

Abstract
Herein we report the use of protein-gold nanoconjugate (PGNs) as probes for elucidating mechanistic events involved in protein homocystamide detection with gold nanoparticles (GNPs), as was previously reported by our laboratory. Three different PGN probes are synthesized by direct adsorption of cytochrome c, albumin, or human serum onto citrate-capped GNPs. The PGNs are subsequently purified and treated to confer N-homocysteinylation. Individual PGN systems are evaluated to assess the effect of modification on (1) surface plasmon resonance (SPR), (2) protein structural conformation, and (3) assembly-association. The degree of PGN assembly and colorimetric signal observed postmodification varies based on the type of conjugated protein. For example, results of time-resolved dynamic light scattering studies indicate that modification of cytochrome c-PGNs yields rapid formation of macroscopic nanoparticle assemblies that eventually precipitate from solution. In contrast, albumin and human serum PGNs exhibit higher stability toward modification. Additionally, findings from circular dichroism studies indicate significant modification-induced denaturation, which is what may initiate assembly via electrosteric destabilization of PGNs. The results of electrophoretic studies appear to confirm that the process of N-homocysteinylation-mediated PGN assembly culminates in covalent interparticle association by disulfide cross-linking among modified proteins.
Introduction
Hyperhomocysteinemia, a condition characterized by elevated serum homocysteine (Hcy) levels, has been identified as an independent risk factor and serological biomarker for cardiovascular disease.1,2 Since Hcy undergoes enzymatic conversion to Hcy thiolactone (HTL) at a rate proportional to serum Hcy concentration, serum HTL levels are also elevated in hyperhomocysteinuric subjects.3,4 One potential pathological consequence of increased in vivo HTL production is selective acylation or N-homocysteinylation of proteins at the ε-amino groups of lysine residues to yield N-Hcy-protein (Scheme 1).5,6 Excessive N-homocysteinylation is an alleged contributor to vascular protein damage, a probable precursory event to formation of early atherosclerotic lesions.7,8 Moreover, recent clinical data suggest that there is a direct correlation between elevated serum N-Hcy-protein levels and increased instances of mortality caused by advanced coronary heart disease.9
Scheme 1.
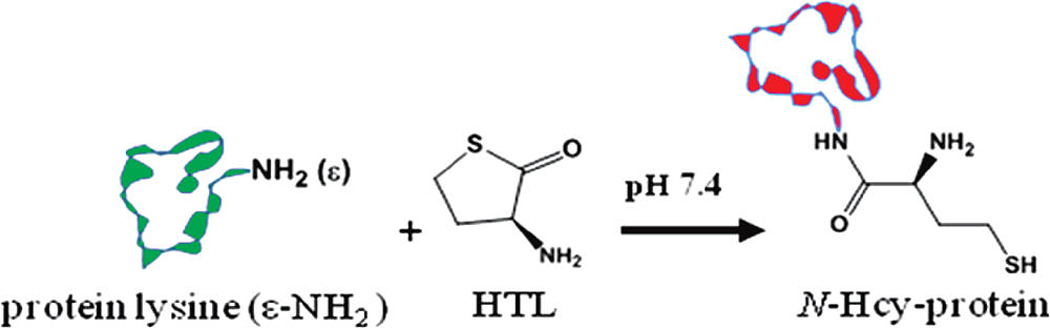
N-Homocysteinylation of a Small Protein Molecule (Green) at an ε-Amino Lysine Residue To Produce N-Hcy-Protein (Red)
Although N-Hcy-protein is complementary to Hcy as a serological biomarker for cardiovascular disease, its significance is diminished in part by a lack of facile clinical screening methods. Conventional high-performance liquid chromatography (HPLC) techniques have been successfully employed for analysis of N-Hcy-protein in serum.10,11 Such methods afford excellent analytical performance but tend to be laborious due to requisite pretreatment steps such as acid hydrolysis, ion exchange column purification, and chromophore labeling. Enzyme-linked immunosorbent assays (ELISA) provide a more rapid and sensitive alternative to HPLC-based methods,12 but mainstream implementation has been hindered by limited availability of N-Hcy-protein antibodies. The aforementioned obstacles lend credence to the need for development of GNP-based sensors for detection of N-Hcy-protein in serum.
The primary objective of colorimetric GNP biochemical sensor design is to control or mediate particle dispersion – assembly by use of specific biomolecules or biomolecular stimuli.13 Dispersed colloidal GNPs having dimensions on the order of 10 nm exhibit intense SPR absorption bands at ~520 nm due to localized surface plasmon resonance (SPR).14 Conversely, interparticle distances decrease upon GNP assembly, thereby facilitating plasmon coupling.15 The resultant decrease in plasmon resonance energy gives rise to broad and red-shifted extended SPR bands. This evolution in SPR absorption makes it possible to directly monitor mediated nanoparticle assembly through a visible change in solution color from red (520 nm) to purple-violet (≥ 620 nm).
Development of viable colorimetric gold nanosensors for N-Hcy protein requires comprehension of underlying mechanistic events involved in N-homocysteinylation-mediated GNP assembly. Fortunately, numerous investigations of biomolecularly mediated GNP assembly have been reported for related serological biomarkers including Hcy, cysteine, and glutathione.13,16–18 Mediated GNP assembly in the presence of aminothiols appears to be contingent on two key mechanistic events: (1) chemisorption of the biomolecules onto the surfaces of GNPs and (2) interparticle association via reversible zwitterionic interactions between head groups of the adsorbed aminothiol molecules. Consequently, the strength of interparticle association can be manipulated simply by varying pH and temperature. In a previous paper, we described colorimetric GNP detection of invitro- derived N-Hcy-serum proteins.19 Interestingly, mediated GNP assembly in this system exhibited characteristics of irreversible association. While it is presumed that protein structural conformation change and thiol binding interactions play an important role in N-homocysteinylation-mediated GNP assembly, the mechanistic details are not yet well understood.
