. Greaves, Lisa; 2007. p. 75s. [Google Scholar]
- 46.Gilbert M, Lamborn K, Lassman AB, et al. Tumor tissue delivery of cilengitide after intravenous administration to patients with recurrent glioblastoma (GBM): preliminary data from NABTC protocol 03-02. 12th Annual Meeting of the Society for Neuro-Oncology; Dallas, TX. 2007. p. 525. [Google Scholar]
- 47.Stupp R, Goldbrunnr R, Neyns B, et al. Phase I/IIa trial of cilengitide (EMD121974) and temozolomide with concomitant radiotherapy, followed by temozolomide and cilengitide maintenance therapy in patients (pts) with newly diagnosed glioblastoma (GBM). In: Grunberg MDSM, editor. 2007 ASCO Annual Meeting Proceedings; Chicago, IL. Greaves, Lisa; 2007. p. 75s. [Google Scholar]
- 48.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 49.Yung WK, Albright RE, Olson J, et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83(5):588–93. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]