

Abstract
β-Lactam-synthesizing enzymes carbapenam synthetase (CPS) and β-lactam synthetase (β-LS) are evolutionarily linked to a common ancestor, asparagine synthetase B (AS-B). These three relatives catalyze substrate acyl-adenylation and nucleophilic acyl substitution by either an external (AS-B) or internal (CPS, β-LS) nitrogen source. Unlike AS-B, crystal structures of CPS and β-LS revealed a putative Tyr-Glu dyad (CPS, Y345/E380; β-LS, Y348/E382) proposed to deprotonate the respective internal nucleophile. CPS and β-LS site-directed mutagenesis (Y345/8A, Y345/8F, E380/2D, E380/2Q, E380A) resulted in the reduction of their catalytic efficiency, with Y345A, E380A, and E382Q producing undetectable amounts of β-lactam product. However, [32P]PPi–ATP exchange assays demonstrated Y345A and E380A undergo the first half-reaction, with the remaining active mutants showing decreased forward commitment to β-lactam cyclization. pH–rate profiles of CPS and β-LS supported the importance of a Tyr-Glu dyad in β-lactam formation and suggested its reverse protonation in β-LS. The kinetics of CPS double-site mutants reinforced the synergism of Tyr-Glu in catalysis. Furthermore, significant solvent isotope effects on kcat (Dkcat) for Y345F (1.9) and Y348F (1.7) maintained the assignment of Y345/8 in proton transfer. A proton inventory on Y348F determined its D(kcat/Km) = 0.2 to arise from multiple reactant-state fractionation factors, presumably from water molecule(s) replacing the missing Tyr hydroxyl. The role of a CPS and β-LS Tyr-Glu catalytic dyad was solidified by a significant decrease in mutant kcat viscosity dependence with respect to the wild-type enzymes. The evolutionary relation and potential for engineered biosynthesis were demonstrated by β-LS acting as a carbapenam synthetase.
The compilation of genome sequence information has revealed genes encoding proteins of natural product biosynthetic pathways that differ in function from their primary metabolic ancestors. The clear evolution of these often well-studied enzymes to new, specialized roles is striking but frequently difficult to deduce from their primary sequence alone (1). Particularly in microorganisms, it is thought that survival under environmental stress will select mutant organisms whose overall fitness has improved (2, 3). The β-lactam-containing secondary metabolites, most widely known for their antibacterial properties, are an instructive example of this evolutionary process.
Streptomyces clavuligerous β-lactam synthetase (β-LS) and Pectobacterium carotorova carbapenam synthetase (CPS)1 catalyze formation of the corresponding β-lactam rings in the clinically important β-lactamase inhibitor clavulanic acid (4) and (5R)-carbapenem-3-carboxylic acid (5), which is believed to be representative of the potent, broad spectrum carbapenem class of antibiotics. These pivotal transformations occur early in the respective biosyntheses. Based on both sequence and structure homology (4, 6), these β-lactam-forming enzymes show evolutionary roots in the broadly distributed asparagine synthetases, class B (AS-B), which convert the essential amino acid aspartate to asparagine. The sequence and structure similarity among AS-B, β-LS, and CPS are reflected in the overall chemistry leading to the synthesis of asparagine, deoxyguanidinoproclava-minic acid (DGPC), and (3S,5S)-carbapenam-3-carboxylate,2 respectively (Scheme 1). In their synthetase domains these three ATP/Mg2+-dependent reactions undergo two distinct chemical steps (or half-reactions): acyl-adenylation and nucleophilic acyl substitution (7–10). Recently, much has been learned about β-LS and CPS by detailed examination of their steady-state kinetics (11, 12). Both proteins exhibit an ordered Bi-Ter kinetic mechanism with ATP the first substrate to bind and PPi the last product to dissociate (7, 10). Additionally, a conformational change involving the opening of a catalytic loop associated with product release is at least partially rate-determining in both enzymes (11, 12). A large forward commitment exists to DGPC and carbapenam formation after acyl-adenylate activation of the respective substrates, implicating both substrate preorganization and the high chemical reactivity of the adenylated intermediate as important catalytic strategies for β-LS and CPS (7, 11). Since these β-lactam-producing enzymes share similar sequence and structure together with other common mechanistic features, it is proposed that this step of clavam and carbapenem biosynthesis is very closely related (13).
Scheme 1.
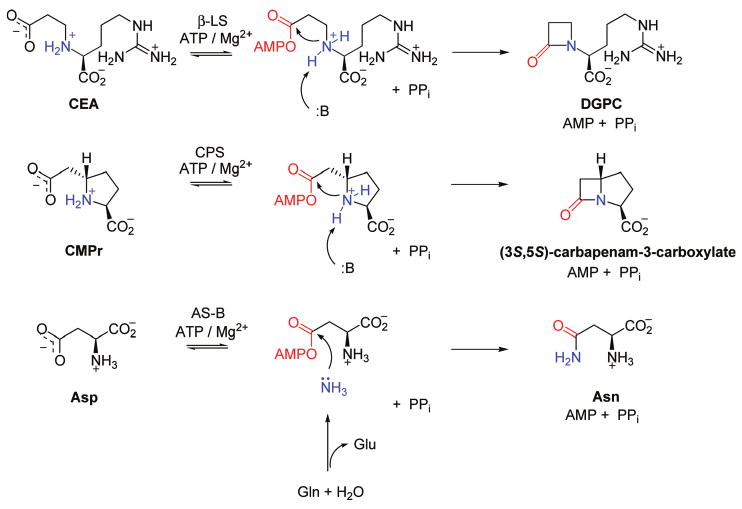
Chemical Reactions of β-LS, CPS, and AS-B: Acyl-Adenylate Activation and Nucleophilic Substitutiona
a The activated acyl group of each substrate is shown in red, and the amine nucleophile in each reaction is shown in blue.
The evolution to β-LS and CPS from their predecessor, AS-B, likely occurred independently through gene duplication and random mutations to enzymes of changed function (14). Crystal structures of β-LS provide hints to how this process occurred. While retaining the two subunits of AS-B (glutaminase and synthetase domains), an essential ammonia tunnel has collapsed and is blocked at the entrance to the synthetase domain of β-LS (15). Moreover, the pseudoglutaminase domain in β-LS lacks the catalytic cysteine essential for glutaminase activity in AS-B (16–18). Kinetic studies on β-LS and CPS reinforced the observation of an inactive glutaminase domain and demonstrated the complete loss of asparagine synthetase activity. Deprived of both a source and a path to transport “nascent” ammonia to an adenylated substrate in the synthetase domain, an intramolecular amine in N2-(2-carboxyethyl)-l-arginine (CEA) and (2S,5S)-carboxymethylproline (CMPr), both β-amino acids, is utilized in β-LS and CPS, respectively. In the case of β-LS, the active site is markedly elongated to accommodate the increased length of the substrate CEA (12 Å) relative to aspartate (5 Å) (19).
A key substitution visualized in overlays of β-LS and CPS with AS-B was a tyrosine residue in the β-lactam-forming enzymes (β-LS, Y348; CPS, Y345), replacing an active site glutamate (E348) found in AS-B (Figure 1). E348 is universally conserved in AS-B enzymes and located at the end of the relatively hydrophobic ammonia tunnel (15). Site-directed mutagenesis of this glutamate to alanine in Escherichia coli AS-B resulted in undetectable asparagine formation and implicated it as the catalytic base responsible for ensuring deprotonation to a neutral ammonia nucleophile leading to Asn formation (unpublished data in ref 20).
Figure 1.
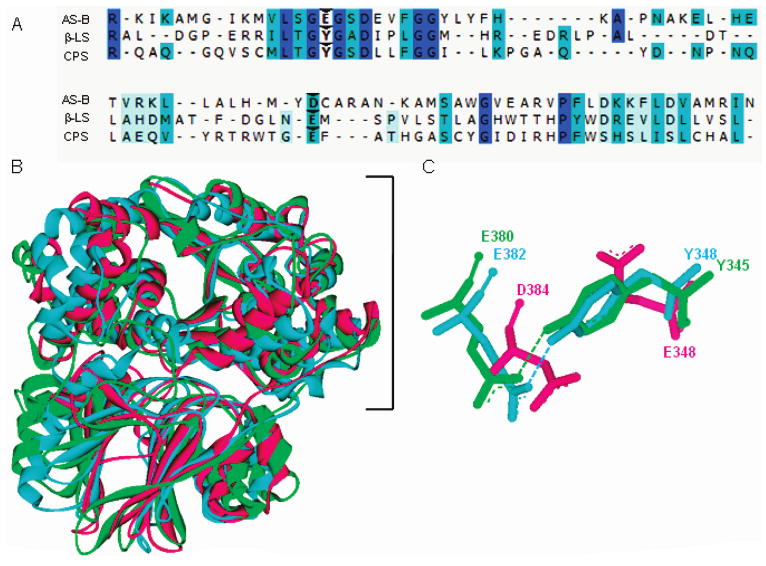
Sequence and structural comparison of homologues AS-B, β-LS, and CPS. Panel A gives a partial sequence alignment of AS-B, β-LS, and CPS with weak to identical matches in amino acid sequence shown as light blue to dark blue, respectively. Carets indicate the conserved carboxylate-containing residues (D384, E382, E380) and the replacement of AS-B E348 with Y348 and Y345 in β-LS and CPS, respectively. Panel B depicts the structural superimposition of AS-B, β-LS, and CPS monomers with the synthetase domains enclosed by a bracket. Panel C illustrates the conserved Tyr-Glu dyads in β-LS and CPS overlaid with the coaligning aspartate and catalytic glutamate in E. coli AS-B. Panels B and C illustrate AS-B, β-LS, and CPS as the colors pink, blue, and green, respectively.
Substitution of this strictly conserved glutamate in primary metabolism to a tyrosine in both β-LS and CPS could account for how AS-B enzymes altered their chemical mechanisms to produce β-lactam rings. While the first half-reaction of AS-B relative to β-LS and CPS is identical, the key difference in the second half-reaction is intermolecular nucleophilic attack by ammonia in AS-B versus intramolecular attack by a secondary amine in the case of β-LS and CPS. A more advanced mechanism of catalysis can be proposed from structural images of β-LS and CPS mediated by way of a Tyr-Glu catalytic dyad (Y348/E382 in β-LS; Y345/E380 in CPS). The dyad glutamate in β-LS and CPS corresponds to a similar amino acid in AS-B, D384. D384, however, is thought to be involved only in binding the α-amine of its aspartate substrate (19). The Tyr-Glu dyad in β-LS and CPS is proposed to deprotonate the secondary amine of their respective substrates, which is bound as its ammonium ion, to allow intramolecular nucleophilic substitution of the acyl-adenylate and ring cyclization. In the case of β-LS, N2-(carboxymethyl)-l-arginine (CMA), the lower homologue of CEA, cannot cyclize to the strained α-lactam. It could, however, undergo acyl-adenylation, which was visualized in high resolution and, thereby, captured an image of the high-energy intermediate generated in the first half-reaction. These pictures indicated that the dyad tyrosine Oη and glutamate Oδ are 2.5 Å apart, and it is estimated that the corresponding hydrogen bound between these two residues is even closer (17).
