

Abstract
Affinity maturation of the immune response and the generation of long-lived bone marrow (BM) plasma cells are hallmarks of CD40-dependent, thymus-dependent (TD) humoral immunity. Through disruption of the tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6)-binding site within the CD40 cytoplasmic domain, we selectively ablated affinity maturation and the generation of plasma cells after immunization. Mutagenesis of both the TRAF6 and TRAF2-TRAF3 sites was essential for arresting germinal center formation in response to immunization. CD40-induced B cell proliferation and early immunoglobulin production occurred even when all TRAF sites were ablated. These studies show that specific CD40-TRAF associations control well defined aspects of humoral immunity. In addition, they define the roles that TRAF-dependent and TRAF-independent pathways play in regulating antigen-driven B cell differentiation.
Binding of CD40 ligand (CD40L, also known as CD154), which is expressed on activated helper T lymphocytes, to its receptor CD40, which is expressed on B lymphocytes, is essential for thymus-dependent (TD) humoral immunity. CD40 engagement triggers clonal B cell expansion, germinal center (GC) formation, production of memory B cells, antibody isotype switching, affinity maturation and the generation of long-lived plasma cells1. Oligomerization of CD40 by CD154 induces cytoplasmic signaling via recruitment of tumor necrosis factor receptor (TNFR)-associated factors (TRAFs) 1, 2, 3, 5 and 6 to specific domains in the CD40 cytoplasmic tail2–4. These adapter molecules, which have no intrinsic enzymatic activities, are believed to activate Ser and Thr kinases, which can then couple the receptor complex to downstream mitogen-activated protein kinase (MAPK) and IκB kinase cascades. Although studies are beginning to resolve how TRAFs act as adapters to canonical signaling pathways following CD40 engagement, none have determined which TRAFs are involved in controlling the complex process of antigen-driven B cell differentiation in vivo.
One means with which to study TRAF function in vivo has been to genetically delete specific TRAF genes in mice. However, mice deficient in TRAF2, TRAF3 or TRAF6 suffer from profound developmental insufficiencies that compromise their use in studies of CD40 function in immunity5–7. Interpretation of data obtained with these mice is further confounded by the fact that TRAFs act as signal-transduction elements for a number of TNFR family and cytokine receptors8. Nonetheless, in vitro studies have shown that B cells from TRAF2- and TRAF6-deficient mice show defects in CD40-induced NF-κB signaling and proliferation6,7,9,10.
An alternative strategy for evaluating the function of TRAF proteins in CD40-induced B cell differentiation has been to selectively mutagenize well defined TRAF-binding sites in the cytoplasmic tail of CD40. Published studies have confirmed the identity of the TRAF2 and TRAF 3 (TRAF2/3)- and the TRAF6-binding sites3,11,12. With the use of B cell lines, site-directed mutagenesis of the TRAF2/3-binding site has shown that this site is important for the induction of c-Jun NH2-terminal kinase (Jnk), NF-κB and certain differentiation functions, including the up-regulation of costimulatory molecules and the induction of immunoglobulin (Ig) germline heavy chain transcripts13–15. Mutagenesis of the TRAF6 site has suggested that this domain may also be involved with the induction of Ig germline transcription; however, it is not clear which downstream kinase cascades are activated by TRAF6 recruitment to the CD40 receptor complex4,12,14.
Adopting the approach described above, we produced transgenic mice that expressed mutant CD40 receptors (X-CD40) in which specific TRAF-binding sites were disrupted. Each of the X-CD40 lines were tested for their ability to propagate a CD40 signal and to develop humoral immune responses upon immunization. Analysis of primary and secondary humoral immune responses in these mice allowed us to define the TRAF-dependent and -independent components of antigen-induced B cell differentiation in vivo. Contrary to what we anticipated based on previous studies in TRAF-deficient mice, we found that loss of TRAF recruitment imparts specific and well defined deficiencies in the development of humoral immunity.
Results
Generation of transgenic X-CD40–expressing mice
The function of TRAF proteins in CD40-dependent humoral immunity was evaluated in a cohort of transgenic mice that expressed mutant CD40 receptors in which specific TRAF-binding sites were disrupted. A chimeric CD40 transgene (generically termed X-CD40) was engineered in which the murine transmembrane and cytoplasmic domains were fused to the extracellular domain of human CD40 and expressed in antigen-presenting cells under the control of the I-Eα promoter16. The human-murine chimeric CD40 molecule was engineered because experiments in vitro suggested that homologous interactions between the murine cytoplasmic domain and murine signaling elements were superior to human CD40 in transducing signals in murine cells (unpublished data). This may be due to substantial sequence divergence between murine and human CD40 in the TRAF6-binding site and membrane-proximal regions of the CD40 tail. We chose to use the human extracellular domain so that selective signaling through the chimeric transgenic receptor could be compared to signaling through endogenous murine CD40. Because murine CD154 binds to human CD40 and signals via this molecule17, we anticipated that the X-CD40 transgenic receptor would function normally in vivo.