Protein nanoconjugates have been used as probes for interrogating various complex biomolecular interactions including protein binding,20 protein conformational change,21,22 cellular uptake,23 drug delivery,24 and immunoaffinity recognition.25 Protein conjugation has also been used as a strategy for increasing colloidal stability,26–28 conferring biochemical activity,29,30 and enhancing biocompatibility31–33 in various nanoparticle systems. In the present study, protein-gold nanoconjugates (PGNs) are used to investigate three mechanistic events believed to be relevant to colorimetric GNP detection of N-Hcy protein: (1) nanoconjugate assembly, (2) protein structural conformation change, and (3) interparticle association (Scheme 2). The findings of these studies could help establish the usefulness of colorimetric GNP-based sensors in facile clinical screening assays.
Scheme 2.
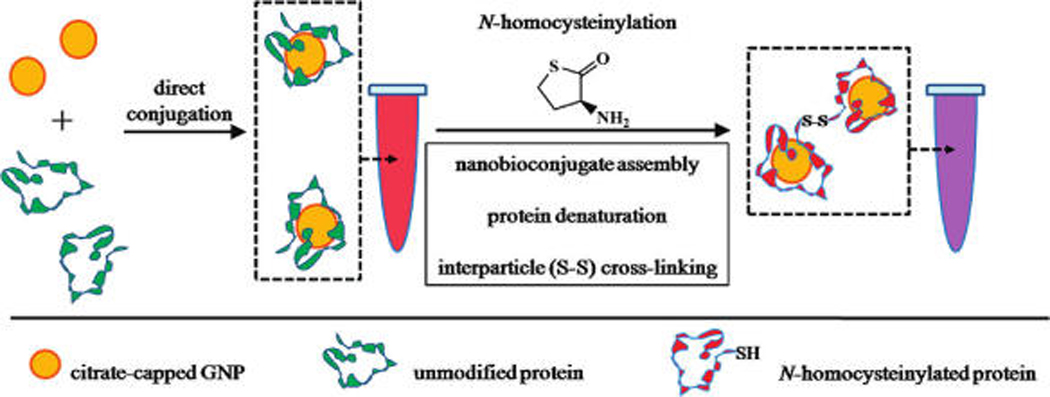
Synthesis of PGN Probes and Proposed Interactions for N-Homocysteinylation-Mediated Assembly
Materials and Methods
Reagents
L-HTL chloride, hydrogen tetrachloroaurate trihydrate, trisodium citrate, ethylenediamine tetraacetic acid (EDTA), tris(2-carboxyethyl)phosphine (TCEP) hydrochloride, and all other reagents used for the preparation of buffers and PGN probe solutions were obtained from Sigma-Aldrich (St. Louis, MO) at the highest purity available and used as received. Ultrapure water (18.2 MΩ · cm) was obtained by use of an Elga model PURELAB Ultra water filtration system.
Synthesis and N-Homocysteinylation of PGN Probes
Citrate-capped GNPs (ca. 13 nm) were synthesized using the Turkevich-Frens method.34,35 Briefly, hydrogen tetrachloroaurate trihydrate was dissolved in water and heated to reflux while stirring. Next, reduction of the gold salt was achieved by the addition of a 40-fold molar excess of trisodium citrate–water solution. The reaction mixture was allowed to reflux for ~25 min until completion of the redox reaction and formation of GNPs, which resulted in a change in solution color from pale yellow to dark red. The concentration of the GNP stock solution was ~1.0 mM, assuming complete reduction of the gold salt. The GNP solutions were purified using a 0.45 µm poly(tetrafluoroethene) syringe filter device and stored at room temperature in an amber glass bottle. Particle size was initially confirmed using UV–vis absorption spectroscopy and transmission electron microscopy (TEM).
PGN probes were prepared via direct adsorption method. Briefly, 4 mL of the GNP stock solution and 2 mL of 80 mg/mL bovine cytochrome c (cyt c) or human serum albumin (HSA) were dissolved in pH 7.4, 100 mM phosphate buffer. The protein–GNP mixture was incubated at 37 °C for at least 12 h to allow maximum surface coverage for protein adsorption. Unbound proteins and low mass material were removed from the PGN solution using centrifugal filtration in Amicon Ultra 100 kDa molecular weight cutoff (MWCO) devices (Millipore, Billerica, MA). The resultant PGN solution was concentrated to 0.5 mL using a 10 kDa MWCO filter device and subjected to five successive 1 mL phosphate buffer wash steps. Lastly, the purified PGNs were reconstituted to the original volume with the desired buffer and stored at 4 °C until use. Human serum-PGN probes were prepared as described above, except 4 mL of the GNP stock solution was added directly to 2 mL of human serum (80 mg/mL protein) purchased from Sigma-Aldrich, St. Louis, MO. Since unconjugated citrate-capped GNPs are unstable and insoluble in the phosphate buffer used in these studies, successful PGN synthesis is indicated by enhanced buffer solubility. In addition, the SPR absorption of PGNs is slightly red-shifted with respect to unconjugated GNPs. Unless otherwise indicated, in-situN-homocysteinylation of PGNs (0.3 mM) was accomplished by incubation with HTL in 100 mM sodium phosphate buffer containing 0.2 mM EDTA at the desired temperature.