The potential functional importance suggested by these observations of a catalytic Tyr-Glu dyad observed in β-LS and CPS is demonstrated by site-directed mutagenesis and kinetic experiments that compare the wild-type enzymes to their respective mutant proteins using pH–rate profiles, 32P-incorporation experiments, solvent isotope effects (SIE), proton inventory, and viscosity variation.
Materials and Methods
Materials
All buffers, coupled enzymes, and most assay components were purchased from Sigma Chemical Co. (St. Louis, MO). NADH, NADP+, and UDP-glucose were obtained from Roche Applied Sciences (Indianapolis, IN). E. coli Rosetta2(DE3) and BL21(DE3) cells were purchased from EMD Biosciences, Inc. (Madison, WI). Plasmid pUC19 was from Invitrogen (Carlsbad, CA), and pCDFDuet-1 was purchased from Novagen (La Jolla, CA). Plasmids pET24a/cps, pET24a/carBC, and pCDFDuet-1/cps were generous gifts from Dr. R.-F. Li of this laboratory (5) (and unpublished experiments). Nitrocefin was kindly provided by K. A. Moshos of this laboratory. D2O (99%) was purchased from Cambridge Isotope Laboratories (Andover, MA), while DCl, NaOD, and hydroxylamine were obtained from Aldrich (Milwaukee, WI). Na432P2O7 was purchased from Perkin-Elmer (Boston, MA), activated charcoal (NoritA) from Fischer Scientific (Pittsburgh, PA), and scintillation fluid (Optifluor) from Packard Instrument Co. (Meridan, CT). The substrates CMPr and CEA, along with the β-LS product DGPC, were synthesized as previously described (11, 21, 22).
Protein Structural and Sequence Overlays
The Accelrys Discovery Studio was used to model the sequence and structures of AS-B (PDB code 1CT9), β-LS (PDB code 1MC1), and CPS (PDB code 1Q19) in Figure 1. MODELER determined protein alignment based on 3D structural similarity. In Figure 7 the hydrogens shown in white were modeled using Accelrys Discovery Studio. MODELER overlaid the Tyr-Glu dyad residues of the apo-β-LS (PDB code 1M1Z) and the structure of β-LS/CMA-AMP/PPi (PDB code 1MBZ).
Figure 7.
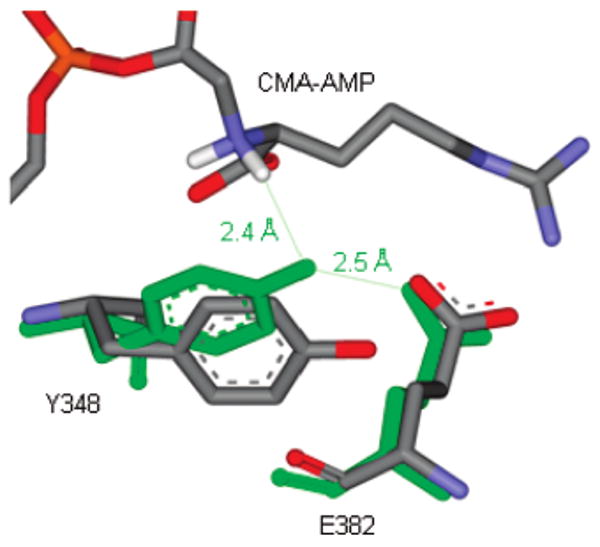
Analysis of Tyr348 rotation in the apo-β-LS and β-LS/CMA-AMP/PPi. The orientation of Tyr348 in the apo-β-LS is colored by element (orange = phosphorus, red = oxygen, blue = nitrogen, gray = carbon, white = hydrogens). The Tyr-Glu dyad residues in the acyl-adenylate image are shown in green with CMA-AMP colored by element as described above. The Oη–H distance from Y348 in the β-LS/CMA-AMP/PPi picture and the hydrogen of the α-nitrogen of N2-(2-carboxymethyl)-l-arginine (CMA), respectively, is estimated at 2.4 Å. The respective Oη–Oδ distance of Tyr348 and Glu382 in the same crystal structure is given as 2.5 Å.
Site-Directed Mutagenesis, Overproduction, and Purification of Mutants in CPS and β-LS
The mutant proteins described in this investigation were constructed, overproduced, and purified as described elsewhere for CPS (12) and β-LS (11). The CPS double mutants Y345A/E380A, Y345F/E380D, and Y345F/E380Q were constructed in the same manner as the single mutants using the mutagenic primers for the various E380 mutants and the template vector of the single mutations Y345F and Y345A. The mutant cps and bls genes were cloned into pET24a and pET29b (Novagen, Madison, WI), respectively, for subsequent overproduction. The presence of the desired mutations in all genes as well as the absence of adventitious mutations was confirmed by complete gene sequencing (DNA Sequence Facility, The Johns Hopkins University, Baltimore, MD).
Circular Dichroism
The structural integrity of the CPS and β-LS active site mutants was verified using circular dichroism (CD) analysis as previously described (11, 12).
[32P]PPi–ATP Exchange Assay
Reaction mixtures of 100 μL at the desired pH were incubated at room temperature for approximately 1–2 h. For CPS, assays were conducted at pH 8.67(μ = 0.15) and contained 10mM MgCl2, 1 mM DTT, 5 mM ATP, 5 mM CMPr, and 2 mM pyrophosphate with 0.02 μCi of [32P]PPi and were initiated by the addition of enzyme (6–9 μM; Y345F, Y345A, E380D, or E380A). β-LS assays were conducted at pH 8.8 (μ = 0.1) and had 12 mM MgCl2, 1 mM DTT, 4 mM ATP,4 mM CEA, and 0.10 mM pyrophosphate with 0.05 μCi of [32P]PPi and initiated with enzyme (3 μM; Y348F, Y348A, E382D, E382Q). E382Q was also tested at pH 8.3 using 9 μM protein and 0.5 μCi of radioactivity. All assays were quenched by the addition of 3% (w/v) perchloric acid followed by 100 mM pyrophosphate and 1% (w/v) activated charcoal. The mixture was vortexed and centrifuged for 5 min (28000g). The pellet was subsequently washed and the radioactivity analyzed as previously reported (11). Each reaction was performed in duplicate or triplicate. Control assays were used to calculate the background for each enzyme and contained all components except substrate (CEA or CMPr).
CPS and β-LS Steady-State Kinetic Assays
PPi and AMP release catalyzed by CPS and β-LS was monitored at 340 nm as the increase in rate of production of NADPH or the rate of NADH oxidation, respectively, in established (23) coupled enzyme assays (10). The AMP assay was used for β-LS (7,11), and the PPi assay was utilized for CPS mutants (10, 12) as previously described for each protein, unless noted otherwise. Reactions were performed at 25.0 ± 0.1 °C using a water-circulating system, carried out in a final volume of 500 μL, and initiated by the addition of enzyme. The established two-buffer system for CPS (100 mM HEPES, 80 mM piperazine, μ = 0.15) (10) and the three-buffer system for β-LS (35 mM HEPES, 35 mM TAPS, 35 mM AMP-HCl, μ = 0.1) (11) were used for all assays with the ionic strength held constant using KCl (24). The second substrate to bind in CPS (CMPr; 0.05–11 mM) and β-LS (CEA; 0.05–8 mM) was varied with ATP held at saturating levels (2–4 mM) for each protein discussed. Control experiments without the second substrate indicated no significant background ATP hydrolysis occurred under the conditions described for CPS and β-LS mutants. Also, the initial velocity conditions of the assays were not limited by the coupling enzymes, and the observed rates for both β-LS and CPS were independent of coupling enzyme concentrations. The kinetic parameters kcat and Km were determined from nonlinear regression using eq 1. The kcat/Km error was propagated from the method of least squares.
Confirmation of β-Lactam Formation in CPS and β-LS Mutant Reactions
Carbapenam formation in CPS mutants was confirmed using an in vivo nitrocefin assay and is described elsewhere (12). The formation of the monobactam product, DGPC, in the β-LS-catalyzed reaction was shown from HPLC assays at pH 8.8 with an authentic DGPC standard and was performed as previously specified (11) with assays containing 1–4 mM ATP, 3–4 mM CEA, 1 mM DTT, 12.5 mM MgCl2, and 16 μM proteins. In addition to pH 8.8, the Y348A mutant was analyzed at pH 7.8 and 7.3 and E382Q at pH 9.5.
pH Dependence of CPS and β-LS Mutant Proteins
At all pH values assays were conducted in duplicate or triplicate. Protein stability was tested by incubating each mutant at the extreme pH values, for at least the duration of the assay, and then measuring the activity. Profiles were plotted as the log kcat or log (kcat/Km) as a weighted function of pH. Based on R2 values, the kcat and kcat/Km parameters were best fit to the appropriate equations and the pKa values determined. Standard error is shown as error bars and is smaller than the data point when not visible.
(A) CPS
CPS single-point mutants were subjected to pH variation in the range of 6.67–10.33. The kinetic constants of each dyad mutant were determined from full Michaelis–Menten profiles. The log kcat and log(kcat/Km) versus pH profiles of Y345F and E380D were fitted to eq 2, and E380Q was best fit to eq 3. CPS dyad mutants Y345A and E380A displayed a linear pH dependence.
(B) β-LS
The pH dependence of the β-LS mutants was studied under the allowable pH range of each protein (pH 6.8–9.8). The pH–(kcat/Km) profiles of E382D were constructed from the linear slope of the Michaelis–Menten hyperbolic curve (low CEA concentrations), which included at least three data points located below the Km,CEA calculated at each pH value. At pH values where the Km,CEA was greater than 1 mM for Y348F (pH 8.3–9.5), this technique was also used to achieve the kcat/Km measurements at high accuracy. The log kcat versus pH dependence of Y348F and E382D was fitted to eq 4. The data for log(kcat/Km) of E382D and Y348F were fitted to eqs 2 and 5, respectively. Y348A showed no reliable pH-dependent fit in the pH range tested.
Solvent Isotope Effects
Preparation of substrates, assay components, and buffers was performed as previously described for solvent isotope effects (SIE) on CPS (12) and β-LS (11). All buffer systems were dissolved in D2O and adjusted to the appropriate pD (pD = pH + 0.4) (25) by the addition of NaOD or DCl and stored under N2(g) immediately before use. Samples dissolved in D2O were stored under N2(g) and covered in parafilm prior to use. The assays were conducted in >95% D2O to determine the SIE on kcat and kcat/Km. The nomenclature of Dkcat and D(kcat/Km) refers to the deuterium solvent isotope effect on kcat and kcat/Km (26), respectively. Error on the SIE values was propagated as previously reported (27).
(A) CPS Y345F and Y345A Mutants
The effects of heavy water on the first- and second-order rate constants of Y345A were determined at pD 8.0 and 9.33, and the corresponding SIEs on Y345F were calculated from the χmax values revealed from the fit of eq 2 to the experimental data (pL = 6.67–10.33). Each assay was performed in duplicate.