Disruption of TRAF binding to the CD40 cytoplasmic tail was accomplished by site-directed mutagenesis, as described15. A mutation was introduced into the cytoplasmic domain that disrupted the binding site for TRAF2 and TRAF3 (Thr255→Ala255, which is referred to hereafter as ΔT2,3, see Fig. 1). Mutation at this site also resulted in the loss of TRAF1 and TRAF5 from the receptor complex, as they associate with TRAF2 and TRAF33. In another construct, mutations were introduced that disrupted the binding site for TRAF6 within the CD40 cytoplasmic domain (Pro213→Gly213 and Glu215→Ala215, referred to hereafter as ΔT6). Finally, mice were produced that expressed X-CD40 with mutations in both the TRAF2/3 and TRAF6 sites (referred to hereafter as ΔT2,3,6). The ΔT2,3,6 mutant CD40 was unable to bind any of the known TRAFs in vitro or in HEK293 cells4. Expression of the X-CD40 transgenes on B cells varied between the different X-CD40–transgenic founder lines used (Fig. 1). Chimeric CD40 expression on other major histocompatibility complex (MHC) class II–bearing cells from each of the X-CD40 mice showed expression of the transgene on dendritic cells (DCs) and MHC class II–bearing cells (Web Fig. 1 online ).
Figure 1. Construction of X-CD40 transgenic mice.

(a) The constructs used to generate X-CD40–transgenic mice consisted of the extracellular domain of human CD40 and the transmembrane and cytoplasmic domains of murine CD40. Amino acids targeted for conversion by site-directed mutagenesis are underlined. (b) X-CD40 expression on T cell–depleted splenocytes in each of the founder lines are shown. Filled histograms, anti–human CD40 staining of the transgenic receptor; open histograms, staining of an isotype-matched control antibody.
Ex vivo studies in B cells from X-CD40 mice
CD154 binding to CD40 on B cells activates the Ikβ kinase (IKK) and MAPK pathways. In vitro studies in B cell lines, as well as studies that used TRAF-deficient primary B cells, have linked TRAF2 and TRAF6 to specific downstream signaling cascades4,6,15,18,19. The impact of TRAF–binding site mutations on CD40 signaling has not been reported in ex vivo hematopoietic cells. To investigate the role played by TRAFs in early CD40 signaling cascades in B cells, the activation of kinase pathways in ex vivo B cells from wild-type (WT) and mutant X-CD40 transgenic mice was evaluated (Fig. 2a–d). Splenic B cells from each of the transgenic lines were isolated and selectively triggered through transgenically expressed X-CD40. All the signaling studies were performed on X-CD40 B cells from mice that were crossed onto the CD40−/− background, so the only CD40 expressed was the X-CD40 transgene. Soluble human CD154 (shCD154) induced NF-κB, Jnk and p38 activation in B cells from the WT X-CD40 mice. The ΔT2,3 mutation prohibited X-CD40–driven phosphorylation of Jnk and p38 (Fig. 2a,b). These results were identical to those data obtained from murine B cell lines in which Jnk activation was prevented by the ΔT2,3 mutation and from B cells in mice that expressed dominant-negative TRAF219,20.
Figure 2. Signaling in X-CD40 transgenic mice relies on TRAF2/3 binding.

(a) Primary cultures of splenic B cells from unimmunized X-CD40 transgenic mice were stimulated in vitro with 1000 ng/ml of shCD154 for various times. Cells were lysed and lysates were immunoblotted (IB) with antibodies to the phosphorylated TpY motif of Jnk2. Both isoforms of phosphorylated Jnk, p54 and p46, were recognized by the primary antibody. (b) Analysis of p38 MAPK activation in primary splenic B cells that were treated with shCD154. Cells were stimulated for various times, then lysed and immunoblotted with an antibody to the phosphorylated TpY motif of p38 MAPK. (c) Analysis of NF-κB activation in primary splenic B cells. Cells were treated with shCD154 for various times, lysed and immunoblotted with an antibody to Ser32-phosphorylated IκBα. For all signaling experiments, data are representative of three or more independent experiments. (d) Measurement of cellular IκBα content in B cells from transgenic mice. The nitrocellulose membrane in c was analyzed by immunoblotting with antibodies to IκBα, as a control for equivalent protein loading.
CD40-mediated activation of NF-κB was measured by phosphorylation of the inhibitory subunit IκBα. Loss of TRAF2/3 recruitment did not completely ablate signaling by this pathway, but rather it imparted a delayed and diminished response (Fig. 2c). No distinguishable effect on NF-κB, Jnk or p38 was observed in cells that contained the ΔT6 mutant X-CD40. Finally, even in the absence of TRAF recruitment—as observed in ΔT2,3,6 mutant mice—phosphorylation of IκBα was apparent, although the kinetics were delayed and the response diminished. Using B cells from each of the X-CD40 mice on the CD40+/+ background, we found that signaling via endogenous murine CD40 appeared normal for all the lines (Web Fig. 2). In addition, signaling studies that used limiting concentrations of shCD154 resulted in similar patterns of phosphorylation (data not shown).
