

Abstract
Autosomal-dominant brachydactyly type E (BDE) is a congenital limb malformation characterized by small hands and feet predominantly as a result of shortened metacarpals and metatarsals. In a large pedigree with BDE, short stature, and learning disabilities, we detected a microdeletion of ∼900 kb encompassing PTHLH, the gene coding for parathyroid hormone related protein (PTHRP). PTHRP is known to regulate the balance between chondrocyte proliferation and the onset of hypertrophic differentiation during endochondral bone development. Inactivation of Pthrp in mice results in short-limbed dwarfism because of premature differentiation of chondrocyte. On the basis of our initial finding, we tested further individuals with BDE and short stature for mutations in PTHLH. We identified two missense (L44P and L60P), a nonstop (X178WextX∗54), and a nonsense (K120X) mutation. The missense mutation L60P was tested in chicken micromass culture with the replication-competent avian sarcoma leukosis virus retroviral expression system and was shown to result in a loss of function. Thus, loss-of-function mutations in PTHLH cause BDE with short stature.
Main Text
Brachydactylies are a family of limb malformations characterized by short hands and/or feet because of aplastic or hypoplastic skeletal elements.1 Brachydactyly type E (BDE [MIM 113300]) is characterized by a general shortening of metacarpals and metatarsals and/or phalanges. The phenotype is variable even within families, ranging from moderate shortening of individual metacarpals to a shortening of all bones in the hands and/or feet. BDE occurs as an isolated trait or as part of more complex syndromic conditions such as hypertension-brachydactyly syndrome [MIM 112410], Turner syndrome, or Albright hereditary osteodystrophy (AHO [MIM 103580]). Isolated BDE has been associated in sporadic cases with microdeletions of 2q37, as well as with mutations in HOXD13 [MIM 142989].2,3 However, the genetic cause of the great majority of BDE cases remains unexplained.
Here we describe a disease gene for BDE in five unrelated families. The pedigrees and the phenotype of affected individuals are shown in Figure 1 and Figure 2, respectively. In family 1, the phenotype was variable, but complete penetrance was observed. All affected presented with clinical features typical for BDE, i.e., shortening of the metacarpals, metatarsals, and phalanges in hands and feet with the fourth and fifth metacarpals being most frequently and most severely affected. In addition, hypoplastic middle and distal phalanges were observed in severe cases (Figures 2A and 2E). In total, six of eight affected individuals presented with short stature (between −2.0 and −2.8 standard deviation [SD] < P3). The growth chart of individual IV-15 is shown in Figure 2I. Learning difficulties were noted in all affected of family 1, but not in the other families. G stimulatory (Gs)-alpha activity in erythrocytes was measured in family 1, but no abnormality was detected. To perform a genetic analysis, we obtained blood samples from all available family members and extracted DNA by standard methods. All participants gave their informed consent for molecular testing. The study was approved by the local ethics committee. All affected of the families described here were screened for mutations in GNAS (formerly GNAS1 [MIM 139320]) and HOXD13, but no mutation was detected. All sequencing experiments were carried out by standard techniques as reported elsewhere.4,5
Figure 1.
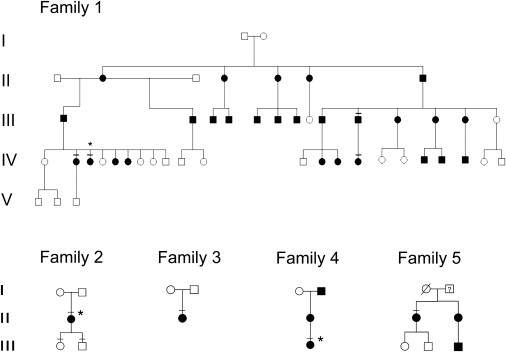
Pedigrees of Families 1–5
Affected individuals are indicated by black symbols. Symbols with crossed lines indicate individuals of whom material was available for testing, and stars indicate individuals of whom clinical or X-ray images are shown in Figure 2.
Figure 2.