(B) β-LS Y348F Mutant
The Dkcat and D(kcat/Km) for Y348F were measured in the plateau region of both kcat and kcat/Km [pH 9.5 (pD 9.9)]. Proton inventory was conducted on kcat/Km of Y348F at different mole fractions of D2O (n = 0,0.35, 0.5, 0.65, 1.0) at pL 9.5 (μ = 0.1). The specificity constant was determined as described above to achieve high accuracy. The pL of each buffer solution was calculated using the relationship specified by Schowen and Schowen (28).
Viscosity Variation
Viscosity dependence of CPS mutants (Y345F, Y345A, E380D, and E380A) was determined at pH 8.0 and of β-LS mutants (Y348F, Y348A, and E382D) at pH 9.0. The viscosities of the solutions were established using a Brook-field viscometer at 25 °C and were performed in triplicate or quadruplicate. Assays were conducted as previously reported for wild-type CPS (12) and β-LS (11) and were performed in duplicate with microviscogen glycerol [0-30% (w/v)] and a macroviscogen control, PEG 8000 [0-6% (w/v)]. The kcat viscosity dependence of the β-LS Y348A mutant was determined at saturating conditions of CEA (3.2 mM) and ATP (2 mM) and conducted in triplicate. All data acquired were plotted as reciprocal relative rates (kcato/kcat) versus relative viscosity (ηrel), and the slopes from the plots are reported as ηkcat and η(kcat/Km) for kcat and kcat/Km, respectively (Supporting Information).
Wild-Type β-LS with (2S,5S)-Carboxymethylproline
(A) [32P]PPi–ATP Exchange Assay. [32P]PPi –ATP exchange assays were performed as described above for β-LS unless noted otherwise. The reactions were run at pH 7.3, 8.3, and 8.8 with a CMPr concentration of 6 mM and 0.02 μCi of [32P]PPi and were initiated with 5 μM wild-type β-LS. (B) Steady-State Kinetic Assays. The steady-state kinetic constants of β-LS using CMPr as its varied substrate (1.5–9 mM) were achieved with the AMP-coupled enzyme assay at saturating levels of ATP (2 mM). (C) Confirmation of Product Formation. Formation of (3S,5S)-carbapenam-3-carboxylate was confirmed using the previously established nitrocefin assay (5, 29) and is described in Supporting Information.
Steady-State Kinetic Analysis
All kinetic data were fit using Kaleidograph 4.0 or Sigma Plot 9.0. Initial velocity patterns were fit to eq 1. The varied substrate is shown as A, and χmax and χmin represent the upper or lower limits of the fits, respectively. The kinetic constants kcat or kcat/Km are given as y.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Results
Construction and Purification of CPS and β-LS Mutants
The singly and doubly mutated active site residues of CPS were constructed using QuikChange site-directed mutagenesis and the pUC19/cps template followed by ligation into pET24a for overproduction in E. coli. β-LS active site mutants were assembled using the PCR overlap extension method and ligated into pET29b for subsequent heterologous expression. The relative purity of all CPS and β-LS purified mutants was determined by SDS–PAGE analysis and found to be >95%. In CPS single-point mutants included Y345F, Y345A, E380D, E380Q, and E380A, while the β-LS mutants were Y348F, Y348A, E382D, and E382Q. The CPS proteins containing double-site mutations were Y345A/E380A, Y345F/E380D, and Y345F/E380Q. The CD spectra of all mutants were little changed from that of wild type, suggesting that minimal global changes in protein structure occurred upon introduction of each new mutation (data not shown).
Characterization of CPS and β-LS Single-Point Mutants
Using the PPi- and AMP-coupled enzyme assays, the kinetic constants for the CPS and β-LS single-point mutants were determined and are summarized in Table 1. For all CPS mutants, except Y345A and E380A in which no AMP release was observed, comparable initial linear rates of formation of AMP and PPi were measured. The β-LS mutants also gave similar values when comparing the AMP and PPi assays, indicating a close correlation of the first and second half-reactions, the only exception being Y348A, which showed increased levels of PPi compared to AMP release (ca. 2-fold increase in kcat).
Table 1.
CPS and β-LS Mutant Kinetic Parameters Compared to Wild-Type Proteinsa
| kcat (s−1) | Km,s (mM) | kcat/Km,s (mM−1 s−1) | −Δkcat | +ΔKm | −Δ(kcat/Km) | |
|---|---|---|---|---|---|---|
| CPS | ||||||
| wild typeb | 0.36 ± 0.01 | 0.35 ± 0.02 | 1.04 ± 0.04 | 1 | 1 | 1 |
| Y345F | 0.17 ± 0.01 | 0.27 ± 0.04 | 0.63 ± 0.08 | 2 | 0.77 | 1.7 |
| Y345A | 0.011 ± 0.001 | 1.7 ± 0.3 | 0.0063 ± 0.0013 | 33 | 5 | 170 |
| E380D | 0.26 ± 0.02 | 0.36 ± 0.06 | 0.72 ± 0.14 | 1.4 | 1 | 1.4 |
| E380Q | 0.020 ± 0.001 | 0.075 ± 0.015 | 0.27 ± 0.06 | 18 | 0.21 | 4 |
| E380A | 0.0067 ± 0.0020 | 0.49 ± 0.02 | 0.014 ± 0.004 | 54 | 1.4 | 74 |
| β-LS | ||||||
| wild type | 0.82 ± 0.01 | 0.044 ± 0.002 | 18.6 ± 0.7 | 1 | 1 | 1 |
| Y348F | 0.155 ± 0.007 | 1.4 ± 0.2 | 0.11 ± 0.02 | 5.3 | 32 | 170 |
| Y348A | 0.0082 ± 0.0002 | 0.19 ± 0.02 | 0.043 ± 0.004 | 100 | 4.3 | 430 |
| E382D | 0.193 ± 0.006 | 2.8 ± 0.2 | 0.069 ± 0.006 | 4.2 | 64 | 270 |
| E382Q | ND | ND | ND |
CPS kinetic parameters were determined at pH 8.0 using the PPi assay. β-LS constants were determined at pH 8.8 with the AMP assay. S designates the varied substrate (2S,5S)-CMPr and CEA in CPS and β-LS, respectively. +Δ represents a fold increase and −Δ a fold decrease relative to wild-type enzymes. ND represents no detectable activity.
The nitrocefin bioassay showed that, while CPS mutants Y345F, E380D, E380Q, Y345F/E380D, and Y345F/E380Q catalyzed the formation of the β-lactam ring in the carbapenam product, (3S,5S)-carbapenam-3-carboxylate, Y345A, E380A, and Y345A/E380A did not show the production of a nitrocefin-positive compound. In β-LS, DGPC formation was directly visualized for all mutant proteins, except E382Q, at the optimal pH of 8.8 by HPLC using an authentic DGPC standard as the reference. Accordingly, E382Q possessed no detectable activity in either the AMP- or PPi-coupled enzyme assay. HPLC analysis of E382Q confirmed the coupled enzyme assay results and revealed that no product was formed at pH 8.8 or 9.5. While Y348F and E382D were active above and below neutral pH, Y348A showed AMP release and DGPC formation only at alkaline pH values with no detectable AMP release at pH ∼7 under initial velocity conditions, which was confirmed by HPLC analysis.
[32P]PPi–ATP Exchange Assay
To test for reversible acyl-adenylate formation during the first chemical transformation, the CPS and β-LS mutants were subjected to [32P]PPi–ATP exchange assays (30). Previously, it was shown that the CPS- and β-LS-catalyzed acyl-adenylation reactions are functionally irreversible and do not exhibit [32P]PPi exchange at their optimal pH values. In CPS and β-LS this results from a large forward commitment to ring cyclization (11). In contrast to the wild-type enzymes, all CPS mutants and β-LS mutants, with the exception of E382Q (inactive), showed [32P]PPi exchange at optimal pH values (Figure 2). No detectable 32P incorporation was observed with the β-LS mutant E382Q (β-LS) under all conditions tested. Additionally, the β-LS mutant Y348A showed appreciable [32P] PPi exchange at pH 7.3 (data not shown), a pH value at which it cannot form detectable amounts of DGPC.
Figure 2.
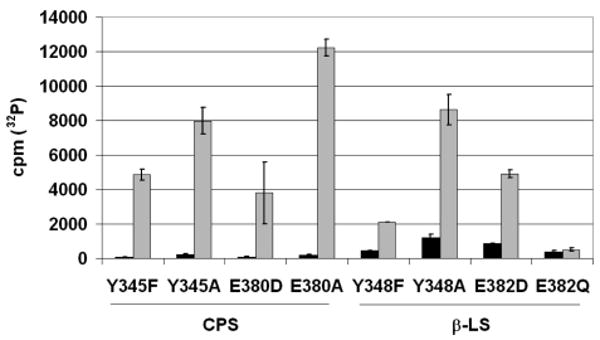
[32P]PPi–ATP exchange assays with CPS and β-LS dyad mutants. The bar graph displays the background [32P]PPi exchange (black) and the corresponding detection in CPS or β-LS assays containing all components (gray). The radioactivity of 32P is given in counts per minute (cpm). Standard errors of the background and complete reactions are shown by the error bars.
pH Dependence
The pH dependence of the kinetic parameters kcat and kcat/Km was determined to identify ionizable groups that are important to catalysis. The pH dependence of the kinetic parameters is summarized in Tables 2 and 3 for CPS and β-LS, respectively. Graphical representations of the data and their best fits are shown in Figures 3 and 4 for CPS and β-LS, respectively. Two ionizable groups are evident from the wild-type CPS pH–(kcat/Km) profile at 7.8 and 10.0 and pH–kcat profile at 7.4 and 9.7 (12), while β-LS shows one ionizable group at 8.1 in the pH–kcat profile and a diprotic system in the pH-dependent profile of kcat/Km at ∼8 and ∼9 (11).
Table 2.
pH Dependence for CPS Dyad Mutantsa
| kcat | kcat/Km | |||
|---|---|---|---|---|
| CPS | pKa | pKb | pKa | pKb |
| WTb | 7.4 ± 0.1 | 9.7 ± 0.1 | 7.8 ± 0.1 | 10.0 ± 0.1 |
| Y345F | 7.67 ± 0.01 | 9.65 ± 0.01 | 7.6 ± 0.1 | 9.5 ± 0.1 |
| Y345A | – | – | – | – |
| E380D | 7.3 ± 0.1 | 9.8 ± 0.1 | 7.63 ± 0.01 | 9.43 ± 0.01 |
| E380Q | 9.46 ± 0.01 | 8.75 ± 0.05 | ||
| E380A | – | – | – | – |
(–) indicates no transition observed in the pH–rate profile. WT represents wild-type CPS.
Values previously reported in ref 12.