Instrumentation
Absorption spectra were acquired using a Shimadzu UV-3101PC UV–vis–NIR equipped with thermoelectric temperature control. Dynamic light scattering (DLS) measurements were acquired using a Malvern (Worcestershire, UK) Zetasizer Nano ZS dynamic light scattering spectrometer with temperature control. All reagents were purified using a 0.1 µm syringe filter device prior to DLS analysis to minimize dust contamination. Modification-mediated assembly of PGNs (8.7 × 10−2mM) was monitored using DLS immediately after the addition of HTL (3.3 × 10−3 mM). Each DLS data point is the composite of 11 consecutive 10 s measurements. Note that only DLS data having monomodal distribution are reported in this work. The range of polydispersity index (PDI) values incurred during DLS measurements was 0.078–0.211, which is within the criteria for acceptable data as determined by the Zetasizer Nano ZS DLS software. Circular dichroism (CD) measurements were obtained by use of an Aviv Model 62DS spectrometer fitted with a 1 mm path length quartz cell and temperature-controlled cell holder. CD spectra were typically acquired in triplicate between 190 and 260 nm. CD response is reported as ellipticity and displayed in units of millidegrees (θ). PGN solutions were typically diluted to ~0.1 mg/mL protein in pH7.4, 10mMphosphate buffer to obtain optimum CD signal. TEM imaging was performed using a JEOL model 100CX transmission electron microscope at 80 kV applied acceleration voltage. TEM specimens were prepared by depositing a thin layer of PGN solution onto carbon-coated copper grids followed by air-drying and staining with uranyl acetate. SDS-PAGE separations were performed by use of a Mini-Protean 3 gel electrophoresis unit with a 1 kV power supply (Bio-Rad Laboratories, Hercules, CA). Standard precast 4–20% Tris HCl polyacrylamide mini-gels were also purchased Bio-Rad. TCEP (50mM) was added to the sample buffer prior to separation to achieve disulfide reduction. Gel separations were stained with Coomassie blue for ~5 h, followed by destaining with water for at least 5 h. Gel images were acquired using a Kodak Gel Logic 200 analyzer purchased from Eastman Kodak Co. (Rochester, NY).
Results and Discussion
SPR Absorption of PGNs
Monitoring the SPR absorption behavior of HSA and serum-PGNs can reveal important insight into colorimetric GNP detection of N-Hcy-protein in serological media. It is noted that unconjugated citrate-capped GNPs undergo salt-induced aggregation when added to 100 mM, pH 7.4 phosphate buffer. However, PGNs remain dispersed and have SPR absorption maxima centered at ~526 nm. In this study, N-homocysteinylation of the PGNs (0.3 mM) is achieved by treatment with 2.5 mM HTL, which results in the appearance of characteristic extended SPR bands at ~620 nm and a change in solution color from red to blue-violet. Representative visible absorption spectra of HSA-PGNs and serum-PGNs are shown in Figure 1. The absorption spectra of HSA and serum-PGNs are similar because albumin is the principal protein component in both systems. However, the relative change in colorimetric signal detected at 620 nm for modified serum-PGNs is about 1.5-fold greater than is detected for modified HSA-PGNs. This outcome is in agreement with the literature, which indicates that serum is more reactive toward N-homocysteinylation than pure HSA.5 Increased reactivity in serum is likely the consequence of numerous other modifiable protein constituents such as transferrin, α2-macroglobulin, and low density lipoprotein.
Figure 1.
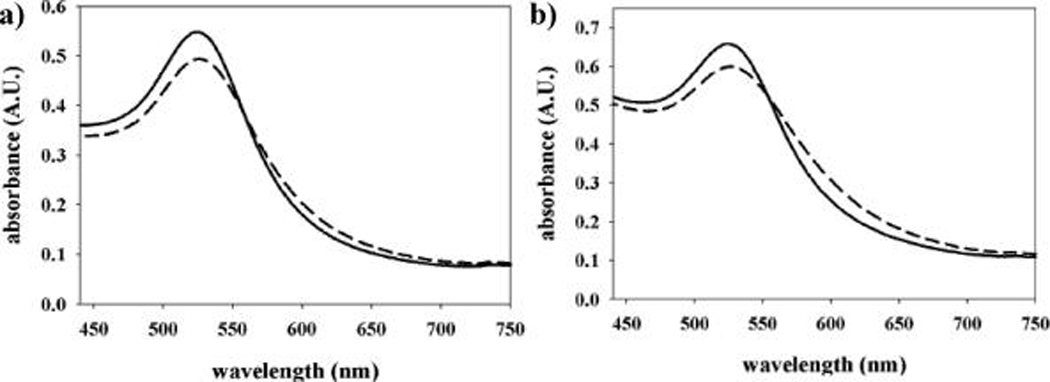
Visible absorption spectra for unmodified (solid line) and 2.5mMHTL-modified (dashed line): (a) HSA-PGN and (b) serum-PGN (0.3 mM) solutions.