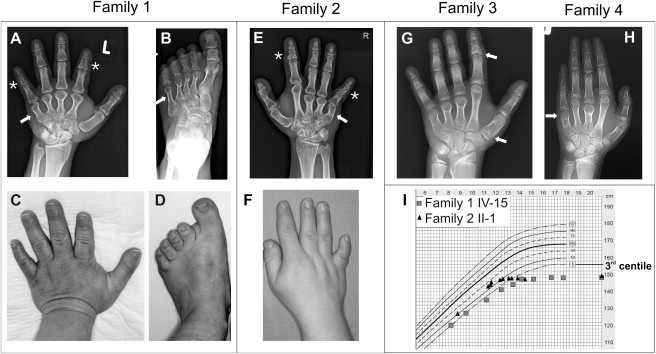
Clinical Phenotypes
(A–D) Family 1, IV-3 (large deletion). Note severe BDE phenotype with small hands (A and C) and feet (B and D) due to shortening of all metacarpals and second to fifth metatarsals (arrows on X-rays). Note additional shortened middle phalanx of digits II and V (A, stars).
(E and F) Family 2, II-1 (missense mutation L60P). Note small hands with shortened third to fifth metacarpals. Middle and distal phalanges of index and fifth finger are short (E, stars).
(G) Family 3, II-1 (missense mutation L44P). Besides shortened metacarpals III–V, note cone-shaped epiphyses at the phalanges (arrows) and premature closure of the metacarpal epiphyses at the age of 9 years. Epiphyses of ulnar and radius appear irregular and flattened.
(H) Family 4, III-1 (nonstop mutation). Note shortened third and fifth metacarpals and abnormal epiphyses prematurely fused to their metaphyses (arrow).
(I) Growth chart of female individuals from family 1 (IV-15) and family 2 (II-1). Note that growth velocity of affected individuals stops prematurely, resulting in small stature.
Because submicroscopic aberrations are a known cause for congenital limb malformations and brachydactylies,6–8 we performed array-based comparative genomic hybridization (array CGH) via a whole-genome oligonucleotide array (244K, Agilent Technologies). With this technology, a microdeletion of about 907.68 kb on chromosome 12p was identified that encompassed six known genes: STK38L (disrupted), ARNTL2, C12orf70, PPFIBP1, MRPS35 [MIM 611995], KLHDC5, and PTHLH (parathyroid hormone-like hormone [MIM 168470]; also known by its product, parathyroid hormone-related protein, PTHRP) (Figure 3; data deposited in the DECIPHER database with ID BER251557). The deletion was confirmed by quantitative real-time PCR (qPCR) and was shown to segregate with the phenotype in the affected family members (position of amplicons indicated by arrows in Figure 3). Of the deleted genes, only PTHLH is known to play a critical role in skeletal development, suggesting that PTHLH was the main candidate gene for BDE. To test this hypothesis, we investigated further individuals with typical BDE by qPCR and by sequencing all known PTHLH isoforms, including introns and UTR regions, to detect deletions as well as point mutations (primer sequences are available on request). With this approach, we identified two missense mutations, a nonstop mutation as well as a nonsense mutation.
Figure 3.
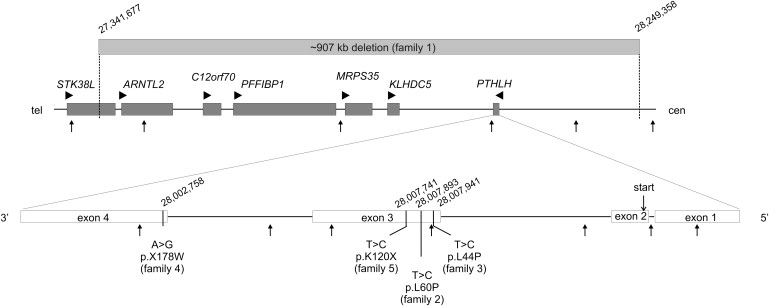
Mutations Associated with BDE
An ∼907 kb genomic microdeletion on chromosome 12p was detected in family 1 affecting six genes (gray bar, top; ISCN2009: array 12p11.23p11.22(27,341,677-28,249,358)x1). Point mutations within the PTHLH gene observed in families 2–5 are indicated (bottom). Genes are shown as dark gray boxes, with transcriptional orientation indicated by arrow heads. Small arrows point to genomic position of qPCR amplicons. Exon structure of PTHLH gene is according to NM_198966. Location of the start codon (ATG) in exon 2 is indicated. Genomic positions are according to hg18.