Engagement of CD40 on ex vivo B cells induces up-regulation of surface molecules, proliferation and immunoglobulin (Ig) secretion1. To assess the role played by TRAFs in these CD40-induced responses, B cells from each of the X-CD40 mice were isolated and activated in vitro. In the presence or absence of TRAF recruitment, up-regulation of the B cell activation marker CD23 was observed in all X-CD40 mutants (Fig. 3a). No alterations in the up-regulation of surface molecules could be attributed to any given mutation. Similar results were seen with the up-regulation of CD80, CD86, CD95 and CD54 (data not shown). In addition, the evaluation of each of these mutations on CD40-induced B cell proliferation in vitro demonstrated no dependence on TRAF recruitment (Fig. 3b and Web Fig. 3). Analysis of cell division with the use of carboxy-fluorescein succinimide ester (CFSE) dilution confirmed that after 4 days of stimulation with CD154, B cells from each X-CD40 mouse line had undergone the same number of cell divisions (data not shown). CD40 and cytokine-induced production of IgM and IgG1 in vitro were similarly unaffected by the lack of TRAF recruitment (Fig. 3c,d). These data show that the early biological responses of B cells to CD40 engagement in vitro are TRAF-independent.
Figure 3. In vitro activation of B cells from X-CD40 mice.

(a) Primary splenic B cells from each of the X-CD40 mice were stimulated in vitro with or without shCD154 (400 ng/ml) for 48 h. Cells were stained with biotinylated antibodies to CD23, which were detected with PE-streptavidin via flow cytometry. Data are the mean fluorescence intensity (MFI) of stained samples from cell cultures. Tg- denotes non-transgenic. (b) Proliferation was measured in splenic B cells treated with IL-4 + IL-5 either with or without shCD154 for 3 days. [3H]thymidine was added to cell cultures at 60 h. Cells were collected at 72 h and [3H]thymidine uptake was measured by scintillation counting. Data are mean±s.e.m. (c) IgM was measured in media samples taken from splenic B cells that were cultured for 5 days with IL-4 + IL-5 in the presence or absence of shCD154. IgM was measured by an isotype-specific ELISA. (d) Primary splenic B cells were cultured for 5 days with IL-4 + IL-5, in the presence or absence of shCD154. IgG1 was measured by an isotype-specific ELISA. All in vitro activation assays are representative of four or more experiments.
Humoral immune responses in X-CD40 mice
Mice that are genetically deficient in CD40 or CD154 as well as mice treated with blocking antibody to CD154 do not develop TD humoral immune responses21–23. All X-CD40 mice were immunized with nitrophenol-conjugated keyhole limpet hemocyanin (NP-KLH) in complete Freund's adjuvant (CFA), and early IgG1 responses were monitored (Fig. 4a). Immunization with NP-KLH allowed us to readily detect antibody produced in response to the hapten NP and also to quantify the development of high-affinity Ig responses. Compared to WT, there was a 50% reduction in the IgG1 anti-NP serum titers in the ΔT6 and ΔT2,3,6 mice. Heightened IgG1 responses were observed in the ΔT2,3 mice, possibly due to the negative regulatory effect of TRAF124. Serum NP-specific IgG1 titers were observed for 60 days after immunization in the absence of TRAF recruitment (that is, in the ΔT2,3,6 mice), albeit in reduced amounts (Fig. 4b). In contrast to total anti-NP IgG1, the production of high-affinity NP-specific IgG1—as measured on day 68 after immunization—was reduced by >95% in the ΔT6 and ΔT2,3,6 mice. These data suggest an essential role played by TRAF6 in Ig affinity maturation.
Figure 4. Humoral immune responses in X-CD40 mice.
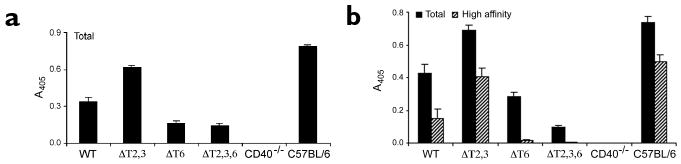
(a) Early IgG1 production was measured in vivo 15 days after intraperitoneal immunization with 100 μg of NP36-KLH that was emulsified in CFA. Circulating antibodies were measured by an NP-specific, isotype-specific ELISA. Data are mean±s.e.m. of three mice per group and are representative of three independent experiments. (b) Affinity maturation of the anti-NP response was measured 68 days after immunization with 100 μg of NP36-KLH emulsified in CFA. Circulating antibodies were quantified by an isotype-specific, NP-specific ELISA. NP25-BSA was used to capture all (total) antigen-specific Igs; NP4-BSA was used to capture only high-affinity Igs. Captured antibodies were detected with enzyme-conjugated anti-IgG1. Data are mean±s.e.m. of three mice per group and are representative of three independent experiments.
