

Abstract
Human purine nucleoside phosphorylase (PNP) is a homotrimer binding tightly to the transition state analogues Immucillin-H (ImmH, Kd = 56 pM) and DATMe-ImmH-Immucillin-H (DATMe-ImmH, Kd = 8.6 pM). ImmH binds with a larger entropic penalty than DATMe-ImmH, a chemically more flexible inhibitor. The testable hypothesis is that PNP conformational states are more relaxed (dynamic) with DATMe-ImmH, despite tighter binding than with ImmH. PNP conformations are probed by peptide amide deuterium exchange (HDX) using liquid chromatography high-resolution Fourier transform ion cyclotron resonance mass spectrometry and by sedimentation rates. Catalytically equilibrating Michaelis complexes (PNP•PO4•Inosine ↔ PNP•Hx•R-1-P) and inhibited complexes (PNP•PO4•DATMe-ImmH and PNP•PO4•ImmH) show protection from HDX at 9, 13 and 15 sites per subunit relative to resting PNP (PNP•PO4) in extended incubations. The PNP•PO4•ImmH complex is more compact (by sedimentation rate) than the other complexes. HDX kinetic analysis of ligand-protected sites corresponds to peptides near the catalytic sites. HDX and sedimentation results establish that PNP protein conformation (dynamic motion) correlates more closely to entropy of binding than to affinity. Catalytically active turnover with saturated substrate sites causes less change in HDX and sedimentation rates than binding of transition state analogues. DATMe-ImmH more closely mimics the transition of human PNP than does ImmH, and achieves strong binding interactions at the catalytic site while causing relatively modest alterations of the protein dynamic motion. Transition state analogues causing the most rigid, closed protein conformation are therefore not necessarily the most tightly bound. Close mimics of the transition state are hypothesized to retain enzymatic dynamic motions related to transition state formation.
Human PNP catalyzes the reversible phosphorolysis of 6-oxypurine nucleosides and 6-oxypurine-2’-deoxynucleosides. A PNP genetic deficiency causes deoxyguanosine accumulation in blood. Since activated T-cells efficiently convert deoxyguanosine to dGTP, an absence of PNP results in the arrest of DNA synthesis and apoptotic cell death. Thus, inhibition of human PNP is a target for the treatment of T-cell proliferative and autoimmune disorders including T-cell leukemia, rheumatoid arthritis, multiple sclerosis and tissue transplant rejection (1–4).
The Immucillins are transition state analogue inhibitors of PNP. Immucillin-H (ImmH) was designed to mimic the early dissociative transition state of bovine PNP and is an imperfect match to the human PNP transition state but still binds tightly (Kd = 56 pM). DATMe-Immucillin-H (DATMe-ImmH) is an acyclic iminoalcohol mimic of the fully dissociative transition state of human PNP. It closely resembles the transition state of human PNP resulting in a Kd = 8.6 pM (5–8) (Table 1). DATMe-ImmH contains more rotational and vibrational degrees of freedom than does ImmH. Thermodynamic analysis of ImmH binding indicates a large and unfavorable entropy change of 7.1 kcal/mol upon binding to E•PO4 (Table 1). In contrast, the entropic penalty for DATMe-ImmH binding is only 2.3 kcal/mol. The thermodynamic differences observed for ImmH and DATMe-ImmH binding have been proposed to reside in increased PNP dynamic freedom (entropy) in the complex with the more flexible DATMe-ImmH inhibitor (20). Here we test the hypothesis directly by HDX and sedimentation coefficients. The HDX analysis permits the identification of conformational dynamics in the protein as influenced by the complexes of E•PO4, E•PO4•Inosine ↔ E•Ribose-1-PO4•Hypoxanthine, E•PO4•ImmH, and E•PO4•DATMe-ImmH (9–14). HDX is the probability of water gaining reactive access to a specific peptide bond during the experimental time period. Exchange is base-catalyzed and therefore contains both geometric access and local pKa elements.
Table 1.
Thermodynamics of Binding ImmH and DATMe-ImmH to human PNP
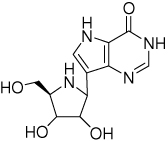
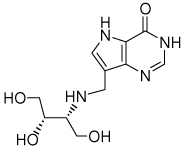
Sedimentation coefficients are related to global protein average cross-section and were measured by analytical centrifugation (15–17). The sedimentation coefficient provides protein conformational information since the sedimentation coefficient, S, depends on the frictional coefficient, ƒ (S = Mb/ƒ), a reflection of molecular size and shape. Small differences in the sedimentation coefficient can be reliably detected with the appropriate internal controls (18–19). The sedimentation coefficient provides a useful global shape parameter to complement the local information provided by HDX.
Thermodynamic analysis of transition state analogues of human PNP indicate that chemically rigid Immucillins (e.g. ImmH) bind human PNP with large and favorable enthalpy terms that are compensated by unfavorable entropy changes. Chemically flexible Immucillins like DATMe-ImmH bind to the enzyme with smaller favorable enthalpy changes but also with smaller unfavorable entropy changes and hence higher binding affinity (20). Increased flexibility in the transition state analogues was proposed to permit increased dynamic flexibility in the enzyme-inhibitor complexes to reduce the system entropic penalty. We test this hypothesis by the independent methods of HDX and sedimentation analysis. The entropic contributions of ImmH and DATMe-ImmH binding to PNP•PO4 correlate with altered dynamics in the PNP protein structures and restricted conformational states. PNP operating at its maximum catalytic potential (kcat) permits more rapid HDX, suggesting a more conformational flexibility with respect to water ↔ peptide bond exchange rate than either of the complexes with transition state analogues.
MATERIALS AND METHODS
Enzyme Purification and Preparation
His-tagged human PNP was expressed and purified to homogeneity as judged by SDS-PAGE according to procedures previously reported (7, 20).