Table 3.
pH Dependence for β-LS Dyad Mutantsa
| kcat | kcat/Km | ||
|---|---|---|---|
| β-LS | pKa | pKa | pKb |
| WTb | 8.07 ± 0.05 | 9.15 ± 0.06 | 7.95 ± 0.08 |
| Y348F | 8.37 ± 0.03 | 7.25 ± 0.05 | |
| Y348A | – | – | – |
| E382D | 8.27 ± 0.08 | 9.5 ± 0.1 | 8.3 ± 0.2 |
(–) indicates no transition observed in the pH–rate profile from no detectable activity below pH 8.3. WT represents wild-type β-LS. Italic type shows the pK values that were further calculated from the observed pH–(kcat/Km) and reflects reverse protonation of the protein.
pKa in the wild-type pH–kcat profile previously reported in ref 11.
Figure 3.
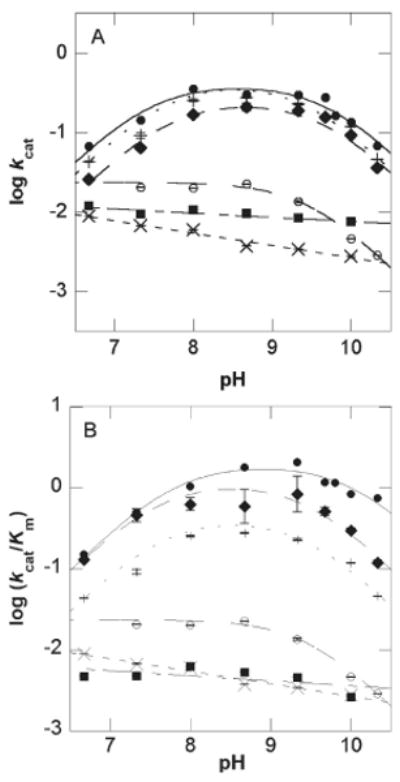
pH dependence of kcat and kcat/Km for wild-type CPS (12) and the active site mutants. Plot of (A) log kcat versus pH and (B) log (kcat/Km) versus pH for wild-type CPS (●), Y345F (◆), Y345A (■), E380D (+), E380Q (○), and E380A (×). Error bars are shown and do not exceed the size of the data points when not visible.
Figure 4.
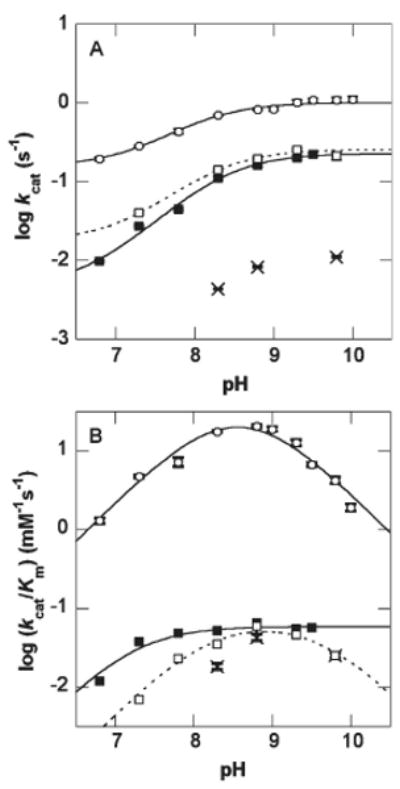
pH dependence of kcat and kcat/Km for wild-type β-LS (11) and the active site mutants. Plot of (A) log kcat versus pH and (B) log (kcat/Km) versus pH for wild-type β-LS (○), Y348F (■), Y348A (×), and E382D (□). Error bars are shown and do not exceed the size of the data points when not visible.
Similar to wild-type CPS, the Y345F and E380D mutants showed bell-shaped curves with unit slopes in the acidic and basic limbs for both kcat and kcat/Km. The Y345A and E380A mutants showed no discernible transition, while the E380Q mutant displayed only a descending limb for both first- and second-order rate constant pH profiles. For β-LS, Y348A had no detectable rate below pH 8.3 under initial velocity conditions. Hence, no reliable curve can be fit to this narrow pH range. From its pH–kcat profile, however, a plateau region above pH 8.8 is evident. The E382D β-LS mutant gave similar pH dependence to that of wild type in both rate versus pH plots. Conversely, Y348F lacked the basic limb of the pH–(kcat/Km) profile, and the acidic limb is shifted ∼1 pK unit lower, relative to wild-type β-LS. Its pH–kcat profile, however, is similar to that of wild type.
For E382D, the pK values were recalculated using the method of Segel (31) because the observed pK values in the bell-shaped curve were only approximately one unit apart. For the same reason wild-type β-LS pK values were also determined by this method. While the best-fit equation of wild-type β-LS kcat/Km data versus pH previously afforded three pK values (R2 = 0.99), the third pK determined is not reliable since it was at or near the highest pH attainable (11). In this instance, wild-type β-LS was fit to a bell-shaped curve (R2 = 0.98) to examine the two pK values within the reliable pH range, which yielded accurate values in the observed diprotic system. In this way, wild-type β-LS is directly comparable to the E382D mutant. Equations 6–8 were used to calculate accurate pK values for wild-type β-LS and E382D (31):
| (6) |
| (7) |
| (8) |
where the maximum rate and apparent pK values in the observed bell-shaped curve are given as χmax,app and pKapp, respectively; the optimal pH of each system is represented as pHopt. The actual pK values are given as pKE1 and pKE2, and the true maximum activities are given as χmax.
SIE on Y345 and Y348 Mutants
Acid–base chemistry is expected to exhibit a significant SIE if chemistry is at least partially rate-determining. Therefore, D2O (99.9%) was employed to determine the effect of deuterium on the reactions catalyzed by CPS and β-LS. SIEs were calculated for the various Y345 (CPS) and Y348 (β-LS) mutants to confirm their role in proton transfer(s) during four-membered ring formation.
Since the rate behavior of the CPS mutant Y345A is essentially independent of pH, SIE measurements were conducted at pL values 8.0 and 9.33. At pD 8.0 and 9.33, respective deuterium solvent isotope effects of 1.18 ± 0.12 and 1.05 ± 0.07 on kcat were calculated, indicating that in this region the Y345A has the same pL profile. Similarly, at pD 8.0 and 9.33, SIE measurements on the second-order rate constant were 1.2 ± 0.2 and 1.1 ± 0.1, respectively. Thus, Y345A does not exhibit a significant SIE on either the first- or second-order rate constants.
To investigate the effect of deuterium on the pK values of the Y345F variant, pD–rate profiles (Figure 5) were constructed at ≥95% D2O. While there is a ΔpK of ∼0.5 for pKb, there is a ΔpK of ∼0.1 for pKa. Dkcat and D(kcat/Km) were determined from the maximum rates of the fitted bell-shaped curves and were calculated to be 1.9 ± 0.3 and 2.3 ± 0.5, respectively. A proton inventory was not attempted with the Y345F variant due to the precision required in rate constants for an isotope effect below 2.0 (0.4–1%) (32).
Figure 5.
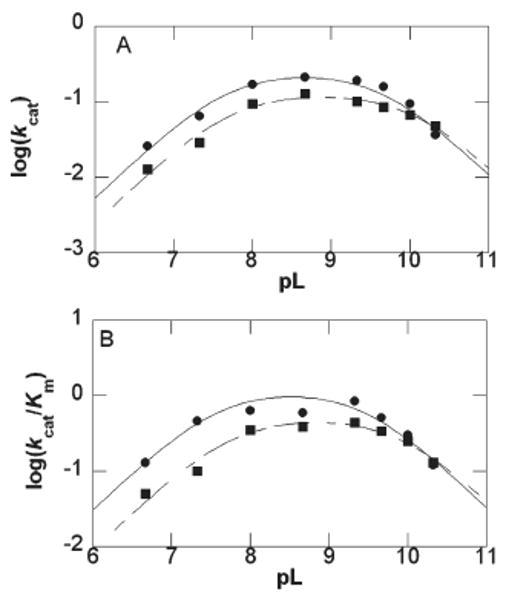
pL dependence of kcat and kcat/Km for CPS mutant Y345F. (A) Plot of log kcat versus (●) pH and pD (■) determined the pK values to be 7.67 ± 0.01, 9.65 ± 0.01 and 7.7 ± 0.1, 10.1 ± 0.1, respectively. (B) Plot of log(kcat/Km) versus (●) pH and pD (■) calculated the pK values to be 7.6 ± 0.1, 9.5 ± 0.1 and 7.7 ± 0.1, 9.9 ± 0.2, respectively. Error bars are shown and do not exceed the size of the data points when not visible.
The pH–rate profiles of the β-LS mutant Y348F exhibit a plateau region above pH 8.8 and 7.8 in the kcat and kcat/Km plots, respectively. Accordingly, SIE measurements were performed at pH 9.5 (pD 9.9), the highest attainable pH without loss in activity, for both kcat and kcat/Km. The effects of heavy water on the Y348F catalyzed reaction gave a normal SIE on kcat of 1.7 ± 0.1 and an inverse D(kcat/Km) of 0.24 ± 0.02. The large, inverse SIE on kcat/Km was further studied at pL 9.5 using the proton inventory technique (33, 34). Since D(kcat/Km) at pD 9.5 was 0.21 ± 0.01, the SIE at pD 9.5 and 9.9 were statistically identical and ensured that the proton inventory was performed in the pL-independent region. The second-order rates (kn) relative to the rate in 100% H2O (k0) were plotted versus n and showed a bowl-shaped dependence (Figure 6). The data were best fit to eq 9, where nk represents kn = 1 divided by k0, and implied that the D(kcat/Km) of ∼0.2 is from multiple reactant-state fractionation factors (φR). Equations 10 and 11 represent one “tight” transition state fractionation factor (φT) and a single φR, respectively (33). Equations 9, 10, and 11 gave R2 values of 0.98, 0.47, and 0.86, respectively, when fitted to the experimental data. Equations 9–11 were derived from the Gross–Butler expression shown in eq 12 (35).
Figure 6.
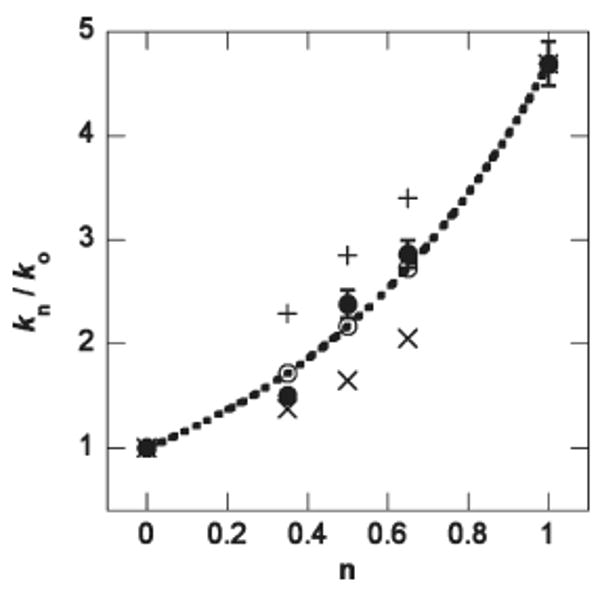
Proton inventory of β-LS dyad mutant Y348F kcat/Km data. The plot of (●) kn/k0 versus mole fraction of D2O (n) is best fit to a line (—) representative of eq 9 in the text. An exact fit to multiple φR (dashed line) is shown as (○). The symbol (+) corresponds to one φT, and (×) shows a fit to one φR. kn and k0 represent the observed rate at n and 100% H2O, respectively. Error bars represent the standard error on the experimental data and do not exceed the size of the data points when not visible.
| (9) |
| (10) |
| (11) |
| (12) |
Additionally, a linear plot can be fit to the natural log of the observed rate (ln kn) versus mole fraction of D2O (n) (R2 = 0.98) supporting many-proton reactant-state fractionation factors (Supporting Information) (33).