Plasma cell differentiation in X-CD40 mice
The terminal differentiation of B cells to plasma cells is a hallmark of TD humoral immune responses. To investigate the role played by TRAFs in triggering terminal B cell differentiation to plasma cells, the number of antibody-secreting cells in the bone marrow of X-CD40 mice 68 days after immunization was determined. In the ΔT6 and ΔT2,3,6 mice, there was a >95% reduction in the number of total plasma cells and high-affinity plasma cells in the bone marrows of immune mice (Fig. 5). These data indicate that the loss of TRAF6 recruitment, but not TRAF2/3 recruitment, abrogated the generation of long-lived bone marrow plasma cells that secrete either low- or high-affinity NP-specific antibodies.
Figure 5. Generation of long-lived bone marrow plasma cells.

(a) Mice were immunized with 100 μg of NP36-KLH emulsified in CFA. Mice were killed 68 days after immunization and bone marrow was collected. Bone marrow–derived lymphocytes were assessed for production of total antigen-specific IgG1 by ELISPOT; NP25-BSA was used as a capture antigen. (b) Generation of high-affinity plasma cells in the bone marrow of the same mice used in a. NP4-BSA was used as a capture antigen. Data are expressed as the number of antibody-secreting cells per 106 input cells and are representative of three or more independent experiments.
The generation of memory B cells in the X-CD40 mice was also evaluated by the adoptive transfer of primed X-CD40 B cells into T cell–primed recipients, which was followed by antigen challenge. Because the ΔT6 mutation impaired affinity maturation, tracking memory B cell responses in recipient mice was problematic. Nonetheless, mice that received B cells from the WT and ΔT2,3 mice, but not the ΔT6 mice, produced high-affinity responses upon challenge (data not shown). Although little can be said with regard to the involvement of TRAF6, TRAF2/3 recruitment was not involved in the generation of memory B cells.
GC formation in X-CD40 mice
GCs are the site of development for TD humoral immune responses, B cell memory and affinity maturation. These processes are dependent on CD40 signaling. To determine whether GC formation was TRAF-dependent, X-CD40 mice were immunized with sheep erythrocytes and GC formation was assessed. Confocal microscopy revealed GCs by immunohistological analysis with the use of a GC-specific marker, peanut agglutinin (PNA). GC B cells (which were PNA+CD19+) were present in the WT, ΔT2,3 and ΔT6 mice, but not in the ΔT2,3,6 mice (Fig. 6a). Similarly, flow cytofluorometric analysis revealed GC B cells as PNA+GL7+ B cells. All X-CD40 mice expressed this GC population except for those with the ΔT2,3,6 mutations (Fig. 6b). Therefore, GC formation in response to immunization is triggered by the recruitment of TRAF2/3 or TRAF6 to the CD40 cytoplasmic domain but cannot occur in the absence of all TRAF recruitment.
Figure 6. GC formation in the X-CD40 mice.

(a) Immunohistological analysis of GC formation in spleens was done 10 days after immunization with sheep erythrocytes. Spleens were thinly sectioned, then stained with fluorochrome-coupled antibodies to CD4 and CD8 (red), PNA (blue) and CD19 (green). Images were captured by confocal microscope; they are representative of two experiments and >30 scanned images of three mice per group. (b) Flow cytofluorometric analysis of GC formation was done in mice immunized intraperitoneally with sheep erythrocytes and killed 10 days after immunization. Splenocytes were stained with fluorochrome-coupled antibodies to B220, PNA and GL7. Fluorescence was quantified by flow cytometry and profiles were gated on B220+ cells. Data are representative of three experiments with three mice per group.
Discussion
The TRAF-binding sites of the CD40 cytoplasmic tail have been implicated as key functional components in CD40 signal transduction. Disruption of TRAF binding by site-directed mutagenesis resulted in specific humoral immune deficiencies in transgenic mice. The data we have presented here establish that CD40-induced B cell growth, up-regulation of surface molecules and early Ig production are not dependent on the recruitment of TRAFs to the cytoplasmic domain of CD40. Later events in the humoral immune response, like GC formation, are dependent on the binding of either TRAF2/3 or TRAF6. Finally, the generation of high-affinity antibody and the generation of long-lived, bone marrow plasma cells specifically require recruitment of TRAF6 to the CD40 cytoplasmic domain.