Preparation of Non-his-tagged Human PNP
Analytical centrifugation experiments were conducted with human PNP lacking the histidine tag. The open reading frame for HsPNP was cloned into the pCR-T7/CT-TOPO vector (Invitrogen) as described previously (7). The resulting plasmid was transformed into BL21-AI E. coli cells (Invitrogen) and grown overnight on LB agar plates containing 100 µg/mL ampicillin. A colony was used to inoculate two 100 mL cultures (LB medium with ampicillin, 100 µg/mL) which were grown overnight at 37 °C and used to inoculate 20 L of LB containing ampicillin. The culture was grown to an OD600 of 0.4–0.6 (3–4 hr) and induced with L-arabinose (0.2%) for 4 hours at 37 °C. Cells were pelleted, resuspended in 300 mL of 5 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl buffer pH 8.0, protease inhibitor (2 tablets EDTA-free protease inhibitor, Roche Diagnostics) and approximately 1 mg each of DNase I (from bovine pancreas, Roche Diagnostics) and lysozyme (from chicken egg white, Sigma-Aldrich). The cells were disrupted twice with a French press. The supernatant from centrifugation (39,000 × g, 30 min) was applied to a 140 mL Q-Sepharose FF column previously equilibrated with 3 column volumes of 50 mM Tris-HCl pH 8.0. Human PNP was eluted with 1 mM NaCl, 50 mM Tris-HCl at pH 8.0 with an AKTA FPLC (GE Healthcare). Fractions with human PNP (SDS-PAGE) were concentrated to approximately 25 mL with an AMICON filtration system. (NH4)2SO4, up to 1 M, was added to the concentrate and the resulting solution loaded onto a 20 mL HiPrep Phenyl FF column (GE Healthcare) previously equilibrated with 50 mM KPO4, pH 7.4. Human PNP was eluted with 1 mM (NH4)2SO4, 50 mM KPO4 at pH 7.4. Fractions containing PNP were pooled, concentrated to approximately 12 mL and loaded unto a 200 mL Superdex size-exclusion column previously equilibrated with 50 mM KH2PO4, pH 7.4. Fractions containing pure human PNP (SDS-PAGE) were concentrated to approximately 20 mL. The enzyme was dialyzed against 50 mM KH2PO4, pH 7.4 (24 hr) and the dialysate retained for ultracentrifugation experiments. The preparations yielded PNP at approximately 6 mg/mL. Hypoxanthine co-purifying with the enzyme was removed by incubating in 100 mM KH2PO4 containing 10 % charcoal (w/v) for 5 minutes followed by centrifugation and filtration to remove the charcoal (21). Small plastic tubes containing 1.8 mL each were frozen rapidly in liquid nitrogen and stored at −80 °C.
Hydrogen Deuterium Exchange (HDX) Automated HDX Platform
Hydrogen/deuterium exchange, low pH quench, proteolysis, and liquid chromatography of peptide fragments were all performed at 0 °C. A Leap robot (HTS PAL, Leap Technologies, Carrboro, NC) was used to automate the HDX experiment (22). Triplicate data sets were collected for 11 different incubation periods: 0 (digestion without exchange), 0.5, 1, 2, 4, 8, 30, 60, 120, 240, and 480 minutes. The LEAP robot was interfaced to a Jasco HPLC/SFC system (Jasco, Easton, MD) for fast desalting chromatography of proteolytic samples (23). Proteolytic peptides or intact PNP (30 µL injection) were separated on a ProZAP™ HP C18 column - HR 1.5 mm particle size, 2.1 mm x 10 mm, and 500 Å pore size (Grace Davidson, Deerfield, IL). Solvent was delivered at 300 µL/min with a 1.5 minute fast gradient (2% to 95% solvent B). Solvent A was H2O/ACN/formic acid, 94.5/5.0/0.5 (v/v), and solvent B was H2O/ACN/formic acid, 5.0/94.5/0.5 (v/v). A post-column splitter reduced the LC eluent to 450 nL/min for efficient microelectrospray ionization (24).
HDX of PNP with Inhibitors and Substrates
Hydrogen/deuterium exchange was performed on samples containing 50 mM sodium phosphate pH 7.4 and the following protein and inhibitor concentrations: 1) 60 µM PNP (20 µM trimer, 60 µM with respect to catalytic sites), 2) 60 µM PNP and 175 µM ImmH, 3) 60 µM PNP and 185 µM DATMe-ImmH, and 4) 60 µM PNP with 300 µM inosine. Exchange was initiated by adding 5 µL of each sample to 45 µL of 50 mM sodium phosphate in D2O, (pH meter reading = 7.8). At the end of the desired exchange period, 50 µL of Protease type XIII (2.0 mg/mL) in 1.5% formic acid was added and incubated for 2 minutes (pH = 2.5) (25). Conditions for intact protein exchange experiments were the same as above, except that 50 µL of 1.5% formic acid (without protease) was added to quench the deuterium exchange and incubated for 2 min prior to injection.
Instrumentation and Data Analysis
Mass spectrometry was performed with a modified LTQ FT-ICR mass spectrometer equipped with a 14.5 T superconducting magnet (Thermo Electron Corp., San Jose, CA) (26). FT-ICR mass spectra of proteolytic peptides were collected at 400 < m/z < 1600 at high mass resolving power (m/Δm50% = 200,000 at m/z 400; m/Δm50% is mass spectral peak full with at half-maximum peak height). Data was extracted from RAW files with custom software package that takes full advantage of high-resolving power and accurate mass measurements (22). All identified peptides had less than a 1.5 ppm mass error. For global PNP exchange experiments, LCMS data was collected at 897 < m/z <910. Based on broadband detection (data not shown), the 40+ charge state provided the highest signal magnitude. Global hydrogen/deuterium exchange data was collected and analyzed with MIDAS software. The number of deuterium incorporated was determined by extracting the average mass of the isotopic distribution for a particular exchange period and subtracting it from the average mass for non-exchanged PNP. Deuterium uptake curves were plotted with Microsoft Excel after fitting the data with maximum entropy method (MEM) software (27). The MEM software allows for the input of the number of total exchangeable hydrogens, the number of hydrogens exchanged for each exchange period, and the standard deviation for each exchange period. The final output provides a best-fit curve and a MEM-derived rate constant distribution. The best-fit curves for each individual peptide for each experiment were plotted together to visualize the change in deuterium uptake upon binding to different ligands.