Although Cyt c is not a major serum protein constituent, Cyt c-PGNs provide an ideal model system for studying N-homocysteinylation. The Cyt c adlayer is highly susceptible to modification because it contains an abundance of lysine residues (~20% w/w),36 the target site for post-translational protein modification by HTL. Also, Cyt c-PGNs tend to exhibit robust colloidal stability due to virtually irreversible electrostatic binding interactions between cationic lysine residues (pK ~ 10.5 at pH 7.4) and the anionic surfaces of citrate-capped GNPs.37
The effect ofN-homocysteinylation on the spectral behavior of bovine Cyt c-PGNs (~0.3 mM) is illustrated in Figure 2. Unmodified Cyt c-PGNs exhibit maximum SPR absorption at 528 nm, whereas modification causes a simultaneous decrease in SPR absorption at 528 nm and the emergence of a broad extended SPR band between 620 and 750 nm. This relatively large shift in the SPR absorption is accompanied by a color change in solution from red to blue-violet. The blue color typically persists for ~1 h and gradually diminishes to colorlessness as a result of near complete precipitation of PGNs as macroscopic assemblies (vide infra).
Figure 2.
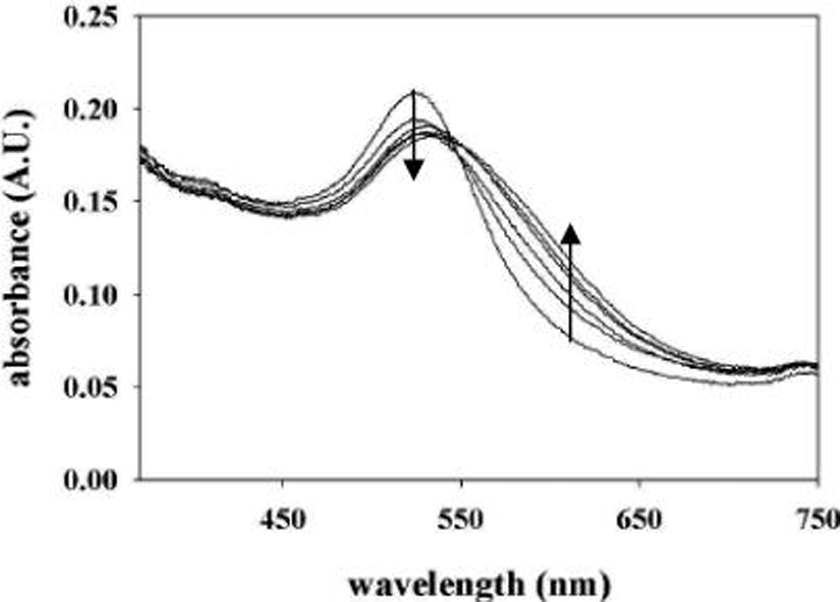
Absorption spectra of bovine Cyt c-PGNs (~0.3 mM) modified with HTL (0–2.5 mM).
Time-Resolved DLS Studies and TEM Imaging
Prior to conducting time-resolved DLS studies, unmodified HSA, serum, and Cyt c-PGNs are evaluated at 25 °C to determine values of average hydrodynamic diameter (Dh). PGNs are expected to be significantly larger than unconjugated citrate-capped GNPs (~17 nm measured by DLS) due to the protein adlayer. Dh values measured for unmodified PGNs were serum (91 nm), HSA (45 nm), and Cyt c (22 nm). These results further substantiate successful PGN synthesis.
Time-resolved DLS spectroscopy provides a means of observing the process of N-homocysteinylation-mediated PGN assembly. Studies involving modification of HSA- and serum-PGNs were conducted at 80 °C to mimic previously reported serum assay conditions.19 Such conditions also help minimize nonspecific association and accelerate the assembly process to allow observation over the course of 30 min. In this study, N-homocysteinylation of PGNs (8.7 × 10−2 mM) was accomplished by treatment with HTL (3.3 × 10−3 mM) in 100 mM, pH 7.4 phosphate buffer.
Time-resolved DLS data acquired for HSA-PGNs (Figure 3a) and serum-PGNs (Figure 3b) indicate similar assembly behavior. Modification of HSA- and serum-PGN assemblies causes a gradual increase in Dh to 70 and 135 nm, respectively. Control samples consisting of HSA- and serum-PGNs are treated with an equivalent volume of phosphate buffer without HTL. The absence of assembly in the control samples is reflected by the stabilization of Dh values for HSA- and serum-PGNs at ~33 and 72 nm, respectively. Note that the addition of either HTL (modification) or buffer solution (control) causes temporary disequilibrium and disruption of nonspecifically associated PGNs as indicated by an inflection in time-resolvedDh plots. Moreover, the formation and subsequent stability of HSA- and serum-PGN assemblies at 80 °C suggests that interparticle association among assembled PGNs is supported by covalent bonding rather than weak nonspecific interactions (vide infra).
Figure 3.