Top: 1 mm represents 5 kb. Bottom: exons, 1 mm represents 10 bp; introns, 1 mm represents 100 bp.
In a 41-year-old female with short metacarpals III–V, short middle phalanges of digits II and V, and short stature (−2.97 SD < P3) (family 2; Figures 2E and 2F; growth chart in Figure 2I), we detected a missense mutation (c.179T>C) in exon 3 that changes the conserved amino acid leucine to proline (p.L60P). A further heterozygous missense mutation (c.131T>C) was detected in family 3, which results in a similar amino acid change (p.L44P). The affected individual presented with normal stature (137 cm at 9 years and 2 months; +0.6 SD) and BDE. X-ray at the age of 9 years showed cone-shaped epiphyses of several phalanges and premature fusion of epiphyses (Figure 2G). Problems with tooth eruption of primary as well as secondary teeth were reported. Bioinformatic analyses of disease potential with PolyPhen9 and MutationTaster predicted both variations to be damaging. Because the conserved amino acid leucine at both positions is located within an alpha helix motif, the changes are likely to break the α-helical structure and thereby disturb the tertiary protein structure. To test the effect of the p.L60P mutation, we performed functional testing in chicken micromass culture.10 Murine wild-type (WT) and mutant Pthlh constructs were overexpressed in chicken limb micromass culture via a retroviral system (RCASBP-A) as described previously.11 Alkaline phosphatase (ALP) activity was measured on day 7 and day 10 as an indicator of late-stage chondrocyte differentiation (Figure 4). Expression of mutant Pthrp resulted in a weaker suppression of ALP activity compared to WT (Figure 4), indicating a loss of function in the mutant.
Figure 4.
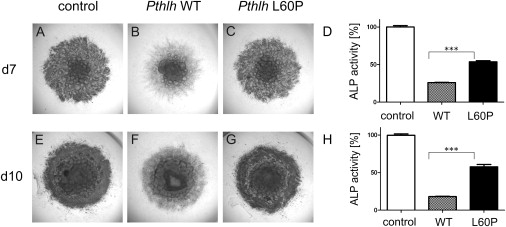
Functional Testing of Missense Mutation L60P in Chicken Micromass System
WT Pthlh (B and F) and L60P mutant (C and H) were retrovirally overexpressed in micromass cultures from chicken limb buds and were grown for 7 days (A–C) and 10 days (D–F) in culture medium. In comparison to the WT, the L60P mutant does not show a strong suppression of ALP activity (D and H). However, ALP activity is reduced compared to the uninfected control (D and H).
Another heterozygous mutation (c.532A>G) was identified in family 4 that converts the stop codon in exon 4 into tryptophan (p.X178WextX∗54), resulting in an extension of the coding region (Figure 3). This 14-year-old female presented with shortened metacarpals III and V, abnormal metacarpal epiphyses prematurely fused to her metaphyses (Figure 2H), and short stature (−2.4 SD < P3). In addition, the nails of the first fingers were hypoplastic. Her mother also had short fifth metacarpals, and the maternal grandfather was reported to have the same phenotype as the index patient (short stature [−3.6 SD < P3] and brachydactyly), but material for testing was not available.
Transcripts lacking in-frame termination codons can be subject to a surveillance mechanism called nonstop decay.12 This mechanism was primarily described in Saccharomyces cerevisiae, and transcripts are degraded 3′ to 5′ by the exosome independently of deadenylation.13,14 Recently, nonstop decay has also been observed as a molecular mechanism in human disease.15 Thus, we propose that the heterozygous mutation in exon 4 results in nonstop-mediated decay of PTHLH mRNA and, consequently, in a reduced amount of PTHRP protein.