Analytical Centrifugation (Sedimentation Velocity)
The rate of sedimentation of PNP•PO4 and the enzyme in complex with both substrates and inhibitors was determined by sedimentation velocity experiments monitored with the interference optics of a Beckman XL-I analytical ultracentrifuge. The experiments were performed with double sector cells in the Ti-60 rotor and a speed of 45,000 rpm. Each sample contained 5 mg/mL PNP in 50 mM potassium phosphate buffer at pH 7.4, and 400 µM inhibitor or 1 mM inosine. Extinction constants were: human PNP ε280 = 31650 M−1 cm−1; Immucillins ε261 = 9540 M−1 cm−1; inosine ε248.5 = 12300 M−1 cm−1 (http://www.expasy.org/tools/protparam/html, 28–29). Each double sector cell was filled with 350 µL of potassium phosphate buffer (left chamber) and 334 µL of PNP-containing sample (right chamber). Up to 200 scans were acquired over the course of a run. Each measurement was acquired in triplicate with a PNP•PO4 internal standard always included. The sedimentation coefficient (S) was calculated from a subset of these scans by use of the program DCDT+ v2.1 (http://www.jphilo.mailway.com). The resulting S values were adjusted to standard conditions (S(20,w)) based on the values v¯ = 0.7373 (from the amino acid composition), solvent density and viscosity calculated with Sednterp v1.06 (Hayes, B., T. Laue & J. Philo, Sedimentation Interpretation Program, 2003, University of New Hampshire). The S values of the complexes were corrected for the increased molecular mass upon addition of ligand prior to taking their ratio with the apo enzyme.
Analytical Centrifugation (Sedimentation Equilibrium)
Sedimentation equilibrium experiments were performed with the interference optics of a Beckman XL-I analytical ultracentrifuge with 6 channel centerpieces in the Ti-60 rotor at 25 °C. Each 6 channel cell was filled with 120 µL of potassium phosphate dialysate in the reference channels and was paired to channels filled with 110 µL of PNP-containing sample. Three samples (PNP•PO4, PNP•PO4•ImmH and PNP•PO4•Inosine) were analyzed at three concentrations (1, 4 and 8 mg/mL). The same enzyme:ligand ratios were used for the ImmH- and inosine-containing samples as in the sedimentation velocity experiments. Data was collected at 22 and 24 h to ensure that equilibrium was attained following equilibration at 9,000 and at 13,000 rpm. The scans obtained at the two speeds were globally analyzed with HeteroAnalysis v1.0.114 (J.L. Cole & J.W. Lary, Analytical Ultracentrifugation Facility, Biotechnology Services Center, University of Connecticut, Storrs, CT 06269) for the weight average molecular weight. The resolved molecular weight and the 95% joint confidence intervals are reported.
RESULTS
Global H/D Exchange for Human PNP
Human PNP is a homotrimer with 289 amino acids per monomer, a subunit molecular weight of 32,148 and 275 exchangeable amide protons per subunit (Figure 1). With the N-terminal His-6 label construct used for HDX, the calculated molecular weight is 36,101. HDX compared PNP•PO4 with an actively catalyzing Michaelis complex (PNP•Ino•PO4 ↔ PNP•Hx•R1P) and two complexes with transition state analogue inhibitors. Global H/D exchange comparing the PNP•PO4 and PNP•PO4•ImmH complexes showed reduced incorporation of deuterium into PNP•PO4•ImmH (Figure 2). After 30 seconds of exchange, 94 amide hydrogens are exchanged in PNP•PO4 compared to 83 for PNP•PO4•ImmH. Therefore, 11 protons are protected from rapid exchange by ImmH binding. After 8 hours of exchange 129 and 114 amide hydrogens are exchanged in the PNP•PO4 and PNP•PO4•ImmH complexes, respectively, with fifteen fewer exchangeable amides in the PNP•PO4•ImmH complex. Relative to PNP•PO4, thirteen amide protons are protected in the PNP•PO4•DATMe-ImmH complex but only nine are protected in the actively reacting Michaelis complex (Figure 3). The reacting Michaelis complex is saturated with reactants to give >95% of the enzyme in the (PNP•Ino•PO4 ↔ PNP•Hx•R1P) complexes. Based binding and crystallographic analysis, all three catalytic sites are occupied with inhibitors or substrate/product with these concentrations (20). Under the conditions used to form the Michaelis complexes, the enzyme catalyzes on-enzyme chemistry at approximately 200 s−1 (21). Global HDX for PNP reveals that the largest protection from amide proton exchange occurs in the PNP•PO4•ImmH complex even though DATMe-ImmH binds more tightly (56 and 8.6 pM, respectively). As shown below, based on peptide analysis, most of the protected amide protons are found near the catalytic sites of PNP (Fig. 4 ).
FIGURE 1.

Amino acid sequence of human PNP indicating the peptides analyzed for the hydrogen/deuterium experiments. Peptides were generated by digestion of PNP at pH 2.5 with Protease type XIII in 1.5% formic acid. Although his-tagged human PNP was used in these experiments, the initial 35 amino acids belonging to the tag are not shown and exchange was not measured in this region. The amino acids shown in red are involved in Immucillin and substrate binding at the catalytic sites.
FIGURE 2.

Comparison of deuterium incorporation into PNP complexes monitored over an 8 hour time period. PNP•PO4 (black spectra); PNP•PO4•ImmH (red spectra). At each time interval more deuterium is incorporated into the PNP•PO4 complex than in the PNP•PO4•ImmH complex. The Blank compares the complexes prior to addition of D2O. In the ms, PNP subunits dissociate and the inhibitor ligands are lost. Thus, the calculated mass (36,101 amu) is close to the determined mass (36,079 amu) and no mass for inhibitor or phosphate is observed. The ImmH concentration gives >97% of catalytic site saturation under these conditions.
FIGURE 3.

Global deuterium uptake as a function of time showing the changes in deuterium uptake for PNP with: phosphate only (blue); the reacting Michaelis complex (red); DATMe-ImmH complex (green); and ImmH complex (orange). Over the 8 hour time period the largest reduction in amide proton exchange is seen for the sample containing ImmH and the least reduction in amide proton exchange is seen for the catalytically active Michaelis complex. These differences are highlighted in the inset which shows the same exchange for the first 2 hours.
FIGURE 4.