The roles played by TRAF molecules as intermediaries in the CD40 signaling cascade have been examined with the use of several independent approaches. The Thr255 residue (target of the ΔT2,3 mutation) was recognized as a critical functional site in CD40 signaling more than 12 years ago25; later it was shown to be essential for the direct binding of TRAF2 and TRAF3 to CD405,26. Because TRAF1 and TRAF5 bind indirectly to TRAF2/312,27, the Thr255→Ala255 mutation also limits their recruitment to CD40. CD40 mutants expressed in B cell lines have confirmed the importance of the TRAF2/3 site in CD40 signaling and transducing biological responses12–15,28,29. As an alternative to disrupting TRAF-binding sites, studies have shown that deletion of the genes encoding TRAFs can exert profound effects on CD40 signaling and biology as well. Specifically, B cells from TRAF2−/− mice show reduced amounts of CD40-induced B cell activation of NF-κB, proliferation and IgG1 production in vivo in response to viral challenge18. Based on these studies, a conspicuous functional phenotype in the ΔT2,3 mice was anticipated. The loss of CD40-induced Jnk activation and reduced NF-κB activation in B cells from ΔT2,3 mice that we observed confirmed the anticipated signaling phenotype of the Thr255→Ala255 mutation as well as that observed in TRAF2 dominant-negative mice. However, the loss of TRAF2/3 (and TRAF1 and TRAF5) recruitment appeared to impart no overt deficiency in humoral immunity in vivo, except in the absence of TRAF6 recruitment, where GC formation was lost. Given that the ΔT2,3 mice manifest a moderate phenotype one must consider the possibility that the severe developmental deficiencies in the TRAF2−/− mice and the global impact of TRAF2−/− deficiency on other TNFR family members (not just CD40) may have contributed substantially to the altered CD40 responsiveness of TRAF2−/− B cells.
The Pro213→Gly213–Glu215→Ala215 mutation disrupts TRAF6 binding to the CD40 cytoplasmic domain4. Disruption of this site ablates p38 activation and represses NF-κB activation via CD40 in HEK293 cells, but not in B cell lines4,11,15; this shows that the signaling role played by TRAF6 may be cell-type specific. Unlike the ΔT2,3 mice, the ΔT6 mice showed no abnormality in CD40 signaling in B cells, but demonstrated substantial defects in the development of late-phase humoral immunity. The data collectively suggest that TRAF6 recruitment may play no role during the activation of naïve B cells, but may play a key role within the GC. It is possible that CD40 engagement and TRAF6 recruitment may signal through factors yet to be identified that promote affinity maturation and B cell differentiation within GC cells. This hypothesis is consistent with the observation that the loss of TRAF6 binding in naïve B cells ex vivo has no effect and the observation that as antigen-activated B cells enter the GC, CD40 is “rewired” to support rescue from Ig-induced apoptosis30.
Another key finding to emerge from these studies is the extent to which B cell responses can proceed in the absence of TRAF recruitment. Data from the ΔT2,3,6 mice showed that CD40-induced NF-κB activation, B cell proliferation, up-regulation of surface molecules and early Ig production occur in the absence of TRAF recruitment. As with the X-CD40 mice, studies that used B cell lines have shown that some CD40-induced events are TRAF-independent14,15. Presently, the identity of the non-TRAF signaling intermediaries are unknown. However, non-TRAF–binding domains are present in the cytoplasmic domain of CD4013,31 that can activate NF-κB and perhaps trigger cellular activation.
With the use of a similar strategy, another group has evaluated the contribution made by CD40 functional domains through the transgenic expression of truncated or site-mutated full-length human CD40 molecules expressed with an Ig promoter32. After crossing onto a CD40−/− background, these mice expressed the human CD40 transgene only in B cells. They found that CD40 triggering induced B cell proliferation, up-regulation of surface molecules and Ig secretion in vitro in the absence of TRAF recruitment32, which was confirmed by the data we obtained with the ΔT2,3,6 mice. They also showed that truncation of the entire cytoplasmic domain ablated these early events, which provides conclusive evidence that non-TRAF–binding domains within the cytoplasmic domain of CD40 are critical for inducing early events. In contrast to the data we obtained with the ΔT2,3 mice, the T254→A254 mice the other group produced showed reduced GC formation and high-affinity antibody production32. Although the reason for these differences remains unresolved, one apparent difference between the studies is the expression of CD40 exclusively in B cells versus all MHC class II–bearing cells. Another issue concerns whether the human cytoplasmic domain is as efficient at recruiting and signaling in murine cells as the murine cytoplasmic domain, due to the sequence differences in and around the TRAF6-binding site. Although some discrepancies exist within the data, taken together these two studies have defined the roles played by TRAF molecules in controlling humoral immunity.
In summary, TRAFs appear to control the terminal phases of CD40-induced B cell differentiation. Although early B cell clonal expansion and early differentiation appear intact in the absence of TRAFs, GC formation, affinity maturation and long-lived humoral immunity are impaired without TRAF recruitment to CD40. The results we present here provide fresh insights into the functional domains of CD40 that control humoral immunity.