In family 5 (individual II-1), we detected a heterozygous nonsense mutation in exon 3 that changes the amino acid lysine into a stop codon (p.K120X). This results in a truncation of the protein within the nuclear localization signal (NLS). Because the mutation is located more than 50 bp away from the 3′ boundary of the second-to-last exon, it may be subjected to nonsense-mediated decay. The four single nucleotide changes were not detected in any of 200 control individuals, thus excluding a rare SNP or copy number variant. Family 5 presented with BDE (shortened metacarpals III–V), short stature, and, in addition, oligodontia. Patient II-1 had 9 and her sister (II-2) had 26 permanent teeth missing. Individual I-1 was reported to have normal hands and feet and normal stature, but clinical information on I-2 was not available. The observation that two out of five families had tooth problems suggests a role for PTHRP in tooth development and/or eruption. In mice, PTHRP was indeed shown to be important for normal tooth development. Philbrick et al.16 investigated PTHrP-knockout mice with a procollagen II-driven transgene that generates PTHrP expression only in cartilage. Besides small stature, these rescued PTHrP-knockout mice presented with a failure of tooth eruption. Although teeth appear to develop normally, they become impacted within the surrounding alveolar bone.16
Overall, we identified 13 affected individuals, of whom 10 were adults. Of the 13 affected, 10 had short stature and 3 were within normal range. The average height of adults with mutations in PTHLH was 150 cm in females and 162 cm in males. The growth velocity of affected individuals appeared to slow down and stop prematurely, resulting in small stature (see also growth curves in Figure 2I). Most likely, this was a result of premature closure of growth plates. This can be observed in abnormal cone-shaped epiphyses of the phalanges and metacarpals, which fuse prematurely with their metaphyses (Figures 2G and 2H). Delayed tooth eruption and/or oligodontia is an additional but not obligatory feature of this condition. Taken together, mutations in PTHLH result in a specific type of skeletal disease that we suggest to name “BDE with short stature, PTHLH type.”
Because only individuals of family 1 presented with learning difficulties in addition to BDE and short stature, the deletion of the five genes distal to PTHLH most likely accounts for this additional phenotype. The deletion in family 1 overlaps with a larger deletion described by Bähring et al.17 in a Japanese BDE case. The latter contains the locus previously assigned for autosomal-dominant brachydactyly and hypertension (MIM 112410).18 The authors claimed that a loss of function due to gene deletions may cause the brachydactyly. However, the critical region proposed for the brachydactyly-relevant gene lies approximately 2 Mb distal to the deletion detected in family 1 and does not include PTHLH.19
Parathyroid hormone-related protein (PTHRP) was initially identified by investigators studying the causes of hypercalcemia of malignancy. Subsequently, PTHRP was discovered to be secreted by many types of cells including chondrocytes, perichondrial cells, and osteoblasts. The initial translation product of PTHRP undergoes several endoproteolytic cleavage steps that give rise to three distinct peptides: the NH2-terminal peptide (residues 1–37), which has PTH-like and growth regulatory activities; the midregion domain (residues 38–93), containing a nuclear localization signal regulating calcium transport and cell proliferation; and the COOH-terminal domain (residues 102–141), also called osteostatin, which is known to modulate osteoclast activity. PTHRP activates the same receptor as PTH, i.e., the PTHRP/PTH receptor (PTHR), leading to a stimulation of both Gs and Gq family of heterotrimeric G proteins. This activation results in a variety of actions ranging from relaxation of smooth muscle cells to regulation of proliferation and differentiation (for review, see 20).