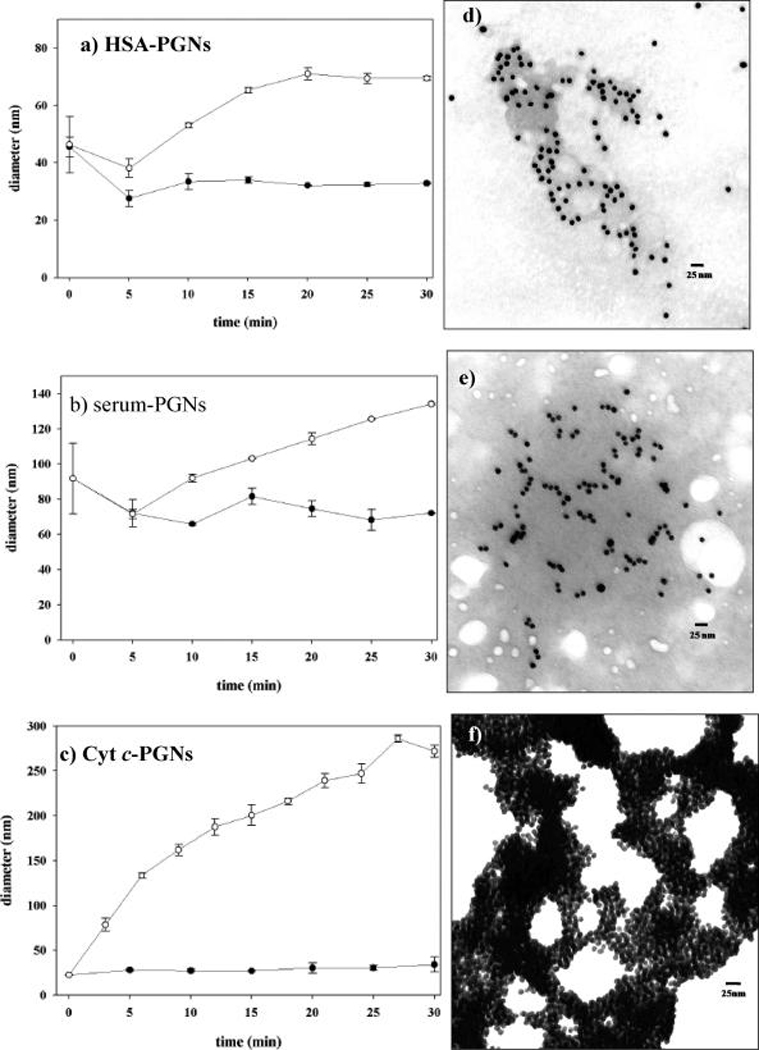
Time-resolved DLS data for unmodified (●) andN-homocysteinylated (○) PGN probes: (a) HSA-PGNs (80 °C), (b) serum-PGNs (80 °C), and (c) Cyt c-PGNs (25 °C). Corresponding TEM images show N-homocysteinylated PGNs (d–f).
Given that Cyt c-PGNs respond more rapidly to N-homocysteinylation than serum protein-based PGNs, time-resolved DLS studies for this model system must be conducted under lower temperature conditions (25 °C) to allow sufficient time for observation of the assembly process. The modification of Cyt c-PGNs induces immediate and comprehensive PGN assembly, which causes a 10-fold increase inDh values within the first 10 min (Figure 3c).Dh values approach 300 nm over the course of 30 min. No appreciable assembly activity is detected in control solutions containing unmodified Cyt c-PGNs (Figure 3c). Representative DLS intensity distribution information along with corresponding time-resolved visible absorption data are provided in Supporting Information.
TEM imaging allows visualization of structures produced by N-homocysteinylation-mediated PGNassembly. Modification of HSA-PGNs (Figure 3d) and serum-PGNs (Figure 3e) results in modest assembly that resembles the small GNP clusters observed in our previous studies.19 Cyt c-PGN assembly results in formation of micrometer-sized superstructures (Figure 3f ). Interestingly, a 3-D structure due to multilayer Cyt c-PGN assembly is indicated in the darker regions of the TEM image. Moreover, the dynamic assembly behavior of modified Cyt c-PGNs is consistent with the literature, which states N-homocysteinylated unconjugated Cyt c readily forms disulfide bonded multimers.5 Control TEM specimens containing unmodified nanoconjugates show well-dispersed PGNs (provided in Supporting Information).Note that some of the spherical nanoconjugates appear to be irregularly shaped due to the protein adlayer.
Modification-Induced Denaturation in PGNs
Recall that HTL can have detrimental effects on protein structure and biological activity. Jakubowski et al. recently used CD spectroscopy to examine the effects of N-homocysteinylation on secondary protein structure in unconjugated Cyt c.38 It was found that Cyt c is particularly vulnerable to modification at certain lysine residues. Nevertheless, only subtle changes in protein structure were detected postmodification.
To the best of our knowledge, investigations of N-homocysteinylation-induced protein structural conformation change using PGN probes have not been reported. CD measurements were acquired for pure PGN systems, HAS and Cyt c, to assess changes in secondary protein structure due to modification. Serum-PGNs are omitted from this discussion due to their complex and inhomogeneous protein composition. Studies were performed at 25 °C using freshly prepared PGN solutions diluted to ~0.1 mg/mL total protein with 10 mM phosphate buffer. Samples were also filtered with MWCO devices to remove low mass reaction byproducts including any unreacted HTL and unbound Hcy prior to acquisition of CD spectra.
Representative CD spectra (Figure 4) exhibit characteristic ellipticity minima at 208 and 222 nm, which correspond to the dominant α-helical structural component of the conjugated proteins. Decreased ellipticity minima are observed postmodification, which signifies a loss of α-helical structure via partial protein denaturation.39 These data suggest that both Cyt c-PGNs and HSA-PGNs are vulnerable to modification-induced changes in protein structure. Processing of the CD data with CDNN version 2 spectral deconvolution algorithm40 provides a more quantitative assessment of changes in secondary protein structure of PGNs (Table 1). These data show a significant decrease in α-helical structure that translates into a 15% and 12% increase in random coil for Cyt c- and HAS-PGNs, respectively. This finding indicates that HSA- and Cyt c-PGNs are far less resistant to modification-induced denaturation than unconjugated HSA5 and Cyt c.38 Thus, one can deduce that modification perturbs the steric stabilization provided by secondary protein structure, thereby contributing to the onset of mediated PGN assembly.