Methods
Construction of chimeric CD40
The oligonucleotides 5′–TTGGATCCATGGTTCGTCT GCCTCTGCAGT–3′ and 5′–ACAGGAATGACCAGCAGGGCTCTCAGCCGATCCTGG GGACCA–3′ were used to amplify the human CD40 ectodomain sequence; phCD40/GemT (Pullen, Danbury, CT) was used as a template. The oligonucleotides 5′–CAGGATCGGCT GAGAGCCCTGCTGGTCATTCCTGTCGT–3′ and 5′–TTATCAAAAGGTCAGCAAG CAGCCA–3′ were used to amplify the mouse cytoplasmic and transmembrane domain sequence that contained an NH2-terminal overlap with the human CD40 ectodomain sequence from a plasmid encoding murine CD40 (a gift of E. Clark, University of Washington, Seattle). These two PCR products were annealed, extended with Taq polymerase and ligated into pGem-T (Promega, Madison, WI) to generate ph-mCD40/GemT. The oligonucleotides 5′–GGTGAAAGCGAATTTCTAGACACCTGGAAC–3′ and 5′–GTTCCAGGTGTCTAGAAATTCGCTTTCACC–3′ were used to generate a silent mutation (underlined) that eliminated the EcoRI site in the mouse CD40 cytoplasmic domain; then the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to generate ph-mCD40/EcoKO/GemT. EcoRI sites were inserted at the 5′ and 3′ ends of the chimeric CD40 coding sequence by PCR to generate ph-mCD40/Eco+/GemT. Amino acid substitutions in the murine CD40 cytoplasmic domain were generated with the use of complementary primers that had the desired base changes and ph-mCD40/Eco+/GemT as a template; the QuikChange site-directed mutagenesis kit was used. Chimeric CD40 was subcloned via the EcoRI sites into vector pDOI-5 for expression under the control of an MHC class II promoter16. All plasmid constructs were verified by automated DNA sequencing.
Generation of transgenic mice
Transgenic mice were generated by the NICHD Transgenic Mouse Development Facility at the University of Alabama at Birmingham under contract number N01-HD-5-3229. All experiments with mice were done according to the principles set out in the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animals Resources, National Research Council, DHHS, publication number (NIH) 86-23 (19850). The experiments were approved by the Institutional Animal Care and Use Committee of Dartmouth College.
Antibodies and reagents
Monoclonal antibodies (mAbs) to Ser32-phosphorylated IκBα were from Cell Signaling Technology (Beverly, MA). Polyclonal rabbit antibodies to the phosphorylated form of Jnk were from Promega. Polyclonal rabbit antibodies to the phosphorylated form of MAPK p38 were from Cell Signaling Technology. mAb S2C6 to human CD40 was a gift of S. Paulie (Stockholm University, Sweden). mAb 1c10 to murine CD40 was from ATCC (Rockville, MD). Staining antibodies to CD23, CD19, CD4, CD8, CD11c and MHC class II were from BD Pharmingen (San Diego, CA). Primary antibodies were detected with horseradish peroxidase (HRP)-conjugated goat anti–rabbit Ig (Vector Labs, Burlingame, CA) or phycoerythrin (PE)-conjugated streptavidin (Pharmingen, Torrey Pines, CA). shCD154 fusion protein was from Immunex (Seattle, WA). Matched antibody sets for IgM and IgG1 ELISA were from Southern Biotechnology (Birmingham, AL). [3H]thymidine (3000 Ci/mmol) was from NEN (Perkin Elmer, Boston, MA). PNA was a gift of T. Waldschmidt (University of Iowa).
Immunoblots
Protein samples were separated by 12% PAGE with SDS. Proteins were transferred to nitrocellulose and membranes were blocked with 1% bovine serum albumin (BSA) and/or 5% milk in TBS with 0.05% Tween-20. Primary antibody incubations were done overnight at 4 °C in blocking buffers that were recommended by the antibody suppliers. After washing, secondary antibody incubations were done over 30-min periods in the same blocking buffers. Blots were developed with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL) and exposed to x-ray film.
B lymphocyte stimulation in vitro
Red blood cell and T cell–depleted splenocytes, which consisted of ∼85% CD19+ cells, were collected from transgenic mice that had been killed first. For signaling assays, cells were cultured at 5×106 per ml for 3–30 min with various stimuli, then lysed in 1% lysis buffer with a mixture of protease and phosphatase inhibitors. Cellular debris was cleared from lysates by centrifugation at 14000g for 10 min before denaturation in gel-loading buffer that contained β-mercaptoethanol and SDS by boiling for 5 min. For biological readouts, cells were cultured at 2×106 per ml for 2–5 days. For the induction of surface markers, 2 days of culture in soluble CD154 was followed by washing cells into staining media with CD23-specific antibodies for staining. For proliferation and Ig secretion, cells were cultured with 10 ng/ml interleukin-4 (IL-4) + IL-5 and various stimuli. Where “untreated” is indicated on figures, cells were cultured in IL-4 + IL-5 with no additional stimuli. Proliferation of cultures was quenched by collection on a Packard Filtermate; the resulting filter plates were analyzed on a Packard TopCount scintillation counter (Packard Bioscience, Meriden, CT). Five-day cultures were transferred to V-bottomed 96-well plates where cells could be made into pellets by centrifugation. Supernatants were then analyzed for Ig content by ELISA.