Viscosity Variation
To test if a conformational change is rate-determining in the mutant CPS and β-LS enzyme-catalyzed reactions as is observed in their respective wild-type enzymes, the viscosity of the assays was increased using the microviscogen glycerol and PEG 8000 as the macroviscogen control (Supporting Information). With the exception of the CPS E380D mutant, the rate of reaction for all of the mutants for both CPS and β-LS was relatively invariant to an increase in solution viscosity. Also, glycerol and PEG 8000 increased the kcat/Km values of β-LS and all its mutants; this effect was attributed to nonspecific effects and will not be discussed further (11, 36).
Carbapenam Synthetase Double-Site Mutants
Since the kinetic parameters of the single-site mutants of CPS were not dramatically altered from those of wild type, double mutations of Y345 and E380 were constructed to further test the hypothesis that these two residues form a catalytic dyad (Table 4). The Y345F/E380D and Y345F/E380Q double mutants exhibited a 61-fold and 33-fold decrease in kcat, respectively, and formed the carbapenam product as assessed by nitrocefin assay. On the other hand, the Y345A/E380A mutant did not form the carbapenam product when similarly assayed and did exhibit PPi release with no corresponding AMP release detected (CMPr at 10 × Km of wild type) when monitored by continuous assay.
Table 4.
Kinetic Parameters of CPS Double-Site Mutantsa
| CPS mutant | kcat (s−1) | −Δ |
Km (mM) |
+Δ |
kcat/Km (mM−1 s−1) |
−Δ |
|---|---|---|---|---|---|---|
| Y345F/E380D | 0.0059 ± 0.0001 | 61 | 0.49 ± 0.06 | 1.4 | 0.012 ± 0.001 | 86 |
| Y345F/E380Q | 0.011 ± 0.001 | 33 | 0.9 ± 0.2 | 2.6 | 0.011 ± 0.001 | 95 |
| Y345A/E380A | 0.0051 ± 0.0004 | 71 | 0.40 ± 0.01 | 1.1 | 0.012 ± 0.001 | 87 |
Steady-state rate constants determined using the PPi coupled enzyme assay at pH 8.0. +Δ refers to the fold change above the Km,CMPr listed for wild-type CPS and −Δ to the fold change below the kcat or kcat/Km,CMPr of wild-type CPS.
β-LS with (2S,5S)-Carboxymethylproline as the Varied Substrate
β-LS was administered the CPS substrate, CMPr, and monitored for the two chemical steps in β-LS catalysis: acyl-adenylation and β-lactam formation. The anticipated β-lactam product of β-LS/ATP and CMPr was the CPS natural product, (3S,5S)-carbapenam-3-carboxylate. At pH 8.3 and 8.8 [32P]PPi– ATP exchange assays revealed an approximate 30-fold increase in radiolabeled ATP compared to control, implying that β-LS binds and activates CMPr above pH 7.3 (Supporting Information). The ability of β-LS to cyclize adenylated CMPr was tested with the AMP-coupled enzyme assay and the nitrocefin assay. The AMP assay confirmed that β-LS cyclizes the acyl-adenylated CMPr to its carbapenam product and gave the following kinetic constants (pH 8.3, μ = 0.1): kcat = 0.026 ± 0.002 s−1, Km,CMPr = 4.9 ± 0.7 mM (for comparison, CPS, pH 8.67, μ = 0.15: kcat = 0.304 ± 0.005, Km,CMPr = 0.17 ± 0.01). The in vivo nitrocefin assay also showed a positive result, confirming the ability of β-LS to produce the expected carbapenam product (Supporting Information).
Discussion
Structural Evidence for a Tyr-Glu Catalytic Dyad
X-ray crystal structures of β-LS revealed a putative short, strong hydrogen bond (<2.5 Å) between active site residues Y348 and E382. In the initial β-LS/CEA/AMP-CPP structure (AMP-CPP, a nonreactive ATP analogue), the hydroxyl oxygen of Y348 is ∼5 Å from the α-amino nitrogen of CEA. Moreover, the β-LS/CMA-AMP/PPi structure of the intermediate acyl-adenylate was also captured and revealed significant movement of Y348 toward the α-amino nitrogen of CEA (Oη−Nα = 3.5 Å). Figure 7 illustrates the favorable positioning of Y348 and estimates the distance between the tyrosine and the proton on the α-amino nitrogen of CMA (Oη−H) to be 2.4 Å in the β-LS/CMA-AMP/PPi snapshot. This image points to Y348 as the catalytic base in the deprotonation of the internal nucleophile, an essential event to initiate β-lactam cyclization. Upon completion of the cycle, the tyrosine residue returns to a distance of ∼5 Å away from the DGPC β-lactam nitrogen in the β-LS/DGPC/AMP/PPi structure. By sequence and structural homology to β-LS it is proposed that CPS also contains a catalytic dyad composed of Y345 and E380.
It is important to note that the corresponding pair of residues in AS-B (E348 and D384) is oriented away from each other, as shown in Figure 1. When comparing the structural overlays of all three proteins, the Oη of the proposed catalytic base Y345/83 is positioned an average of 4.4 Å away from where the closer carboxyl oxygen of E348 in AS-B was originally observed. This finding revealed a significant residue and geometry modification in CPS and β-LS and foreshadowed the importance of the Tyr-Glu dyad in the β-lactam-forming proteins.
Kinetic Characterization of Mutant Proteins by pH Variation
Each residue of the putative catalytic dyad was systematically varied and kinetically characterized to investigate its role (37) in the wild-type CPS and β-LS catalyzed reaction. Substitution of alanine for both Y345/8 and E380 allowed the effects of chemistry and sterics to be probed. The hydrogen bond between the dyad tyrosine and glutamate was explored by mutating Y345/8 to phenylalanine, thereby maintaining the aromatic ring of the side chain but removing the ability to hydrogen bond with E380/2. Also, varying E380/2 to an aspartate and glutamine allowed the chemistry of the dyad to be investigated while retaining steric bulk in the active site. The kinetic parameters, kcat, Km, and kcat/Km, of each of the purified active site variants were compared to those of the wild-type CPS and β-LS enzymes.
(A) CPS Mutants
All CPS single-site mutants, except Y345A and E380A, produced the carbapenam product. As predicted by the cycle of β-LS X-ray crystal structures (6, 17, 19), the aromatic ring of Y345 is important for substrate binding. This is best illustrated by the fact that the Y345A mutant exhibited a 5-fold increase in Km of CMPr and a 165-fold decrease in specificity constant compared to wild-type CPS (Table 1). The role of the phenolic hydroxyl of Y345 in β-lactam formation was also tested by replacing this residue with phenylalanine. pH-dependent kcat and kcat/Km plots retained the bell-shaped curve of wild-type CPS with similar pK values, indicating that the Y345F-catalyzed reaction remains governed by acid–base chemistry.
By variation to an aspartate and glutamine, the role of E380 in the catalysis of β-lactam formation was elucidated. The pH–rate profiles revealed that while E380D retained a functional general base with a pKa of ∼7.4 for kcat, the E380Q variant displayed a plateau in the acidic region. Also, the acidic region of the log (kcat/Km) versus pH profiles displayed an ionizable group with a pKa of ∼7.7 for E380D but became pH-independent for E380Q.
(B) β-LS Mutants
All β-LS mutants produced the monobactam product, with the exception of E382Q. If the dyad glutamate was only involved in binding of the CEA guanidinium moiety, however, glutamine would also be able to form a hydrogen bond with the guanidino group, thus retaining catalytic function. Since E382Q resulted in no detectable activity, the essential role of E382 to both binding and catalysis was demonstrated. The active E382 mutant, E382D, showed a dramatic increase in Km,CEA, along with the Michaelis constant of its hydrogen-bonding partner, Y348, when replaced with phenylalanine (Table 1). While the E382D mutant demonstrated similar pH behavior to that of wild-type β-LS, Y348F showed a loss of the basic limb in the kcat/Km versus pH profile, suggesting that the ionizable phenol of tyrosine is important to binding and catalysis. The fold change in kcat of the Y348A mutant is the most pronounced of all the dyad mutants and substantiates the essential role of Y348 in β-LS catalysis. All β-LS mutants followed the same trend as wild type in their pH–kcat profiles, supporting the previously published conclusion that K443 is the titratable group observed (11).
(C) Summary of Y345/8 and E380/2 pH Behavior
Overall, the pH dependence of the CPS β-lactam-forming mutants Y345F, E380D, and E382D demonstrated similar pH dependence relative to wild-type enzyme. From the retention and lack of an acidic limb in the pH–rate profiles of the E380D and E380Q mutants, respectively, it is proposed that E380 is the general base with a pKa of 7.4, seen in the pH–rate profiles of wild-type CPS. Similarly, the mutation of E382 in β-LS to a nonionizable residue that retains the ability to hydrogen bond, E382Q, obliterates all activity and bolsters its significance in catalysis and CEA binding. Because Y348F lacks a basic limb in its pH–(kcat/Km) profile, it is proposed to be the general base with a pKa of 9.15 observed in the corresponding wild-type profile.
Reverse Protonation Mechanism in β-LS
The log (kcat/Km) versus pH bell-shaped curve of wild-type β-LS displayed ionizable groups at pK ∼8 and ∼9. Since the pK values are only one unit apart, it was previously proposed that β-LS utilized a reverse protonation mechanism (11, 38). Reinvestigation of this hypothesis supports the proposal that the active form of β-LS is in the reverse protonated state from that observed in the apparent pH–(kcat/Km) profile of wild type. For wild-type β-LS, the observed pH–(kcat/Km) profile shows one ionizable group that needs to be protonated with a pKb of 7.95 and another with a pKa of 9.15 that needs to be deprotonated, suggesting reverse protonation. Calculation from the pKapp values to actual values in E382D also revealed the same type of mechanism (Table 3).
β-LS meets the three overall requirements of a reverse-protonation mechanism: (1) a low proportion of the active form of β-LS is present at optimal pH (10 −1.2 = 0.06) (39); (2) the accurate maximum activity calculated for β-LS is below the diffusion limit (3.46 × 105M−1 s−1) (39); and (3) the ascending and descending limbs of the wild-type β-LS pH profile of kcat/Km is mostly driven by a large change in Km,CEA and not kcat (40). The latter supports the notion CEA preferentially binds to one enzyme form at the pH optimum (40).