Stereo tube diagram of the three-dimensional structure of the human PNP trimer in the presence of ImmH and phosphate (Panel A) or in the presence of DATMe-ImmH and phosphate (Panel B). ImmH and phosphate are shown in yellow in all catalytic sites in Panel A. Exchange sites based on H/D exchange properties are color-coded from the peptide H/D exchange data. Grey represents peptides with no change in deuterium uptake as a consequence of Immucillin binding. Blue represents peptides strongly protected from H/D exchange as a consequence of Immucillin binding. Green peptides indicate no sequence coverage. Most exchange occurs at the surfaces exposed to solvent and most reductions in exchange occur at the catalytic/binding sites. In B, DATMe-ImmH and phosphate are shown in yellow in all catalytic sites. Exchange sites based on H/D exchange properties are color-coded as above from the peptide H/D exchange data. Most exchange in the presence of DATMe-ImmH and phosphate occurs at the surfaces exposed to solvent and most reductions in exchange occurs at the catalytic/binding sites.
Peptide H/D Exchange
The peptide exchange protocol identified 59 proteolytic peptides, representing 95% of the primary sequence, by Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry (30–31). HDX into peptides reveals specific peptides with different exchange rates (Fig. 5; Fig. 1 and Table 4 in Supporting Information). Peptides V79 – H86, V195 – E205, V208 – G232 and I241 – D248 showed the greatest HDX variability of the PNP complexes. The exchange rate constant distribution derived from the H/D time course data (Fig. 5; Fig. 1 and Table 4 Supporting Information) is plotted on the x-axis and the y-axis is scaled such that the total area under the curve corresponds to the total number of exchangeable amide protons in the peptide. Rate constants too fast or slow to be determined from the experimental data are represented by horizontal lines to the left or right respectively (30). Deuterium exchange into these four peptides is least for the PNP•PO4•ImmH complex followed by the PNP•PO4•DATMe-ImmH complex, is greater for the reacting Michaelis complex and is most rapid for the binary complex of PNP•PO4.
FIGURE 5.

Deuterium uptake as a function of time for peptides V79 – H86, V195 – E205, V208 – G232 and I241 – D248 showing changes in deuterium uptake for PNP with: phosphate only (blue); the catalytically active Michaelis complex (red); DATMe-ImmH complex (green); and ImmH complex (orange). Highlighted in red in the peptide sequence are the residues implicated in nucleoside and/or binding.
Peptide V79-H86
About half of the seven exchangeable amide protons in this peptide are exchanged over 8 hr and most are exchanged rapidly (Figures 5; Fig. 1 and Table 4 in Supporting Information). The catalytically equilibrating Michaelis complex is no different from PNP•PO4 in terms of the solvent exchange for V79-H86. The PNP•PO4•DATMe-ImmH complex prevents exchange of an additional proton whereas the PNP•PO4•ImmH complex prevents exchange of two additional protons. Peptide V79-H86 includes Arg84 and His86 both of which form ionic or hydrogen bond interactions with bound phosphate in PNP•PO4•ImmH. In PNP•PO4•DATMe-ImmH, His86 is closer and Arg84 is less involved in hydrogen bonding to the phosphate molecule (Fig. 6, Fig. 7). This difference is proposed to provide increased V79-H86 dynamic motion in the PNP•PO4•DATMe-ImmH complex and accounts for the altered amide proton exchange for this peptide.
FIGURE 6.

Ribbon diagram showing a catalytic site of human PNP in the presence of ImmH and phosphate. Blue labels (residues protected during H/D exchange); dark yellow (ribbon structure of monomer); bronze (ribbon structure of neighboring monomer); cyan (carbon backbone); blue (nitrogen atoms); red (oxygen atoms); yellow (sulfur atom); gold (phosphorus atom). F159* is protected from the neighboring subunit. Dashed lines indicate hydrogen bonds or ionic interactions and distances are shown in angstroms (from PDB code: 1rr6).
FIGURE 7.

Ribbon diagram showing a catalytic site of human PNP in the presence of DATMe-ImmH and phosphate. Blue labels (residues protected during H/D exchange); dark yellow (ribbon structure of monomer); orange (ribbon structure of neighboring monomer); cyan (carbon backbone); blue (nitrogen atoms); red (oxygen atoms); yellow (sulfur atom); gold (phosphorus atom). F159* is protected from the neighboring subunit. Dashed lines indicate hydrogen bonds or ionic interactions and distances are shown in angstroms (from PDB code: 2oyd). In the DATMe-ImmH complex loop 50–65 is in its closed form bringing H64 and G34 in catalytic contact to be protected (Figure 10). These residues are not protected in the ImmH complex (Figure 8).
Peptide V195-E205
Amino acids Phe200 and Glu201 from V195-E205 are in contact with the purine base (Fig. 6, Fig. 7). Glu201 is in H-bond contact with N1, a leaving-group interaction, and Phe200 is in a stacking interaction with the purine base (32). In the x-ray structure of ImmH and phosphate with PNP, a catalytic water molecule mediates a second contact to Glu201 through the 6-oxygen atom of the purine base (PDB code: 1rr6). This peptide is reasonably ordered even in the absence of a purine base since only 3 of the 9 amide protons in V195-E205 are exchanged in PNP•PO4 (Table 4 Supporting Information). The catalytically active Michaelis complex reduces proton exchange to two while the DATMe-ImmH and ImmH complexes reduces protons exchanged to 1 and 0.5 (exchange in half the peptides detected by ms), respectively (Table 4 Supporting Information).
Peptide V208-G232
Six of the 23 exchangeable amide protons in V208-G232 are exchanged in PNP•PO4 (Table 4 Supporting Information). This peptide retains the same solvent access in catalytically active PNP and the same extent of amide proton exchange. Exchanged protons decrease to 3.5 with ImmH bound and to 4 with DATMe-ImmH bound (Figure 5 and Table 4 in Supporting Information). Peptide V208-G232 contains Met219 and Ser220, both of which form hydrogen bond contacts within the catalytic site (Fig. 6 and Fig. 7). The carbonyl oxygen of Met219 forms a hydrogen bond with the ribose ring while the side chain hydroxyl of Ser220 forms a hydrogen bond with bound phosphate.
Peptide I241-D248
Asn243 forms two of the three hydrogen bonds between PNP and the purine base, important contacts for leaving group activation (Fig. 6, Fig. 7). Of the seven exchangeable amide protons in I241-D248, 4.5 are exchanged in the PNP•PO4 complex. Remarkably, this value is unchanged for the catalytically equilibrating Michaelis complex (Fig. 5; Table 4 in Supporting Information). PNP•PO4•ImmH and the DATMe-ImmH complexes protect this peptide, permitting exchange of only two amide protons. Thus, Asn243 (I241-D248) contact to the purine base, known to exist in both hypoxanthine and nucleoside complexes, remains dynamic during catalysis when the catalytic sites are continually saturated with hypoxanthine and inosine. Thus, motion for the peptide containing Asn243 is dynamic in catalysis but is sequestered from solvent when transition state analogues are bound.