In developing bones, PTHLH is expressed in the distal ends of the cartilage anlagen and in the perichondrium. It diffuses away from its site of production and binds to its receptor (PTHR) located on prehypertrophic chondrocytes. PTHRP binding to its receptor results in Gs activation, which in turn keeps chondrocytes proliferating through suppression of the cyclin-cdk inhibitor p57, thereby increasing the pool of proliferating nondifferentiated chondrocytes.20 The production of Pthrp is under control of Ihh, as shown by various genetic experiments.21 Together, Ihh and Pthrp form a feedback loop that regulates the onset of hypertrophic differentiation and thus endochondral bone development.22 The effects of Pthrp on chondrocyte differentiation are also mediated via the regulation of the phosphorylation of Sox9, a transcription factor important for chondrocyte differentiation, and by suppressing Runx2, a transcription factor essential for osteoblast differentiation.23,24
The important function of PTHRP and its pathway in endochondral bone formation are demonstrated by various gene inactivation experiments in the mouse and by human mutations. Inactivation of Pthlh in the mouse results in a lethal short-limbed chondrodysplasia. Growth plate chondrocytes in these mice undergo the normal sequence of proliferation and hypertrophy, except that they show an accelerated maturation and thus become hypertrophic much sooner than WT cells.25–27 Conversely, overexpression of Pthlh in proliferating chondrocytes through a transgenic strategy results in profound delay of maturation.28 In addition, inactivation of Pthlh leads to low bone density in mice.29 However, we found no evidence for osteoporosis in the families presented here. Mice with inactivated Pthrp/Pth receptor show a very similar phenotype as Pthlh−/− mice, indicating that the Pthrp/Pth receptor mediates most of the action of Pthrp on growth plate chondrocytes.30 Loss-of-function mutations in the PTHRP/PTH receptor in humans result in a recessive lethal chondrodysplasia, Blomstrand dysplasia (MIM 215045), whereas activating mutations in the receptor are the cause for Jansen metaphyseal dysplasia (MIM 156400).
The primary mediator of PTHRP/PTH receptor signaling in chondrocytes is GNAS. Haploinsufficiency of Gnas, the downstream effector of Pthrp, in mice results in premature differentiation of growth plate chondrocytes, thus resembling the phenotype described for Pthrp/Pth receptor as well as for Pthlh-deficient mice.31 Mutations in GNAS in humans cause Albright hereditary osteodystrophy (AHO), a genetically heterogenous phenotype that includes pseudohypoparathyroidism type 1a (PHP1a [MIM 13580]), type 1c (PHP1c [MIM 612462]), and pseudopseudohypoparathyroidism (PPHP [MIM 612463]), the normocalcemic variant of PHP. Mutations of the GNAS gene on chromosome 20q13.2 have been shown to be associated with PHP1a and PPHP.5 GNAS encodes the Gs-alpha subunit of the Gs protein complex. The Gs proteins transduce extracellular signals from transmembrane receptors to intracellular downstream effector proteins via second messengers like cyclic AMP and activation of protein kinase A (PKA). Interestingly, GNAS appears to be imprinted: loss-of-function mutations on the maternal allele cause PHP1A, whereas mutations on the paternal allele result in PPHP. AHO is associated with a variety of clinical symptoms such as short stature, obesity, and mental retardation. The skeletal phenotype, however, is rather uniform and strikingly similar to the BDE phenotype described here. Other than in association with mutations in GNAS, BDE is observed in a large number of other conditions, some of which are caused by mutations in molecules that are known to interact with the PTHRP pathway. Mutations in IHH, for example, cause acrocapitofemoral dysplasia (MIM 607778), a condition with brachydactyly and cone-shaped epiphyses.32 Short metacarpals are also a feature of basal cell nevus syndrome (MIM 109400), which is caused by mutations in PTCH1 (MIM 601309), the receptor for Hedgehog proteins,33 and cleidocranial dysplasia (CCD [MIM 119600]), a condition caused by mutations in RUNX2 (MIM 600211), a direct regulator of IHH and a downstream target of PTHRP.34 These results suggest that the IHH-PTHRP pathway is of particular importance for cartilage differentiation and growth in the metacarpals and support the concept of molecular disease families based on phenotypic similarities and interacting signaling pathways.1
Our results thus add another condition to the PTHRP pathway disease family. The observation that heterozygous loss-of-function mutations in PTHLH result in a growth defect that is very similar to that observed in GNAS mutations suggests a particular importance for this component of the pathway in the growth of bones of the hands and feet. Other conditions with similar phenotypes, such as acromicric dysplasia (MIM 102370) and acrodysostosis (MIM 101800), may be caused by mutations in the same pathway.