The penalty of having a small proportion of an enzyme in the active form in reverse protonation mechanisms is offset by a kinetic advantage to proton transfer (41). Additionally, the substrate, CEA, presumably has a much higher affinity for the minor enzyme form (reverse protonation) than the major (normal protonation) (42). The difference in binding energy could be used to induce the reaction, since the acid–base combination would be significantly more potent (42). Not surprisingly, reverse protonation mechanisms are increasingly implicated in enzyme reactions involving acid–base chemistry (39–41, 43–47) and may explain, in part, why the measured Km,CEA of wild-type β-LS is significantly lower than the Km,CMPr of CPS.
The loss of the pKa ∼9 limb in the Y348F pH–(kcat/Km) profile designated Y348 as the ionizable group that must be deprotonated for optimal binding. It is proposed that E382 is the residue that needs to be protonated in the wild-type pH–(kcat/Km) profile and has a pKb of 7.95. In addition to the inactive E382Q mutant, the assignment of E382 as the reversely protonated partner of Y348 is also supported by structural evidence of a short hydrogen bond between the dyad residues (17).
The pKa of 7.25 determined from the pH–(kcat/Km) profile of Y348F further supports a functional Tyr-Glu catalytic dyad in wild-type β-LS. The value is close to the general acid pKb value visible in wild-type β-LS (7.95) and presumably represents E382 in the Y348F plot. In this way, the glutamate is now ionized to act as a general base to deprotonate a water molecule proposed to replace the hydroxyl group on the wild-type Y348. Since the replacement of tyrosine with phenylalanine would disrupt the short, Tyr-Glu hydrogen bond seen in wild-type β-LS, a less favorable binding and catalytic mechanism is likely. This is shown in a 3100- and 5000-fold decrease in kcat/Km, of the respective proteins Y348F and Y348A, when compared to the actual maximum activity (346 mM−1 s−1) calculated for wild-type β-LS. This mechanism for the Y348F mutant would utilize a normal protonation mechanism and could explain the large increase in Km,CEA of the phenylalanine variant of Y348. The proposed mechanism for the functional catalytic dyad in β-LS along with a graphical representation of the reverse protonated pH dependence is shown in Scheme 2.
Scheme 2.
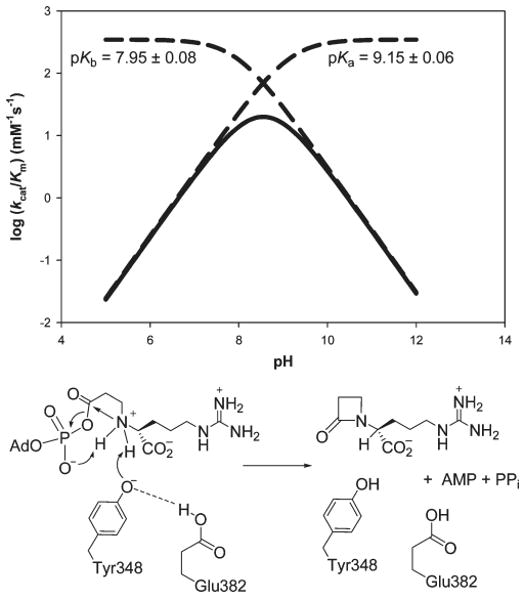
β-LS Reverse-Protonated Tyr-Glu Catalytic Dyad and Its Role in DGPC Formation
[32P]PPi –ATP Exchange Assay
No 32P incorporation was previously observed in wild-type β-LS (11) or CPS (10) and was attributed to a large forward commitment to catalysis in the step following acyl-adenylation (β-lactam formation), thus rendering acyl-adenylation effectively irreversible at optimal pH (7, 11). Under the same conditions used for wild type, all β-LS and CPS mutants tested showed 32P incorporation at pH 8.7–8.8, apart from the β-LS mutant E382Q. For the product-forming mutants (CPS, Y345F and E380D; β-LS, Y348F, Y348A, and E382D), it is likely that reversible acyl-adenylation was now detected because the forward rate of β-lactam formation was decreased by these active site substitutions. It is important to note that while the CPS mutants Y345A and E380A do not form the final carbapenam product, significant levels of [32P]PPi exchange demonstrated that the proteins do undergo the acyl-adenylate reaction.
First Half-Reaction CPS Mutants
From the product formation studies and [32P]PPi–ATP exchange reactions, it is proposed that the alanine mutants of CPS, Y345A and E380A, stop at the first chemical reaction, acyl-adenylation. Using the PPi-coupled enzyme assay, pH–rate studies on CPS mutants Y345A and E380A were investigated to study the changes in protonation state involved in the acyl-adenylate half-reaction. Since these mutants do not form the β-lactam product, the pH profiles were expected to lack a general base but retain the pKb of ∼10, proposed to be K443, a residue important for ATP binding and orientation. Due to the large rate decrease of the alanine mutations, it is possible that the K443 contribution to the pH dependence is masked. Addition of hydroxylamine to the in vitro reactions of Y345A and E380A displayed the release of AMP (Supporting Information) and confirmed the conclusions from the [32P]PPi –ATP exchange and coupled enzyme assays that acyl-adenylation had taken place. Together, the data implicate Y345 and E380 as critical catalytic residues in the second half-reaction of wild-type CPS.
Carbapenam Synthetase Double-Site Mutants
Since the side chain Oη of Y345 and Oδ of E380 are less than 3 Å apart in the CPS crystal structures (17), their proximity suggests that these residues act synergistically to promote catalysis. Accordingly, the Y345F/E380D double mutant exhibited a 61-fold decrease in kcat, 12-fold greater than the product of the effects of the two single mutants, indicating synergism of their damaging effects on kcat in the double mutant (48). Also, the Y345F/E380Q double mutant exhibited a slightly synergistic effect with a 32-fold decrease in kcat, 1.3-fold greater than the product of the two single mutants (48). Synergistic effects of two mutations on kcat can occur under three sets of conditions: (1) when two mutated residues interact anticooperatively, introducing strain into the transition state, to facilitate the same rate-limiting step; (2) when extensive unfolding of the double mutant occurs, beyond simple additive damage of the two single mutations; and (3) when the two mutations affect noninteracting residues which facilitate the same non-rate-limiting step (48). Condition 2 can be immediately dismissed because CD analysis of the two double mutants indicates that substantial unfolding of the proteins does not occur (data not shown). Condition 3 can also be eliminated because product formation assays, SIE measurements, and pH dependency studies indicate that Y345 and E380 facilitate the same rate-limiting step. Therefore, quantitative interpretation implies condition 1 holds where the Tyr-Glu dyad acts cooperatively in the rate-determining step of β-lactam formation (Scheme 3).
Scheme 3.
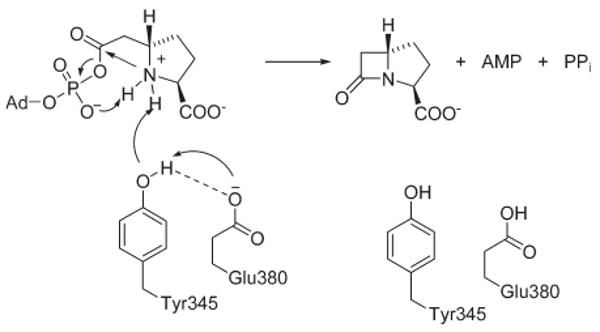
Role of the CPS Tyr-Glu Cataytic Dyad in (3S,5S)-Carbapenam Formation
Solvent Isotope Effects on Y345 and Y348 Mutant Proteins
SIE analysis on the tyrosine dyad residue in β-LS and CPS supported its role in proton transfer during β-lactam formation. In terms of the first chemical event in both enzymes, acyl-adenylate activation is not expected to exhibit a significant SIE. Creation of this acyl-AMP intermediate involves the migration of charge from the substrate carboxylate (CMPr or CEA) to the PPi phosphate oxygen but does not result in a change of protonation state. β-Lactam formation, however, involves acid–base chemistry to deprotonate the secondary amine of the adenylated substrates to allow nucleophilic attack onto the activated carboxyl and, therefore, does result in the transfer of protons in the transition state. Accordingly, a significant SIE on wild-type CPS and β-LS indicates that β-lactam formation is at least partially rate-determining.
(A) CPS Y345 Mutants: Y345A and Y345F
Since Y345A only undergoes the first chemical step of acyl-adenylation, it was not expected to exhibit a significant SIE on kcat. The lack of a SIE on kcat or kcat/Km for Y345A underscores the rate-determining nature of β-lactam formation in wild-type CPS. Dkcat and D(kcat/Km) values of ∼1 for the first half-reaction are similarly in good agreement with the chemistry of acyl-adenylate formation.
Comparison of the kinetic parameters of the CPS Y345F reaction in H2O with those measured in D2O from full pD–rate profiles revealed SIE values of Dkcat 1.9 ± 0.3 and D(kcat/Km) 2.3 ± 0.5. An unusual equilibrium isotope effect (ΔpKa ∼ 0.1) of the ascending limb in the pL–rate profiles could be indicative of a water molecule hydrogen bonded to the general base E380. In D2O, D3O+ is a stronger acid than H3O+ in H2O making the donation of D+ easier than H+ (49). A pKa value of a functional group interacting with a water molecule will be lower in D2O than H2O and will not exhibit a normal ΔpKa shift of 0.5 (32, 50). Thus, the retained catalytic activity of the Y345F mutant could result from a network of water molecules, which substitute for the ablated hydroxyl in this variant (32, 49). A Mg2+-bound water molecule has a φR ∼ 1 (0.9) (49) and would not be expected to contribute appreciably to an observed solvent isotope effect. Therefore, the expected Dkcat would now be 1.6 and 1.7 for a two- and three-proton model, respectively. Additionally, hydronium and hydroxide ions have low reactant-state fractionation factors and could alter a normal SIE to a value less than that of wild type.
(B) β-LS Y348 Mutant: Y348F
In β-LS, the Dkcat of the Y348F mutant (1.7 ± 0.1) is significantly higher than that observed in wild-type β-LS (1.38 ± 0.04) and suggests that acid–base chemistry of β-lactam formation is comparatively more rate-determining, further implicating Y348 in catalysis. The smaller than expected SIE in wild-type β-LS derives from a large forward commitment to β-lactam formation along with a partially rate-determining conformational change that suppressed the observed SIE (11). Surprisingly, a large inverse D(kcat/Km) of 0.24 ± 0.02 was observed in Y348F. In wild-type β-LS the increased kcat/Km in D2O compared to that of H2O [D(kcat/Km) ∼ 0.7] was attributed to the active site “flap” squeezing down on the substrates after CEA binding and possibly to a low-barrier hydrogen bond(s) (11). To probe the origin of the increased effect of D2O on the specificity constant of Y348F relative to wild type, a proton inventory was performed in the pL-independent region of Y348F. The best-fit curve (R2 = 0.98) calculated multiple φR responsible for the observed inverse SIE (Figure 6). Because hydroxide and hydronium ions are predicted to have low reactant-state fractionation factors (φR ∼ 0.4–0.7) (51), the inverse SIE observed from replacement of tyrosine with phenylalanine is presumably from a network of water molecules that replace the omitted hydroxyl moiety of tyrosine. Therefore, the SIE observed on the first- and second-order rate constants of the Y348F mutant further supports its role as a catalytic base during DGPC formation.