Affinity maturation
Mice were given intraperitoneal immunizations of 100 μg of NP36-KLH (molar ratio of hapten to KLH) emulsified in CFA. On days 15 and 68 after immunization, serum was collected from peripheral blood. Circulating antibodies were measured by an isotype-specific, antigen-specific ELISA. NP25-BSA was used to capture all antigen-specific Igs, whereas NP4-BSA was used to capture only high-affinity Igs. Captured antibodies were detected with enzyme-conjugated rabbit anti-IgG1. Data are mean±s.e.m. of three mice per group.
ELISPOT assay
Bone marrow anti-NP–specific IgG1a–secreting cells were enumerated by an NP-specific ELISPOT assay. Bone marrow–derived B cells were plated at 2.4×106 cells/well in NP-BSA–coated HA96 well plates (Millipore, Bedford, MA) and threefold serial dilutions were made before incubation. Plates were incubated for 5 h at 37 °C within a 5% CO2 incubator. After incubation, plates were washed in 1× PBS and 0.5% Tween in 1× PBS. Antibody-secreting cells were detected with biotinylated anti–mouse IgG1 followed by HRP-streptavidin (Amersham Life Sciences, Piscataway, NJ). ELISPOTS were developed by a chromagen substrate, AEC (Sigma, St. Louis, MO) in DMF (Fischer Scientific, Fair Lawn, NJ) and 0.1 M sodium acetate buffer at pH 4.8–5.0. Immediately before development of ELISPOTS, 3% H2O2 was added to chromagen substrate. ELISPOTS were enumerated via a dual-axis light-dissecting microscope.
GCs
Mice were given intraperitoneal immunizations with sheep erythrocytes without adjuvant. Ten days after immunization, mice were killed and spleens were sectioned for confocal microscopic analysis or T cell–depleted for flow cytometric analysis. GCs were characterized as GL7+PNA+CD19+ B cells that stained near to CD4+CD8+ T cell zones in the spleen. Flow cytometric profiles were gated on B220+ B cells.
Acknowledgments
Confocal microscopy and flow cytometry was done at the Herbert C. Englert Cell Analysis Laboratory, which was established by equipment grants from the Fannie E. Rippel Foundation, the NIH Shared Instrument Program and Dartmouth Medical School and is supported, in part, by Core Grant CA 23108 from the National Cancer Institute to the Norris Cotton Cancer Center. Supported by NIH grant AI26296 (to R. J. N.) and the American Cancer Society (L. D. E.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.Banchereau J, et al. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 2.Kuhne MR, et al. Assembly and regulation of the CD40 receptor complex in human B cells. J Exp Med. 1997;186:337–342. doi: 10.1084/jem.186.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pullen SS, et al. CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998;37:11836–11845. doi: 10.1021/bi981067q. [DOI] [PubMed] [Google Scholar]
- 4.Pullen SS, Dang TT, Crute JJ, Kehry MR. CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J Biol Chem. 1999;274:14246–14254. doi: 10.1074/jbc.274.20.14246. [DOI] [PubMed] [Google Scholar]
- 5.Cheng G, Baltimore D. TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-κB activation. Genes Dev. 1996;10:963–973. doi: 10.1101/gad.10.8.963. [DOI] [PubMed] [Google Scholar]
- 6.Lomaga MA, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh WC, et al. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 8.Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001;13:389–400. doi: 10.1016/s0898-6568(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 9.Nakano H, et al. Targeted disruption of Traf5 gene causes defects in CD40- and CD27- mediated lymphocyte activation. Proc Natl Acad Sci USA. 1999;96:9803–9808. doi: 10.1073/pnas.96.17.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Cheng G, Baltimore D. Immunity. Vol. 5. 1996. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses; pp. 407–415. [DOI] [PubMed] [Google Scholar]
- 11.Jalukar SV, Hostager BS, Bishop GA. Characterization of the roles of TNF receptor-associated factor 6 in CD40-mediated B lymphocyte effector functions. J Immunol. 2000;164:623–630. doi: 10.4049/jimmunol.164.2.623. [DOI] [PubMed] [Google Scholar]
- 12.Leo E, et al. Differential requirements for tumor necrosis factor receptor-associated factor family proteins in CD40-mediated induction of NF-κB and Jun N-terminal kinase activation. J Biol Chem. 1999;274:22414–22422. doi: 10.1074/jbc.274.32.22414. [DOI] [PubMed] [Google Scholar]