Hydrodynamic Changes by Analytical Centrifugation
Sedimentation equilibrium studies estimates mass and confirmed that PNP is a tightly associated trimer and does not dissociate under conditions used here for HDX analysis. Thus, the molecular weights determined for PNP•PO4, equilibrating PNP•PO4•Inosine and PNP•PO4•ImmH are identical within experimental error (Table 2) and consistent with the calculated trimer weight values including bound ligand when present.
Table 2.
Calculated molecular weights determined by analytical ultracentrifugation sedimentation equilibrium
| Complex | Apparent Molecular Weight (Da)a |
|---|---|
| PNP + PO4 | 94,268 (92,425, 96,126) |
| PNP + PO4 + Inosine | 94,261 (92,667, 95,846) |
| PNP + PO4 + ImmH | 96,147 (94,236, 98,058) |
The resolved molecular weight and the 95% joint confidence intervals are reported. The results demonstrate no change in oligomer status as a function of ligand binding.
Sedimentation velocity analysis of PNP in paired samples tracked by the interference optics established an increased sedimentation rate (hydrodynamically more compact shape) with a change of 0.68 ± 0.05% in S(20,w) for the catalytically active Michaelis complex compared to PNP•PO4. The PNP•PO4•DATMe-ImmH complex displayed a similar sedimentation difference of 0.95 ± 0.38% relative to PNP•PO4 indicating similar hydrodynamic properties for equilibrating PNP•PO4•Inosine and PNP•PO4•DATMe-ImmH. However, both complexes are more hydrodynamically compact than is PNP•PO4. The PNP•PO4•ImmH complex sedimented the most rapidly to give a 2.21 ± 0.23% increase in S(20,w) relative to PNP•PO4 and is therefore the most compact of these species (Table 3). The conformational state(s) for PNP•PO4•ImmH indicate decreased diffusion and/or reactivity of peptide amide bonds with water and a more compact, stable architecture of the PNP complex with this transition state analogue.
Table 3.
Percent difference S(20,w) from PNP•PO4 (control) determined by analytical ultracentrifugation sedimentation velocity a
| Complex | Percent Difference S(20,w) |
|---|---|
| PNP + PO4 + Inosine | 0.68 ± 0.05 |
| PNP + PO4 + DATMe-ImmH | 0.95 ± 0.38 |
| PNP + PO4 + ImmH | 2.21 ± 0.23 |
Each sample was centrifuged in triplicate with PNP•PO4 always included as an internal standard. The individual and average S(20,w) values were determined from runs summarized in Supporting Information Table 5. All complexes sediment more rapidly than PNP•PO4 alone (the internal control). The PNP•PO4•Inosine and PNP•PO4•DATMe-ImmH sediment within experimental errors of each other but both sediment significantly slower than PNP•PO4•ImmH.
DISCUSSION
Amide hydrogens are involved in hydrogen bonds forming the secondary structure of PNP and contacts to catalytic site ligands. Thus, their exchange rates are a reflection of structure, stability, catalytic site dynamics and local pKa values for reacting water molecules (30–31, 33–35). Ligand binding to protein induces conformational changes reflected in the rate of amide hydrogen exchange with deuterated solvent. With PNP, exchange of specific peptides is useful to probe local solvent access (dynamic motion) during catalytic function and in inhibitor complexes. Dynamic motion is linked to proton exchange at peptide bonds since solvent-exposed amide protons exchange before the first time point in the protocol used here (eg. Fig. 2).
Sedimentation velocity can be interpreted to define the time-averaged shape and size of the PNP trimeric protein (36–37). HDX is also a time-dependent process, both for the chemical exchange rate (usually fast) and for the probability of exposing specific regions of the protein to solvent (both fast and slow components).
Dynamic Structures of PNP Complexes
The HDX experiments show both transition state analogue complexes to be less dynamic than the fully active enzyme (Fig. 5). Transition state inhibitor binding protects amide hydrogens in the vicinity of the PNP catalytic sites, but the actions are different for ImmH and DATMe-ImmH. ImmH causes greater protection from amide proton exchange than the complex formed with DATMe-ImmH, despite the 6.5-fold tighter binding by DATMe-ImmH. Thus, the complex with ImmH permits less dynamic access to solvent than does DATMe-ImmH. Catalytic site filling causes structurally significant changes in PNP geometry, predicting changes in backbone solvent access (Fig. 8).
FIGURE 8.

Superimposition of Apo PNP (blue), PNP-ImmH (yellow) and PNP-DATMe-ImmH (magenta) monomeric structures. The DATMe-ImmH structure shows the movement of loop 50–65 to a closed position which brings H64 in catalytic contact with the phosphate molecule while the Apo and ImmH structures show this loop in an open position.
Ligand Binding Induces More Condensed PNP States
Substrate and inhibitor binding to the symmetric PNP trimer result in an increased sedimentation rate, which when interpreted together with the H/D exchange data, reveals a less dynamic and more compact protein-ligand structure. Since the active PNP complex (PNP•PO4•Ino ↔ PNP•R1P•Hx) is involved in catalysis (200 s−1) during the experiments, the observed HDX rate represents a statistical average of the substrate-bound and product-bound enzyme. Since both reactants and products are present at fixed concentrations during the exchange experiment, the effects from substrate and reactant cannot be distinguished. However, the small change in the sedimentation coefficient and modest protection from HDX relative to PNP•PO4 demonstrates that PNP stays flexible and dynamic while fully engaged in enzymatic catalysis. It is remarkable that PNP•PO4•DATMe-ImmH, a complex bound with an 8.6 pM transition state analogue inhibitor reveals hydrodynamic properties similar to the equilibrating complexes.