(C) CPS Y345F and β-LS Y348F Mutant Activities
Supported by a small equilibrium isotope effect of CPS Y345F and proton inventory of β-LS Y348F, the higher activity of the tyrosine to phenylalanine relative to alanine mutants for both CPS and β-LS could be explained by the presence of a coordinated water molecule mimicking the hydroxyl moiety of tyrosine. This is reinforced by the presence of active site water molecules coordinated to Mg2+ ions in β-LS crystal structures (17). If bulk water molecules filled the void in the Y345/8 alanine mutants, it would provide a less hydrophobic environment than that of the phenylalanine mutants (52, 53) and could result in comparatively better binding as seen with β-LS.
Viscosity Dependence
The importance of the CPS and β-LS dyad mutants in the ring cyclization step was further supported by a dramatic decrease in the viscosity dependence of kcat in all of the mutations tested except E380D in CPS. While the loss of one methylene in the E382D variant of β-LS results in a significant decrease in viscosity dependence, the active site of CPS is more tolerant to its corresponding glutamate mutant. Viscosity analysis of β-lactam-forming mutants Y345/8F, E382D, and Y348A determined that a conformational change is only 0–21% rate-determining relative to their corresponding wild-type enzymes. As a result, the viscosity data confirmed that the internal chemical step of β-lactam formation is more rate-determining in Y345F, Y348F, Y348A, and E382D than in wild-type enzymes, further establishing their essential role in the chemistry during β-lactam cyclization (54). Since the alanine mutants of the CPS Tyr-Glu dyad only catalyze up to acyl-adenylation, the lack of viscosity dependence on kcat reflects the view that the rate-determining conformational change occurs after β-lactam synthesis to assist in product release.
β-LS as a CPS
Based on structurally conserved active site residues and similar kinetic mechanisms, the interesting question whether β-LS is capable of creating a carbapenam and, therefore, become a carbapenam synthetase in addition to its normal role in synthesizing the monocyclic β-lactam ring of DGPC was addressed. Indeed, β-LS cyclized CMPr with most of the energetic penalty observed to result from less favorable binding (Table 5). When compared to the same chemical reaction in the native system, the β-LS-catalyzed carbapenam formation gave a kcat value of only 10-fold lower than CPS. The relatively minor ∼300-fold loss in specificity compared to CPS is a promising outcome encouraging efforts toward the engineering of β-LS to synthesize a variety of new, potentially medicinally useful compounds.
Table 5.
β-Lactam Synthetase as a Carbapenam Synthetase Compared to the Wild-Type CPS and β-LS Catalytic Reactionsa
| varied substrate | kcat (s−1) | −Δ | Km (mM) | +Δ | |
|---|---|---|---|---|---|
| β-LS | CEA | 0.695 ± 0.007 | 1 | 0.040 ± 0.001 | 1 |
| CPS | CMPr | 0.304 ± 0.005 | 2.3 | 0.17 ± 0.01 | 4.3 |
| β-LS | CMPr | 0.026 ± 0.002 | 27 | 4.9 ± 0.7 | 123 |
−Δ shows a fold decrease in kcat and +Δ a fold increase in Km,CMPr of β-LS and CPS at pH 8.3 and 8.67, respectively, relative to β-LS and its natural substrate, CEA, at pH 8.3.
Conclusion
The universally conserved glutamate in the active sites of AS-Bs has been replaced by tyrosine in CPS and β-LS. These β-lactam-synthesizing enzymes are more closely related to their AS-B parents than they are to each other, suggesting, perhaps, independent evolution from their highly conserved ancestors. All of these enzymes share a common first half-reaction of substrate adenylation to create an intermediate of high chemical potential more than sufficient for amide bond formation or strained four-membered ring formation. The second half-reactions differ, however, and the appearance of the Tyr-Glu dyad has been shown here to be important to carrying out the new task of β-lactam synthesis from an adenylated intermediate. When considering the acidity of the substrate nucleophiles in AS-B, CPS, and β-LS, the pKa of ammonia in water is 9.3, while the α-amino group of arginine is slightly above 9.0. The secondary amine of the CPS and β-LS substrates is likely little altered from these values. Such a small difference in reference pKa values does not explain the evolutionary choice of tyrosine in CPS and β-LS over glutamate in AS-B. Notwithstanding, the Tyr-Glu catalytic dyad can be seen to generate a phenoxide well matched in pKa to deprotonate the nitrogen nucleophile of the adenylated β-amino acids to effectively carry out synthesis.
The substrate binding regions among the three enzymes illustrate why CPS and β-LS utilize a different nucleophile activation mechanism than AS-B. X-ray crystal structures illustrate a strikingly similar ATP binding pocket and imply that its shape and position are maintained in the three enzymes (6, 10, 15, 19). Beyond this conserved cofactor binding motif, however, the active sites of CPS and β-LS have been remodeled to accommodate and bind their larger substrates, one bearing a compact, hydrophobic prolyl ring and the other a remote, charged guanidine group, with the corresponding lengths: AS-B (Asp, 5 Å), CPS (CMPr, 8 Å), and β-LS (CEA, 12 Å)(6,19). Evolution of the Glu to Tyr engaged in a catalytic dyad in CPS and β-LS serves to reposition the general base proximal to the substrate α-amino group to now mediate β-lactam closure. In β-LS, the reverse protonation of this dyad enhances its catalytic power for this reaction. These simple amino acid changes have maintained the fundamental catalytic features of AS-Bs but redirected them to a new synthetic outcome. Catalytic efficiency, on the other hand, is characteristically poor compared to its primary metabolic counterparts. As a consequence, reaction rate differences are relatively small in kinetic studies of mutants. Nonetheless, it can be recognized that a general base manifested in the Tyr-Glu dyad, substrate preorganization, acyl-adenylation, and a kinetically coupled protein conformational change all play coordinated roles in the two-step process of transforming a β-amino acid to a β-lactam.
The continued progression of antibiotic resistance reinforces the need to understand the process by which β-lactam-synthesizing enzymes operate. The widespread use of β-lactam antibiotics for more than five decades has led to the emergence of β-lactamases and aggressive resistance to this class of pharmaceuticals. The engineering of native biosynthetic pathways to produce products with improved clinical properties would allow their efficient production by fermentation technology. The importance of this effort was reemphasized recently by the combination of the β-lactamase inhibitor clavulanic acid with a commercial carbapenem, which demonstrated good activity against extensively drug-resistant tuberculosis (55). The comparatively small penalty exacted by β-LS to take its noncognate CPS substrate to the more strained bicyclic β-lactam-containing product and the intrinsic stereochemical flexibility of CPS (10) augur well for the prospects of such experiments.
Supplementary Material
Acknowledgments
We are indebted to M. J. Bodner for a gift of (2S,5S)-carboxymethylproline and to K. A. Moshos for providing the nitrocefin assay reagent. Drs. R. F. Li and M. F. Freeman graciously donated the pET24a/carBC and pET24a/carBC_bls constructs, respectively. We thank Professor B. Gerratana (University of Maryland) and Professor A. Mildvan (The Johns Hopkins University) for insightful discussions of the results and Dr. R. F. Li (The Johns Hopkins University) for guidance with the nitrocefin assays.
Footnotes
Abbreviations: CPS, carbapenam synthetase; β-LS, β-lactam synthetase; AS-B, asparagine synthetase, class B; CEA, N2-(2-carboxyethyl)-l-arginine; CMA, N2-(2-carboxymethyl)-l-arginine; CMPr, (2S,5S)-carboxymethylproline; PPi, pyrophosphate; ATP, adenosine 5′-triphosphate; AMP, adenosine 5′-monophosphate; NADH, nicotinamide adenine dinucleotide; AMP-HCl buffer, 2-amino-2-methyl-1-propanol hydrochloride buffer; TAPS, N-tris(hydroxymethyl)-3-aminopropane-sulfonic acid.
The designation of a carbapenem refers to presence of a C2–C3 double bond in the bicyclic β-lactam, while the CPS product, a carbapenam, is the corresponding saturated bicyclic β-lactam.
The Y345/8 and E380/2 notations represent the dyad residues in CPS (Y345, E380) and β-LS (Y348, E382).