Figure 4.
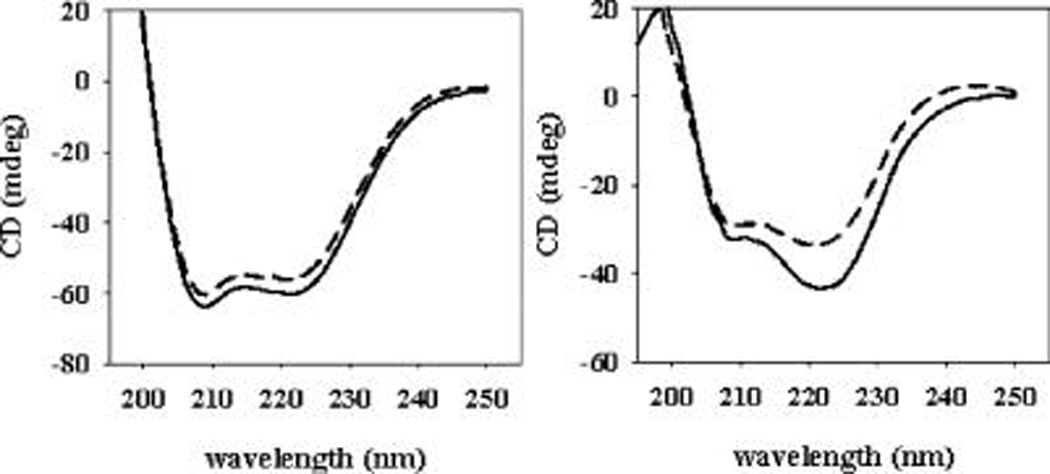
Representative CD spectra for unmodified (solid curve) and modified (dashed curve) HSA- and Cyt c-PGNs, respectively.
Table 1.
Effect of Modification on Secondary Structural Components in Cyt c- and HSA-PGNsa
| PGN species | α-helix (%) | β-strand antiparallel (%) | β-strand parallel (%) | β-turn (%) | random coil (%) |
|---|---|---|---|---|---|
| Cyt c | 33 | 8 | 9 | 17 | 33 |
| N-Hcy-Cyt c | 17 | 15 | 16 | 20 | 48 |
| HSA | 72 | 2 | 3 | 11 | 14 |
| N-Hcy-HSA | 46 | 6 | 6 | 15 | 26 |
Values do not sum to 100% because of rounding errors incurred during CD spectral deconvolution.
Evaluation of Interparticle Association
In addition to the steric protection provided by the protein adlayer, further stabilization is achieved through electrostatic interactions, e.g., repulsion between basic Cyt c-PGNs. However,N-homocysteinylation of proteins neutralizes the positive charge of ε-amino lysine residues (pK ~ 11) upon conversion to N-Hcy-lysine (pK ~ 7).5 The previously mentioned destabilizing effects collectively disrupt the electrosteric environment and initiate PGN assembly. Consequently, interparticle spacing among PGNs is decreased, thereby facilitating disulfide cross-linking between N-homocysteinylated PGNs.
In order to confirm the presence of disulfide cross-linking, assembled PGNs resulting from N-homocysteinylation were subjected to SDS-PAGE under both nonreducing and reducing conditions. TCEP, a non-thiol-containing reducing agent, was added to the sample buffer to achieve cleavage of disulfide bonds. SDS-PAGE is employed for facile detection of interparticle association and dissociation based on size exclusion. Briefly, the stacking region of a gradient gel contains large pores (4% polyacrylamide) that can accommodate assembled PGNs, whereas the remainder of the gel contains progressively smaller pores (4–20% polyacrylamide) that can only be accessed by dissociated PGNs.
Representative SDS-PAGE separations are shown in Figure 5. The horizontal lines in the gel images approximate the lower boundary of the stacking region. Lanes a and b contain assembled HSA-PGNs (0.3mM) resulting from N-homocysteinylation with 10 and 20 mM HTL, respectively. Note that more material is retained in the stacking region of lane b because the degree of PGN assembly is proportional to HTL concentration. Corresponding experiments were also conducted under reducing conditions (50 mM TCEP). Treatment of assembled HSA-PGNs with TCEP results in a decrease in the amount of material in the stacking region of the gel (lanes c and d) due to cleavage of interparticle disulfide bonds, which allows dissociated PGNs to permeate the smaller pores of the resolving gel. The effect of disulfide cleavage is more noticeable in analogous experiments performed with serum-PGNs (lanes e–h). It is noted that assembled Cyt c-PGNs have insufficient electrophoretic mobility to be separated by SDS-PAGE, presumably due to a lack of binding with SDS in the sample buffer. Nevertheless, these data help to confirm that the robust association characteristics of assembled N-homocysteinylated PGNs are attributable to interparticle disulfide cross-linking.
Figure 5.