Thermodynamics and PNP Dynamics
Thermodynamic analysis of ImmH and DATMe-ImmH binding to the first catalytic site of the PNP•PO4 complex revealed that although ImmH binds 6.5-fold weaker to the enzyme it has a substantially larger favorable enthalpy (−21.2 kcal/mol) and larger entropy penalty (7.1 kcal/mol) than does DATMe-ImmH binding (ΔH = −17.5 kcal/mol; -TΔS = 2.3 kcal/mol) (Table 1) (20). Entropic penalties can arise from increased inhibitor order when bound, solvent reorganization and increased protein order parameters. If we assume that solvent and inhibitor order changes are similar in both inhibitor interactions, the result requires that the protein be more disordered in the more tightly bound DATMe-ImmH complex. HDX and sedimentation rate are independent and direct tests of this hypothesis.
The HDX and hydrodynamic data are consistent with varied protein disorder upon transition state analogue binding to human PNP. PNP•PO4•ImmH is the more rigid complex, suggesting that the 7.1 kcal/mol entropy penalty upon binding comes primarily from protein organization or solvent reorganization. The more flexible DATMe-ImmH would be expected to generate an equivalent or larger entropic penalty if the entropy term is altered solvent plus inhibitor order parameters on binding. Since DATMe-ImmH binds more tightly than ImmH but only with a 2.3 kcal/mol entropy penalty, we propose that the large entropy term with ImmH is increased protein order. This proposal is confirmed by both the HDX and sedimentation velocity experiments.
Distinguishing Transition State Analogue Complexes from Catalytic Complexes
Why do transition state analogues produce more condensed enzyme states than catalytically functioning enzyme? The PNP transition state is short-lived (lasting ~10−14 s), while each catalytic turnover on the enzyme requires 5 × 10−3 sec (21, 38). Formation of the short-lived transition state requires a specific geometry of catalytic site elements. The transition-state-promoting geometry is formed only rarely by realizing coincident and optimized distances between catalytic site elements to the leaving group, the ribose and the phosphate nucleophile of the reactants. When all of these interactions are simultaneously optimized, passage over the transition state barrier occurs. The nearly instantaneous geometry that forms the transition state generates the local dynamic conformational change for only a few picoseconds or less and is a transient, not a long-lived state. The results suggest that this state arises from loose dynamic states rather than a ‘clamped down’ conformation that exists for a significant part of the 5 msec catalytic cycle. Transition path sampling of the PNP reaction indicates a reaction coordinate lifetime of approximately 70 fsec, occupying less than 10−9 of the catalytic cycle (38).
When a transition state analogue is bound to human PNP, the transient, favorable interactions that form the transition state become stable, thermodynamic interactions. The complexes create a stable state from a closely related but very short-lived (psec) state that normally exists on the reaction coordinate. Protein flexibility is necessary to allow catalytic site groups the dynamic motions required to search for and locate the transition state. When transition state analogues are bound, the enzyme forms favorable hydrogen bond and ionic interactions that serve to convert the short-lived dynamic excursions that form the transition state into long-lived thermodynamic interactions. These structures approximate the dynamic excursions involved in transition state barrier crossing. Protein evolution favors formation of the geometric arrangement that promotes barrier crossing. Transition state analogues capture this dynamic state, stabilize it into a thermodynamic state, improve peptide backbone packing within the enzyme and provide large binding energy for transition state analogue complexes.
H/D Exchange in Related Systems
Solvent HDX with hypoxanthine guanine phosphoribosyltransferase alone, complexed with a transition state analogue and the equilibrating Michaelis complexes revealed that transition state analogue binding strengthened the interactions between the trimeric subunits and tightened the catalytic site loops relative to the Michaelis complex (39). HDX in PNP from calf spleen bound to substrate, product and the transition state analogue ImmH showed the most protection from HDX in the transition state analogue complex (40). In both studies, the differences in HDX among the different protein complexes supported decreased protein dynamics with bound transition state analogue, but were not confirmed by independent methods. The present study links reduced dynamics from HDX to both thermodynamic and hydrodynamic experiments.
Negative Cooperativity
Immucillins bind to the three catalytic sites of human PNP in a negatively cooperative manner. The catalytic sites are linked by a loop containing Tyr200 that contributes to the catalytic sites of the neighboring subunits. Binding is weaker and more entropy-driven as the second and third sites fill (20). Binding a transition state analogue to the first catalytic site of human PNP causes a conformational change that is transmitted throughout the trimer and is greater than that for the second and third site binding. The large molar excess of reactants and inhibitors used here, assure that all three sites are filled during these experiments. HDX with calf spleen PNP exchange established that filling one of the three catalytic sites with ImmH resulted in similar HDX at all three subunits (40). The physiological relevance of this effect is that filling a single subunit on the enzyme renders the enzyme catalytically inactive (41).
Substrate analogue or product binding to human PNP is not cooperative and occurs with independent catalytic site interactions (20). Crystallographic analysis of the enzyme in complex with transition state analogues, substrates and products reveals that all three sites can be saturated with the ligands used here (42).
Flexibility of Transition State Analogues and PNP Dynamics
DATMe-ImmH binds human PNP with a smaller entropic penalty than ImmH, resulting in a more dynamic protein structure but tighter binding with the DATMe-ImmH analogue. HDX of individual peptides indicates that the three protected amide exchange differences in the ImmH and DATMe-ImmH complexes reside in the peptides V79 – H86, V195 – E205, V208 – G232 and A262 – F275. The first three of these peptides are in contact with phosphate, ribose and the purine base in the catalytic site. Crystal structures of these PNP complexes demonstrate that phosphate binding is connected to binding of the transition state analogue by forming hydrogen bonds between the phosphate oxygens and the hydroxyl groups of the inhibitors. Thus, inhibitor binding also influences peptides only in contact with the phosphate. Bound DATMe-ImmH incorporates chemical flexibility to the hydroxyl groups interacting with the phosphate, and therefore allows more motion of peptides bridging the phosphate and ribosyl groups. Although A262 – F275 has no direct contact to catalytic site ligands, they reside in an alpha-helix that begins with H257, an important contact to the 5’-hydroxyl groups of inosine as substrate and ImmH as transition state analogue. With the acyclic structure of the ribocation mimic of DATMe-ImmH, the increased dynamic motion of His257 and its connected alpha helix is readily explained.
Geometric, electrostatic and kinetic analysis of DATMe-ImmH reveals it to be a better mimic of the human PNP transition state than ImmH. With DATMe-ImmH the enzyme is less restricted to reach a conformation similar to the transition state than it is when ImmH is bound. PNP adopts a stiffer overall geometry (and a large entropic penalty) in order to fit ImmH into the active site. The more flexible geometry of DATMe-ImmH and its extended distance between the leaving group interactions and the ribocation mimic allows optimal binding while retaining increased dynamic flexibility around the catalytic sites, and a therefore a smaller protein entropic penalty in binding.