Supporting Information Available: Tables of all primers used to obtain the CPS and β-LS mutants, a table of CPS and β-LS viscosity dependence results, UV–vis data with CPS Y345A and E380A with and without hydroxylamine, proton inventory linear plot, [32P]PPi–ATP exchange data, and nitrocefin assay result along with the corresponding expression SDS–PAGE gel from reactions with β-LS/ATP and CMPr. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cane DE. Perspectives: Biosynthetic pathways. Biosynthesis meets bioinformatics. Science. 2000;287:818–819. doi: 10.1126/science.287.5454.818. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Clardy J. One pathway, many products. Nat Chem Biol. 2007;3:353–355. doi: 10.1038/nchembio0707-353. [DOI] [PubMed] [Google Scholar]
- 3.Fischbach MA, Walsh CT, Clardy J. The evolution of gene collectives: How natural selection drives chemical innovation. Proc Natl Acad Sci U S A. 2008;105:4601–4608. doi: 10.1073/pnas.0709132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmann BO, Li R, Townsend CA. beta-Lactam synthetase: A new biosynthetic enzyme. Proc Natl Acad Sci U S A. 1998;95:9082–9086. doi: 10.1073/pnas.95.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li RF, Stapon A, Blanchfield JT, Townsend CA. Three unusual reactions mediate carbapenem and carbapenam biosynthesis. J Am Chem Soc. 2000;122:9296–9297. [Google Scholar]
- 6.Miller MT, Gerratana B, Stapon A, Townsend CA, Rosenzweig AC. Crystal structure of carbapenam synthetase (CarA) J Biol Chem. 2003;278:40996–41002. doi: 10.1074/jbc.M307901200. [DOI] [PubMed] [Google Scholar]
- 7.Bachmann BO, Townsend CA. Kinetic mechanism of the beta-lactam synthetase of Streptomyces clavuligerus. Biochemistry. 2000;39:11187–11193. doi: 10.1021/bi000709i. [DOI] [PubMed] [Google Scholar]
- 8.Boehlein SK, Stewart JD, Walworth ES, Thirumoorthy R, Richards NG, Schuster SM. Kinetic mechanism of Escherichia coli asparagine synthetase B. Biochemistry. 1998;37:13230–13238. doi: 10.1021/bi981058h. [DOI] [PubMed] [Google Scholar]
- 9.Tesson AR, Soper TS, Ciustea M, Richards NG. Revisiting the steady state kinetic mechanism of glutamine-dependent asparagine synthetase from Escherichia coli. Arch Biochem Biophys. 2003;413:23–31. doi: 10.1016/s0003-9861(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 10.Gerratana B, Stapon A, Townsend CA. Inhibition and alternate substrate studies on the mechanism of carbapenam synthetase from Erwinia carotovora. Biochemistry. 2003;42:7836–7847. doi: 10.1021/bi034361d. [DOI] [PubMed] [Google Scholar]
- 11.Raber ML, Freeman MF, Townsend CA. Dissection of the stepwise mechanism to beta-lactam formation and elucidation of a rate-determining conformational change in beta-lactam synthetase. J Biol Chem. 2009;284:207–217. doi: 10.1074/jbc.M805390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnett SO, Gerratana B, Townsend CA. Rate-limiting steps and role of active site lys443 in the mechanism of carbapenam synthetase. Biochemistry. 2007;46:9337–9345. doi: 10.1021/bi0618464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen SE, Paradkar AS. Biosynthesis and molecular genetics of clavulanic acid. Antonie Van Leeuwenhoek. 1999;75:125–133. doi: 10.1023/a:1001755724055. [DOI] [PubMed] [Google Scholar]
- 14.Stone MJ, Williams DH. On the evolution of functional secondary metabolites (natural products) Mol Microbiol. 1992;6:29–34. doi: 10.1111/j.1365-2958.1992.tb00834.x. [DOI] [PubMed] [Google Scholar]
- 15.Larsen TM, Boehlein SK, Schuster SM, Richards NG, Thoden JB, Holden HM, Rayment I. Three-dimensional structure of Escherichia coli asparagine synthetase B: A short journey from substrate to product. Biochemistry. 1999;38:16146–16157. doi: 10.1021/bi9915768. [DOI] [PubMed] [Google Scholar]
- 16.Boehlein SK, Richards NG, Schuster SM. Glutamine-dependent nitrogen transfer in Escherichia coli asparagine synthetase B. Searching for the catalytic triad. J Biol Chem. 1994;269:7450–7457. [PubMed] [Google Scholar]
- 17.Miller MT, Bachmann BO, Townsend CA, Rosenzweig AC. The catalytic cycle of beta-lactam synthetase observed by x-ray crystallographic snapshots. Proc Natl Acad Sci U S A. 2002;99:14752–14757. doi: 10.1073/pnas.232361199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnizer HG, Boehlein SK, Stewart JD, Richards NG, Schuster SM. gamma-Glutamyl thioester intermediate in glutaminase reaction catalyzed by Escherichia coli asparagine synthetase B. Methods Enzymol. 2002;354:260–271. doi: 10.1016/s0076-6879(02)54022-2. [DOI] [PubMed] [Google Scholar]
- 19.Miller MT, Bachmann BO, Townsend CA, Rosenzweig AC. Structure of beta-lactam synthetase reveals how to synthesize antibiotics instead of asparagine. Nat Struct Biol. 2001;8:684–689. doi: 10.1038/90394. [DOI] [PubMed] [Google Scholar]
- 20.Richards NG, Kilberg MS. Asparagine synthetase chemotherapy. Annu Rev Biochem. 2006;75:629–654. doi: 10.1146/annurev.biochem.75.103004.142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Townsend CA. New reactions in clavulanic acid biosynthesis. Curr Opin Chem Biol. 2002;6:583–589. doi: 10.1016/s1367-5931(02)00392-7. [DOI] [PubMed] [Google Scholar]
- 22.Lloyd MD, Merritt KD, Lee V, Sewell TJ, Wha-Son B, Baldwin JE, Schofield CJ, Elson SW, Baggaley KH, Nicholson NH. Product-substrate engineering by bacteria: Studies on clavaminate synthase, a trifunctional dioxygenase. Tetrahedron. 1999;55:10201–10220. [Google Scholar]
- 23.Vanpelt JE, Northrop DB. Purification and properties of gentamicin nucleotidyltransferase from Escherichia coli—Nucleotide specificity, pH optimum, and the separation of 2 electrophoretic variants. Arch Biochem Biophys. 1984;230:250–263. doi: 10.1016/0003-9861(84)90106-1. [DOI] [PubMed] [Google Scholar]
- 24.Ellis KJ, Morrison JF. Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 1982;87:405–426. doi: 10.1016/s0076-6879(82)87025-0. [DOI] [PubMed] [Google Scholar]
- 25.Lumry R, Smith EL, Glantz RR. Kinetics of carboxypeptidase action. 1. Effect of various extrinsic factors on kinetic parameters. J Am Chem Soc. 1951;73:4330–4340. [Google Scholar]
- 26.Cleland WW, O'Leary MH, Northrop DB. Isotope Effects on Enzyme-Catalyzed Reactions. University Park Press; Baltimore, MD: 1977. [Google Scholar]
- 27.Mukherjee A, Smirnov VV, Lanci MP, Brown DE, Shepard EM, Dooley DM, Roth JP. Inner-sphere mechanism for molecular oxygen reduction catalyzed by copper amine oxidases. J Am Chem Soc. 2008;130:9459–9473. doi: 10.1021/ja801378f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schowen KB, Schowen RL. Solvent isotope effects of enzyme systems. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 29.Sykes RB, Wells JS. Screening for beta-lactam antibiotics in nature. J Antibiot. 1985;38:119–121. doi: 10.7164/antibiotics.38.119. [DOI] [PubMed] [Google Scholar]
- 30.McElroy WD, DeLuca M, Travis J. Molecular uniformity in biological catalyses. The enzymes concerned with firefly luciferin, amino acid, and fatty acid utilization are compared. Science. 1967;157:150–160. doi: 10.1126/science.157.3785.150. [DOI] [PubMed] [Google Scholar]
- 31.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley; New York: 1993. Effect of pH; pp. 914–917. [Google Scholar]
- 32.Schowen KBJ. Transition States in Biochemical Processes. Plenum; New York: 1978. Solvent hydrogen isotope effects; pp. 225–283. [Google Scholar]
- 33.Venkatasubban KS, Schowen RL. The Proton Inventory Technique. CRC Crit Rev Biochem. 1985;17:1–41. doi: 10.3109/10409238409110268. [DOI] [PubMed] [Google Scholar]
- 34.Schowen RL. Mechanistic deductions from solvent isotope effects. Prog Phys Org Chem. 1972;9:275–332. [Google Scholar]
- 35.Kresge AJ. Solvent isotope effect in H2O-D2Omixtures. Pure Appl Chem. 1964;8:243–258. [Google Scholar]
- 36.Blacklow SC, Raines RT, Lim WA, Zamore PD, Knowles JR. Triosephosphate isomerase catalysis is diffusion controlled. Appendix: Analysis of triose phosphate equilibria in aqueous solution by 31P NMR. Biochemistry. 1988;27:1158–1167. doi: 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- 37.Plapp BV. Site-directed mutagenesis: A tool for studying enzyme catalysis. Methods Enzymol. 1995;249:91–119. doi: 10.1016/0076-6879(95)49032-9. [DOI] [PubMed] [Google Scholar]
- 38.Cleland WW. Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv Enzymol Relat Areas Mol Biol. 1977;45:273–387. doi: 10.1002/9780470122907.ch4. [DOI] [PubMed] [Google Scholar]
- 39.Price NE, Cook PF. Kinetic and chemical mechanisms of the sheep liver 6-phosphogluconate dehydrogenase. Arch Biochem Biophys. 1996;336:215–223. doi: 10.1006/abbi.1996.0551. [DOI] [PubMed] [Google Scholar]
- 40.Knuckley B, Bhatia M, Thompson PR. Protein arginine deiminase 4: Evidence for a reverse protonation mechanism. Biochemistry. 2007;46:6578–6587. doi: 10.1021/bi700095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sims PA, Larsen TM, Poyner RR, Cleland WW, Reed GH. Reverse protonation is the key to general acid-base catalysis in enolase. Biochemistry. 2003;42:8298–8306. doi: 10.1021/bi0346345. [DOI] [PubMed] [Google Scholar]
- 42.Mock WL. Theory of enzymatic reverse-protonation catalysis. Bioorg Chem. 1992;20:377–381. [Google Scholar]
- 43.Mock WL, Stanford DJ. Arazoformyl dipeptide substrates for thermolysin. Confirmation of a reverse protonation catalytic mechanism. Biochemistry. 1996;35:7369–7377. doi: 10.1021/bi952827p. [DOI] [PubMed] [Google Scholar]
- 44.Joshi MD, Sidhu G, Pot I, Brayer GD, Withers SG, McIntosh LP. Hydrogen bonding and catalysis: a novel explanation for how a single amino acid substitution can change the pH optimum of a glycosidase. J Mol Biol. 2000;299:255–279. doi: 10.1006/jmbi.2000.3722. [DOI] [PubMed] [Google Scholar]
- 45.Mock WL, Cheng H. Principles of hydroxamate inhibition of metalloproteases: Carboxypeptidase A. Biochemistry. 2000;39:13945–13952. doi: 10.1021/bi001497s. [DOI] [PubMed] [Google Scholar]
- 46.Vocadlo DJ, Wicki J, Rupitz K, Withers SG. A case for reverse protonation: Identification of Glu160 as an acid/base catalyst in Thermoanaerobacterium saccharolyticum beta-xylosidase and detailed kinetic analysis of a site-directed mutant. Biochemistry. 2002;41:9736–9746. doi: 10.1021/bi020078n. [DOI] [PubMed] [Google Scholar]
- 47.Woodyer R, Wheatley JL, Relyea HA, Rimkus S, van der Donk WA. Site-directed mutagenesis of active site residues of phosphite dehydrogenase. Biochemistry. 2005;44:4765–4774. doi: 10.1021/bi047868c. [DOI] [PubMed] [Google Scholar]
- 48.Mildvan AS, Weber DJ, Kuliopulos A. Quantitative interpretations of double mutations of enzymes. Arch Biochem Biophys. 1992;294:327–340. doi: 10.1016/0003-9861(92)90692-p. [DOI] [PubMed] [Google Scholar]
- 49.Quinn DM, Sutton LD. Theoretical basis and mechanistic utility of solvent isotope effects. In: Cook PF, editor. Enzyme Mechanism from Isotope Effects. CRC Press; Boston, MA: 1991. pp. 73–126. [Google Scholar]
- 50.Schowen RL. Isotope Effects on Enzyme-Catalyzed Reactions. University Park Press; Baltimore, MD: 1977. pp. 64–99. [Google Scholar]
- 51.Quinn DM, Sutton LD. In: Enzyme Mechanism from Isotope Effects. Cook PF, editor. CRC Press; Boston, MA: 1991. pp. 73–126. [Google Scholar]
- 52.Makhatadze GI, Privalov PL. Heat capacity of proteins. I. Partial molar heat capacity of individual amino acid residues in aqueous solution: hydration effect. J Mol Biol. 1990;213:375–384. doi: 10.1016/S0022-2836(05)80197-4. [DOI] [PubMed] [Google Scholar]
- 53.Eisenberg D. Three-dimensional structure of membrane and surface proteins. Annu Rev Biochem. 1984;53:595–623. doi: 10.1146/annurev.bi.53.070184.003115. [DOI] [PubMed] [Google Scholar]
- 54.Brouwer AC, Kirsch JF. Investigation of diffusion-limited rates of chymotrypsin reactions by viscosity variation. Biochemistry. 1982;21:1302–1307. doi: 10.1021/bi00535a030. [DOI] [PubMed] [Google Scholar]
- 55.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, Blanchard JS. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science. 2009;323:1215–1218. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.