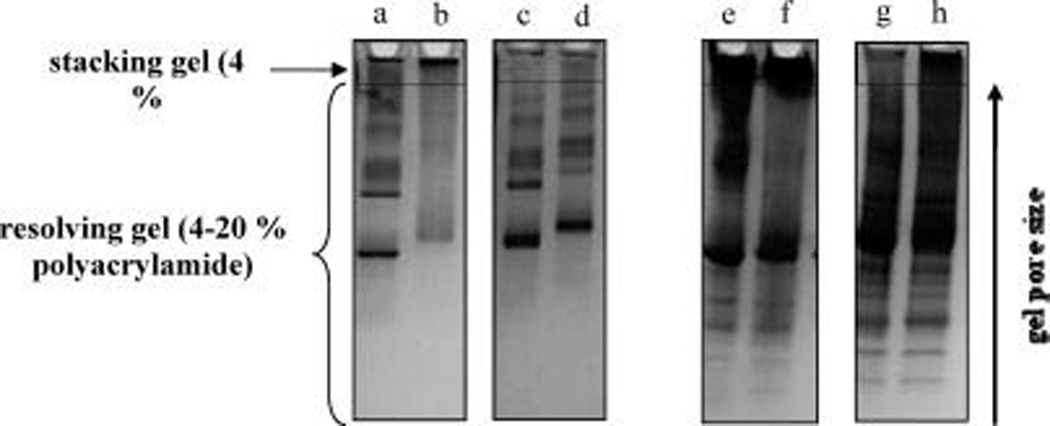
SDS-PAGE separations of HSA-PGN assemblies resulting from modification with 10 and 20mM HTL before (lanes a and b, respectively) and after treatment with 50 mM TCEP (lanes c and d, respectively). Identical separations performed with serum-PGNs assemblies are shown in lanes e–h.
Conclusions
We have described investigations of mechanistic events that occur upon N-homocysteinylation of PGN probes. In particular, SPR absorption, protein structural conformation, and interparticle association were studied to better understand modification-mediated GNP assembly as it pertains to colorimetric detection of N-Hcy-protein. Modification resulted in PGN assembly, which was indicated by the appearance of extended SPR bands at and beyond 620 nm. Time-resolved DLS studies revealed that modification of HSA- and serum-PGNs resulted in a modest 2-fold increase in Dh values within 30 min at 80 °C. In contrast, modification of Cyt c-PGNs yielded an impressive 10-fold increase in Dh values within 30 min at 25 °C. The loss of α-helical structural component was detected postmodification, which led to significant increases in the random coil component in both HSA- and Cyt c-PGNs. Electrophoretic separation of modification-derived PGN assemblies under reducing conditions helped confirm that interparticle association is likely the result of disulfide cross-linking among conjugated N-Hcy-protein molecules. On the basis of these findings, there are at least three relevant mechanistic events caused by modification: (1) protein denaturation, (2) PGN assembly, and (3) interparticle disulfide cross-linking. We believe that this work provides motivation for further studies of GNP-based colorimetric assays for detection of N-homocysteinylation in serological samples.
Supplementary Material
Acknowledgment
A.T.G. acknowledges support from Procter & Gamble Company, Dow Chemical Company, and the National Organization for the Professional Advancement of Black Chemist and Chemical Engineers (NOBCChE). R.M.S. acknowledges support from National Institutes of Health (award number NIH R01 EB2044). I.M.W. acknowledges NIH award RO1 GM079670 and the Philip W. West Endowment for support of this work. The authors also thank Dr. Jennifer Rood, Dr. Claude Bouchard, and Dr. Tuomo Rankinen (Pennington Biomedical Research Center-Louisiana State University System) for helpful discussions.
Footnotes
Supporting Information Available: Additional experimental data include TEM images, time-resolved UV–vis absorption spectra, and DLS intensity distribution profiles. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PWF, Wolf PA. N. Engl. J. Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 2.Clarke R, Daly L, Robinson K, Naughten E, Cahalane S, Fowler B, Graham I. N. Engl. J. Med. 1991;324:1149–1155. doi: 10.1056/NEJM199104253241701. [DOI] [PubMed] [Google Scholar]
- 3.Chwatko G, Boers GHJ, Strauss KA, Shih DM, Jakubowski H. FASEB J. 2007;21:1707–1713. doi: 10.1096/fj.06-7435com. [DOI] [PubMed] [Google Scholar]
- 4.Jakubowski H. Biochemistry. 1997;36:11077–11085. doi: 10.1021/bi970589n. [DOI] [PubMed] [Google Scholar]
- 5.Jakubowski H. FASEB J. 1999;13:2277–2283. [PubMed] [Google Scholar]
- 6.Jakubowski H. J. Nutr. 2000;130:377S–381S. doi: 10.1093/jn/130.2.377S. [DOI] [PubMed] [Google Scholar]
- 7.Mercie P, Garnier O, Lascoste L. Apoptosis. 2000;5:403–411. doi: 10.1023/a:1009652011466. [DOI] [PubMed] [Google Scholar]
- 8.Austin RC, Lentz SR, Werstuck GH. Cell. Death Differ. 2004;11:S56–S64. doi: 10.1038/sj.cdd.4401451. [DOI] [PubMed] [Google Scholar]
- 9.Yang X, Gao Y, Zhou J. Clin. Chim. Acta. 2006;364:230–234. doi: 10.1016/j.cccn.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Jakubowski H. Anal. Biochem. 2008;380:257–261. doi: 10.1016/j.ab.2008.05.049. [DOI] [PubMed] [Google Scholar]