Acknowledgments
We thank the graduate students Nicola Diny and Yu Qian who assisted us in this project during their human genetics module and Mareen Schmidt-von Kegler for technical assistance in micromass experiments. We are grateful to the patients and their families for participating in this study. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG; KL 2158/2-1) to E.K., K.D., and S.M. The authors declare no conflict of interest.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
GenBank, http://www.ncbi.nlm.nih.gov/
Ensembl Genome Browser, http://www.ensembl.org/index.html
MutationTaster, http://www.mutationtaster.org
References
- 1.Mundlos S. The brachydactylies: A molecular disease family. Clin. Genet. 2009;76:123–136. doi: 10.1111/j.1399-0004.2009.01238.x. [DOI] [PubMed] [Google Scholar]
- 2.Wilson L.C., Leverton K., Oude Luttikhuis M.E., Oley C.A., Flint J., Wolstenholme J., Duckett D.P., Barrow M.A., Leonard J.V., Read A.P. Brachydactyly and mental retardation: An Albright hereditary osteodystrophy-like syndrome localized to 2q37. Am. J. Hum. Genet. 1995;56:400–407. [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson D., Kan S.H., Oldridge M., Trembath R.C., Roche P., Esnouf R.M., Giele H., Wilkie A.O. Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am. J. Hum. Genet. 2003;72:984–997. doi: 10.1086/374721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodman F.R., Mundlos S., Muragaki Y., Donnai D., Giovannucci-Uzielli M.L., Lapi E., Majewski F., McGaughran J., McKeown C., Reardon W. Synpolydactyly phenotypes correlate with size of expansions in HOXD13 polyalanine tract. Proc. Natl. Acad. Sci. USA. 1997;94:7458–7463. doi: 10.1073/pnas.94.14.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahrens W., Hiort O., Staedt P., Kirschner T., Marschke C., Kruse K. Analysis of the GNAS1 gene in Albright's hereditary osteodystrophy. J. Clin. Endocrinol. Metab. 2001;86:4630–4634. doi: 10.1210/jcem.86.10.7946. [DOI] [PubMed] [Google Scholar]
- 6.Dathe K., Kjaer K.W., Brehm A., Meinecke P., Nürnberg P., Neto J.C., Brunoni D., Tommerup N., Ott C.E., Klopocki E. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 2009;84:483–492. doi: 10.1016/j.ajhg.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klopocki E., Schulze H., Strauss G., Ott C.E., Hall J., Trotier F., Fleischhauer S., Greenhalgh L., Newbury-Ecob R.A., Neumann L.M. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am. J. Hum. Genet. 2007;80:232–240. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klopocki E., Ott C.E., Benatar N., Ullmann R., Mundlos S., Lehmann K. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J. Med. Genet. 2008;45:370–375. doi: 10.1136/jmg.2007.055699. [DOI] [PubMed] [Google Scholar]
- 9.Sunyaev S., Ramensky V., Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000;16:198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- 10.Seemann P., Schwappacher R., Kjaer K.W., Krakow D., Lehmann K., Dawson K., Stricker S., Pohl J., Plöger F., Staub E. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Invest. 2005;115:2373–2381. doi: 10.1172/JCI25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan B.A., Fekete D.M. Manipulating gene expression with replication-competent retroviruses. Methods Cell Biol. 1996;51:185–218. doi: 10.1016/s0091-679x(08)60629-9. [DOI] [PubMed] [Google Scholar]
- 12.Wagner E., Lykke-Andersen J. mRNA surveillance: The perfect persist. J. Cell Sci. 2002;115:3033–3038. doi: 10.1242/jcs.115.15.3033. [DOI] [PubMed] [Google Scholar]
- 13.van Hoof A., Frischmeyer P.A., Dietz H.C., Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- 14.Frischmeyer P.A., van Hoof A., O'Donnell K., Guerrerio A.L., Parker R., Dietz H.C. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- 15.Chatr-Aryamontri A., Angelini M., Garelli E., Tchernia G., Ramenghi U., Dianzani I., Loreni F. Nonsense-mediated and nonstop decay of ribosomal protein S19 mRNA in Diamond-Blackfan anemia. Hum. Mutat. 2004;24:526–533. doi: 10.1002/humu.20117. [DOI] [PubMed] [Google Scholar]