- 13.Hsing Y, Hostager BS, Bishop GA. Characterization of CD40 signaling determinants regulating nuclear factor-κB activation in B lymphocytes. J Immunol. 1997;159:4898–4906. [PubMed] [Google Scholar]
- 14.Leo E, Zapata JM, Reed JC. CD40-mediated activation of IgCγ1 and IgCε germ-line promoters involves multiple TRAF family proteins. Eur J Immunol. 1999;29:3908–3913. doi: 10.1002/(SICI)1521-4141(199912)29:12<3908::AID-IMMU3908>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 15.Manning EM, Pullen SS, Souza DJ, Kehry MR, Noelle RJ. Cellular responses to murine CD40 in a mouse B cell line may be TRAF dependent or independent. Eur J Immunol. 2002;32:39–49. doi: 10.1002/1521-4141(200201)32:1<39::AID-IMMU39>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 16.Kouskoff V, Fehling HJ, Lemeur M, Benoist C, Mathis D. A vector driving the expression of foreign cDNAs in the MHC class II- positive cells of transgenic mice. J Immunol Meth. 1993;166:287–291. doi: 10.1016/0022-1759(93)90370-m. [DOI] [PubMed] [Google Scholar]
- 17.Mazzei GJ, et al. Recombinant soluble trimeric CD40 ligand is biologically active. J Biol Chem. 1995;270:7025–7028. doi: 10.1074/jbc.270.13.7025. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen LT, et al. TRAF2 deficiency results in hyperactivity of certain TNFR1 signals and impairment of CD40-mediated responses. Immunity. 1999;11:379–389. doi: 10.1016/s1074-7613(00)80113-2. [DOI] [PubMed] [Google Scholar]
- 19.Sutherland CL, Krebs DL, Gold MR. An 11-amino acid sequence in the cytoplasmic domain of CD40 is sufficient for activation of c-Jun N-terminal kinase, activation of MAPKAP kinase-2, phosphorylation of IκBα, and protection of WEHI-231 cells from anti-IgM-induced growth arrest. J Immunol. 1999;162:4720–4730. [PubMed] [Google Scholar]
- 20.Lee SY, et al. TRAF2 is essential for JNK but not NF-κB activation and regulates lymphocyte proliferation and survival. Immunity. 1997;7:703–713. doi: 10.1016/s1074-7613(00)80390-8. [DOI] [PubMed] [Google Scholar]
- 21.Castigli E, et al. CD40-deficient mice generated by recombination-activating gene-2-deficient blastocyst complementation. Proc Natl Acad Sci USA. 1994;91:12135–12139. doi: 10.1073/pnas.91.25.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foy TM, et al. In vivo CD40-gp39 interactions are essential for thymus-dependent humoral immunity. II. Prolonged suppression of the humoral immune response by an antibody to the ligand for CD40, gp39. J Exp Med. 1993;178:1567–1575. doi: 10.1084/jem.178.5.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, et al. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 24.Tsitsikov EN, et al. TRAF1 is a negative regulator of TNF signaling: Enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–657. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- 25.Inui S, et al. Identification of the intracytoplasmic region essential for signal transduction through a B cell activation molecule, CD40. Eur J Immunol. 1990;20:1747–1753. doi: 10.1002/eji.1830200819. [DOI] [PubMed] [Google Scholar]
- 26.Hu HM, O'Rourke K, Boguski MS, Dixit VM. A novel RING finger protein interacts with the cytoplasmic domain of CD40. J Biol Chem. 1994;269:30069–30072. [PubMed] [Google Scholar]
- 27.Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 28.Hostager BS, Hsing Y, Harms DE, Bishop GA. Different CD40-mediated signaling events require distinct CD40 structural features. J Immunol. 1996;157:1047–1053. [PubMed] [Google Scholar]
- 29.Tsukamoto N, Kobayashi N, Azuma S, Yamamoto T, Inoue J. Two differently regulated nuclear factor κB activation pathways triggered by the cytoplasmic tail of CD40. Proc Natl Acad Sci USA. 1999;96:1234–1239. doi: 10.1073/pnas.96.4.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siepmann K, Skok J, van Essen D, Harnett M, Gray D. Rewiring of CD40 is necessary for delivery of rescue signals to B cells in germinal centres and subsequent entry into the memory pool. Immunology. 2001;102:263–272. doi: 10.1046/j.1365-2567.2001.01162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanissian SH, Geha RS. Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. Immunity. 1997;6:379–87. doi: 10.1016/s1074-7613(00)80281-2. [DOI] [PubMed] [Google Scholar]
- 32.Yasui T, et al. Dissection of B cell differentiation during primary immune responses in mice with altered CD40 signals. Int Immunol. 2002;14:319–329. doi: 10.1093/intimm/14.3.319. [DOI] [PubMed] [Google Scholar]