CONCLUSIONS
Unfavorable entropy changes accompany the enthalpy-driven binding of Immucillins to human PNP. The large unfavorable entropic change upon ImmH binding is caused by a condensed protein dynamic structure, protecting more amide hydrogens from exchange than in a complex with a more powerful, but more conformationally flexible inhibitor. The condensed state of the ImmH complex is global and thus observed by changes in the sedimentation rate. ImmH is not an optimal transition state analogue for human PNP and induces rigidity in the protein. The DATMe-ImmH transition state analogue is more closely matched to the transition state of human PNP and is a flexible molecule. The ΔG of DATMe-ImmH binding is greater than that of ImmH complex because of a smaller entropic penalty of binding. That change in entropic penalty is manifested as a more flexible, dynamic complex with the bound DATMe-ImmH complex. PNP protein dynamics are more active in the equilibrating Michaelis complex than the complexes of transition state analogues. Transition state analogues cause protein conformational collapse around an architecture closely related to the transition state while reacting substrate complexes exhibit dynamic motion throughout the reaction cycle.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Agnes Rinaldo-Matthis for providing the crystal structure coordinates for the human PNP•PO4•DATMe-ImmH complex and Dr. Suwipa Saen-oon for assisting in the generation of the catalytic site figures.
Footnotes
This work was supported by NIH R01 grants GM41916 and GM78359, NSF Division of Materials Research grant DMR 0654118, Florida State University, and the National High Magnetic Field Laboratory in Tallahassee, Florida.
SUPPORTING INFORMATION AVAILABLE
Additional Supporting Figure 1 and Table 4 and Table 5 are provided as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Giblett ER, Ammann AJ, Wara DW, Sandman R, Diamond LK. Nucleoside-phosphorylase deficiency in a child with severly defective T-cell immunity and normal B-cell immunity. Lancet. 1975;1:1010–1013. doi: 10.1016/s0140-6736(75)91950-9. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell BS, Mejias E, Daddona PE, Kelly WN. Purinogenic immunodeficiency diseases: selective toxicity of deoxyribonucleosides for T cells. Proc. Natl. Acad. Sci. U.S.A. 1978;75:5011–5014. doi: 10.1073/pnas.75.10.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ullman B, Gudas LJ, Clift SM, Martin DW., Jr Isolation and characterization of purine-nucleoside phosphorylase-deficient T-lymphoma cells and secondary mutants with altered ribonucleotide reductase: genetic model for immunodeficiency disease. Proc. Natl. Acad. Sci. U.S.A. 1979;76:1074–1078. doi: 10.1073/pnas.76.3.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korycka A, Blonski JZ, Robak T. Forodesine (BCX-1777, Immucillin H)-a new purine nucleoside analogue: mechanism of action and potential clinical application. Mini Reviews in Medicinal Chemistry. 2007;7:976–983. doi: 10.2174/138955707781662636. [DOI] [PubMed] [Google Scholar]
- 5.Kline PC, Schramm VL. Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry. 1993;32:13212–13219. doi: 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- 6.Evans GB, Furneaux RH, Lewandowicz A, Schramm VL, Tyler PC. Exploring structure-activity relationships of transition state analogues of human purine nucleoside phosphorylase. J. Med.Chem. 2003;46:3412–3423. doi: 10.1021/jm030145r. [DOI] [PubMed] [Google Scholar]
- 7.Lewandowicz A, Schramm VL. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 8.Taylor EA, Clinch K, Kelly PM, Li L, Evans GB, Tyler PC, Schramm VL. Acyclic ribooxacarbenium ion mimics as transition state analogues of human and malarial purine nucleoside phosphorylases. J. Am. Chem. Soc. 2007;129:6984–6985. doi: 10.1021/ja071087s. [DOI] [PubMed] [Google Scholar]
- 9.Wagner G, Wüthrich K. Amide proton exchange and surface conformation of the basic pancreatic trypsin inhibitor in solution. J. Mol. Biol. 1982;160:343–361. doi: 10.1016/0022-2836(82)90180-2. [DOI] [PubMed] [Google Scholar]
- 10.Katta V, Chait BT. Hydrogen/deuterium exchange electrospray ionization mass spectrometry: A method for probing protein conformational changes in solution. J. Am. Chem. Soc. 1993;115:6317–6321. [Google Scholar]
- 11.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F, Blanchard JS, Tang XJ. Amide hydrogen exchange/electrospray ionization mass spectrometry studies of substrate and inhibitor binding and conformational changes of E. coli dihydropicolinate reductase. Biochemistry. 1997;36:3755–3759. doi: 10.1021/bi963065g. [DOI] [PubMed] [Google Scholar]
- 13.Woodward C. Advances in protein hydrogen exchange by mass spectrometry. J. Am. Soc. Mass Spectrom. 1999;10:672–674. [Google Scholar]
- 14.Zhou B, Zhang ZY. Application of hydrogen/deuterium exchange mass spectrometry to study protein tyrosine phosphatase dynamics, ligand binding, and substrate specificity. Methods. 2007;42:227–233. doi: 10.1016/j.ymeth.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen JC, Lebowitz J, Demeler B. Analytical ultracentrifugation of complex macromolecular systems. Biochemistry. 1994;33:13155–13163. doi: 10.1021/bi00249a001. [DOI] [PubMed] [Google Scholar]
- 16.Cole JL, Hansen JC. Analytical ultracentrifugation as a contemporary biomolecular research tool. J. Biomol. Tech. 1999;10:163–174. [PMC free article] [PubMed] [Google Scholar]
- 17.Scott DJ, Schuck P. A brief introduction to the analytical ultracentrifugation of proteins for beginners. In: Scott DJ, Harding SE, Rowe AJ, editors. Analytical Ultracentrifugation. Cambridge, UK: Royal Society of Chemistry; 2005. pp. 1–25. [Google Scholar]
- 18.Richards EG, Schachman HK. A differential ultracentrifuge technique for measuring small changes in sedimentation coefficients. J. Am. Chem. Soc. 1957;79:5234–5235. [Google Scholar]