- 16.Philbrick W.M., Dreyer B.E., Nakchbandi I.A., Karaplis A.C. Parathyroid hormone-related protein is required for tooth eruption. Proc. Natl. Acad. Sci. USA. 1998;95:11846–11851. doi: 10.1073/pnas.95.20.11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bähring S., Nagai T., Toka H.R., Nitz I., Toka O., Aydin A., Mühl A., Wienker T.F., Schuster H., Luft F.C. Deletion at 12p in a Japanese child with brachydactyly overlaps the assigned locus of brachydactyly with hypertension in a Turkish family. Am. J. Hum. Genet. 1997;60:732–735. [PMC free article] [PubMed] [Google Scholar]
- 18.Schuster H., Wienker T.E., Bähring S., Bilginturan N., Toka H.R., Neitzel H., Jeschke E., Toka O., Gilbert D., Lowe A. Severe autosomal dominant hypertension and brachydactyly in a unique Turkish kindred maps to human chromosome 12. Nat. Genet. 1996;13:98–100. doi: 10.1038/ng0596-98. [DOI] [PubMed] [Google Scholar]
- 19.Bähring S., Kann M., Neuenfeld Y., Gong M., Chitayat D., Toka H.R., Toka O., Plessis G., Maass P., Rauch A. Inversion region for hypertension and brachydactyly on chromosome 12p features multiple splicing and noncoding RNA. Hypertension. 2008;51:426–431. doi: 10.1161/HYPERTENSIONAHA.107.101774. [DOI] [PubMed] [Google Scholar]
- 20.Kronenberg H.M. PTHrP and skeletal development. Ann. N Y Acad. Sci. 2006;1068:1–13. doi: 10.1196/annals.1346.002. [DOI] [PubMed] [Google Scholar]
- 21.St-Jacques B., Hammerschmidt M., McMahon A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vortkamp A., Lee K., Lanske B., Segre G.V., Kronenberg H.M., Tabin C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 23.Guo J., Chung U.I., Yang D., Karsenty G., Bringhurst F.R., Kronenberg H.M. PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev. Biol. 2006;292:116–128. doi: 10.1016/j.ydbio.2005.12.044. [DOI] [PubMed] [Google Scholar]
- 24.Huang W., Chung U.I., Kronenberg H.M., de Crombrugghe B. The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc. Natl. Acad. Sci. USA. 2001;98:160–165. doi: 10.1073/pnas.011393998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amizuka N., Warshawsky H., Henderson J.E., Goltzman D., Karaplis A.C. Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J. Cell Biol. 1994;126:1611–1623. doi: 10.1083/jcb.126.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karaplis A.C., Luz A., Glowacki J., Bronson R.T., Tybulewicz V.L., Kronenberg H.M., Mulligan R.C. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 27.Miao D., He B., Karaplis A.C., Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J. Clin. Invest. 2002;109:1173–1182. doi: 10.1172/JCI14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weir E.C., Philbrick W.M., Amling M., Neff L.A., Baron R., Broadus A.E. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc. Natl. Acad. Sci. USA. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miao D., Su H., He B., Gao J., Xia Q., Zhu M., Gu Z., Goltzman D., Karaplis A.C. Severe growth retardation and early lethality in mice lacking the nuclear localization sequence and C-terminus of PTH-related protein. Proc. Natl. Acad. Sci. USA. 2008;105:20309–20314. doi: 10.1073/pnas.0805690105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanske B., Karaplis A.C., Lee K., Luz A., Vortkamp A., Pirro A., Karperien M., Defize L.H., Ho C., Mulligan R.C. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 31.Bastepe M., Weinstein L.S., Ogata N., Kawaguchi H., Jüppner H., Kronenberg H.M., Chung U.I. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc. Natl. Acad. Sci. USA. 2004;101:14794–14799. doi: 10.1073/pnas.0405091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hellemans J., Coucke P.J., Giedion A., De Paepe A., Kramer P., Beemer F., Mortier G.R. Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am. J. Hum. Genet. 2003;72:1040–1046. doi: 10.1086/374318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet J. Rare Dis. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mundlos S., Otto F., Mundlos C., Mulliken J.B., Aylsworth A.S., Albright S., Lindhout D., Cole W.G., Henn W., Knoll J.H. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]