- 11.Uji Y, Motmiya Y, Hanyu N, Ukaji F, Okabe H. Clin. Chem. 2002;48:941–944. [PubMed] [Google Scholar]
- 12.Undas A, Perla J, Lacinski M, Trzeciak W, Kazmierski R, Jakubowski H. Stroke. 2004;35:1299–1304. doi: 10.1161/01.STR.0000128412.59768.6e. [DOI] [PubMed] [Google Scholar]
- 13.Zhao W, Brook MA, Li Y. ChemBioChem. 2008;9:2363–2371. doi: 10.1002/cbic.200800282. [DOI] [PubMed] [Google Scholar]
- 14.Brust M, Kiely CJ. Colloids Surf., A. 2002;202:175–186. [Google Scholar]
- 15.Grant CD, Schwartzberg AM, Norman TJ, Zhang JZ. J. Am. Chem. Soc. 2002;125:549–553. doi: 10.1021/ja028532y. [DOI] [PubMed] [Google Scholar]
- 16.Lim IS, Ip W, Crew E, Njoki PN, Mott D, Zhong C, Pan Y, Zhou S. Langmuir. 2007;23:826–833. doi: 10.1021/la062334t. [DOI] [PubMed] [Google Scholar]
- 17.Zhang FX, Han L, Israel LB, Daras JG, Maye MM, Ly NK. Analyst. 2002;127:462–465. doi: 10.1039/b200007e. [DOI] [PubMed] [Google Scholar]
- 18.Lim IIS, Mott D, Ip W, Njoki PN, Pan Y, Zhou S, Zhong C-J. Langmuir. 2008;24:8857–8863. doi: 10.1021/la800970p. [DOI] [PubMed] [Google Scholar]
- 19.Gates AT, Fakayode SO, Lowry M, Ganea GM, Murugeshu A, Robinson JW, Strongin RM, Warner IM. Langmuir. 2008;24:4107–4113. doi: 10.1021/la7033142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brewer SH, Glomm WR, Johnson MC, Knag MK, Franzen S. Langmuir. 2005;21:9303–9307. doi: 10.1021/la050588t. [DOI] [PubMed] [Google Scholar]
- 21.Chah S, Hammond MR, Zare RN. Chem. Biol. 2005;12:323–328. doi: 10.1016/j.chembiol.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Yokoyama K, Welchons DR. Nanotechnology. 2007;18 doi: 10.1088/0957-4484/19/37/375101. [DOI] [PubMed] [Google Scholar]
- 23.Chithrani BD, Chan WCW. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 24.Lu W, Zhang Y, Tan YZ, Hu KL, Jiang XG, Fu SK. J. Controlled Release. 2005;107:428–448. doi: 10.1016/j.jconrel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 25.Zhang CX, Zhang Y, Wang X, Tang ZM, Lu ZH. Anal. Biochem. 2003;320:136–140. doi: 10.1016/s0003-2697(03)00353-1. [DOI] [PubMed] [Google Scholar]
- 26.Keating CD, Kovaleski KM, Natan MJ. J. Phys. Chem. B. 1998;102:9404. [Google Scholar]
- 27.Feng J, Zhoa G, Xu J, Chen H. Anal. Biochem. 2005;342:280–286. doi: 10.1016/j.ab.2005.04.040. [DOI] [PubMed] [Google Scholar]
- 28.Yang T, Li Z, Wang L, Guo C, Sun Y. Langmuir. 2007;23:10533–10538. doi: 10.1021/la701649z. [DOI] [PubMed] [Google Scholar]
- 29.Srivastava S, Verma A, Frankamp BL, Rotello VM. Adv. Mater. 2005;17:617–621. [Google Scholar]
- 30.Katz E, Willner I, Wang J. Electroanalysis. 2004;16:19–44. [Google Scholar]
- 31.Mattoussi H, Mauro JM, Goldman ER, Anderson GP, Sundar VC, Mikulec FV, Bawendi MG. J. Am. Chem. Soc. 2000;122:12142. [Google Scholar]
- 32.Goldman ER, Anderson GP, Tran PT, Mattoussi H, Charles PT, Mauro JM. Anal. Chem. 2002;74:841. doi: 10.1021/ac010662m. [DOI] [PubMed] [Google Scholar]
- 33.Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nat. Biotechnol. 2003;21:47. doi: 10.1038/nbt767. [DOI] [PubMed] [Google Scholar]
- 34.Kimling J, Maier M, Okenve B, Kotaidis V, Ballot H, Plech A. J. Phys. Chem. B. 2006;110:15700–15707. doi: 10.1021/jp061667w. [DOI] [PubMed] [Google Scholar]
- 35.Frens G. Nat. Phys. Sci. 1973;241:20–22. [Google Scholar]
- 36.Nakashim T, Higa H, Matsubar H, Benson AM, Yasunobu KT. J. Biol. Chem. 1966;241 1166-&. [PubMed] [Google Scholar]
- 37.Gomes I, Santos NC, Oliveira LMA, Quintas A, Eaton P, Pereira E, Franco R. J. Phys. Chem. C. 2008;112:16340–16347. [Google Scholar]
- 38.Perla-Kajan J, Marczak L, Kajan K, Pawel S, Twardowski T, Jakubowski H. Biochemistry. 2007;46:6225–6231. doi: 10.1021/bi602463m. [DOI] [PubMed] [Google Scholar]
- 39.Shang L, Wang Y, Jiang J, Dong S. Langmuir. 2007;23:2714–2721. doi: 10.1021/la062064e. [DOI] [PubMed] [Google Scholar]
- 40.Bohm G, Muhr R, Jaenicke R. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.