- 19.Cole JL, Lary JW, Moody TP, Laue TM. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods in Cell Biology. 2008;84:143–179. doi: 10.1016/S0091-679X(07)84006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards AA, Mason JM, Clinch K, Tyler PC, Evans GB, Schramm VL. Altered enthalpy-entropy compensation in picomolar transition state analogues of human purine nucleoside phosphorylase. Biochemistry. 2009;48:5226–5238. doi: 10.1021/bi9005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghanem M, Saen-oon S, Zhadin N, Wing C, Cahill SM, Schwartz SD, Callender R, Schramm VL. Tryptophan-free human pnp reveals catalytic site interactions. Biochemistry. 2008;47:3202–3215. doi: 10.1021/bi702491d. [DOI] [PubMed] [Google Scholar]
- 22.Kazazic S, Emmett MR, Blakney GT, Hendrickson CL, Marshall AG. Automated hydrogen deuterium exchange with high resolution ft-icr ms analysis and enhanced automated data reduction. Proceedings of the 54th American Society for Mass Spectrometry Annual Conference on Mass Spectrometry and Allied Topics; Indianapolis, IN. 2006. [Google Scholar]
- 23.Zhang H, Bou-Assaf GM, Emmett MR, Marshall AG. Fast reversed-phase liquid chromatography to reduce back exchange and increase throughput in H/D exchange monitored by FT-ICR mass spectrometry. Journal of the American Mass Spectrometry Society. 2009;20:520–524. doi: 10.1016/j.jasms.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emmett MR, Caprioli RM. Micro-electrospray MS, ultra-high-sensitivity analysis of peptides and proteins. J. Am. Soc. Mass Spectrom. 1994;5:605–613. doi: 10.1016/1044-0305(94)85001-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Kazazic S, Schaub TM, Tipton JD, Emmett MR, Marshall AG. Enhanced digestion efficiency, peptide ionization efficiency, and sequence resolution for protein hydrogen/deuterium exchange monitored by fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2008;80:9034–9041. doi: 10.1021/ac801417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaub TM, Hendrickson CL, Horning S, Quinn JP, Senko MW, Marshall AG. High performance mass spectrometry: fourier transform ion cyclotron resonance at 14.5t. Analytical Chemistry. 2008;80:3985–3990. doi: 10.1021/ac800386h. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Li W, Logan TM, Li M, Marshall AG. Human recombinant [C22A] FK506-binding protein amide hydrogen exchange rates from mass spectrometry match and extend those from NMR. Protein Sci. 1997;6:2203–2217. doi: 10.1002/pro.5560061015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim M, Ren Y, Otter BA, Klein RS. Synthesis of “9-deazaguanosine” and other new pyrrolo[3,2-d]pyrimidine C-nucleosides. J. Org. Chem. 1983;48:780–788. [Google Scholar]
- 29.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research. 3rd ed. Oxford, U.K: Clarendon Press; 1986. [Google Scholar]
- 30.Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrometry Review. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 31.Marshall AG, Hendrickson CL, Shi SDH. Scaling MS plateaus with high-resolution FT-ICRMS. Analytical Chemistry. 2002;74 doi: 10.1021/ac022010j. 253A–259A. [DOI] [PubMed] [Google Scholar]
- 32.Núñez S, Wing C, Antoniou D, Schramm VL, Schwartz SD. Insight into catalytically relevant correlated motions in human purine nucleoside phosphorylase. J. Phys. Chem. 2006;A110:463–472. doi: 10.1021/jp051277u. [DOI] [PubMed] [Google Scholar]
- 33.Wang F, Li W, Emmett MR, Hendrickson CL, Marshall AG, Zhang Y, Wu L, Zhang Z. Conformational and dynamic changes of yersinia protein tyrosine phosphatase induced by ligand binding and active site mutation and revealed by H/D exchange and electrospray ionization fourier transform ion cyclotron resonance mass spectrometry. Biochemistry. 1998;37:15289–15299. doi: 10.1021/bi981481q. [DOI] [PubMed] [Google Scholar]
- 34.Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, English JM, Greig M, He Y, Jacques SL, Lunney EA, McTigue M, Molina D, Quenzer TA, Wells PA, Yu X, Zhang Y, Zou A, Emmett MR, Marshall AG, Zhang H, Demetri G. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. U.S.A. 2009;106:1542–1547. doi: 10.1073/pnas.0812413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frantom PA, Zhang H, Emmett MR, Marshall AG, Blanchard JS. Mapping of the allosteric network in the regulation of α-isopropylmalate synthase from Mycobacterium tuberculosis by the feedback inhibitor L-leucine: solution-phase H/D exchange monitored by FT-ICR mass spectrometry. Biochemistry. 2009;48:7457–7464. doi: 10.1021/bi900851q. [DOI] [PubMed] [Google Scholar]
- 36.Laue TM, Stafford WF. Modern applications of analytical ultreacentrifugation. Annu. Rev. Biophys. Biomol. Struct. 1999;28:75–100. doi: 10.1146/annurev.biophys.28.1.75. [DOI] [PubMed] [Google Scholar]
- 37.Howlett GJ, Minton AP, Rivas G. Analytical ultracentrifugation for the study of protein association and assembly. Curr. Opin, Chem. Biol. 2006;10:430–436. doi: 10.1016/j.cbpa.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 38.Saen-oon S, Quaytman-Machleder S, Schramm VL, Schwartz SD. Atomic detail of chemical transformation at the transition state of an enzymatic reaction. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16543–16548. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang F, Shi W, Nieves E, Angeletti RH, Schramm VL, Grubmeyer C. A transition state analogue reduces protein dynamics in hypoxanthine-guanine phosphoribosyltransferase. Biochemistry. 2001;40:8043–8054. doi: 10.1021/bi010203f. [DOI] [PubMed] [Google Scholar]
- 40.Wang F, Miles RW, Kicska G, Nieves E, Schramm VL, Angeletti RH. Immucillin-H binding to purine nucleoside phosphorylase reduces dynamic solvent exchange. Prot. Sci. 2000;9:1660–1668. doi: 10.1110/ps.9.9.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry. 1998;37:8615–8621. doi: 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]
- 42.Mao C, Cook WJ, Zhou M, Federov AA, Almo SC, Ealick SE. Calf spleen purine nucleoside phosphorylase complexed with substrates and substrate analogues. Biochemistry. 1998;37:7135–7146. doi: 10.1021/bi9723919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.