

Abstract
The mechanism by which the serine-threonine kinase glycogen synthase kinase-3β (GSK3β) affects survival of renal epithelial cells after acute stress is unknown. Using in vitro and in vivo models, we tested the hypothesis that GSK3β promotes Bax-mediated apoptosis, contributing to tubular injury and organ dysfunction after acute renal ischemia. Exposure of renal epithelial cells to metabolic stress activated GSK3β, Bax, and caspase 3 and induced apoptosis. Expression of a constitutively active GSK3β mutant activated Bax and decreased cell survival after metabolic stress. In contrast, pharmacologic inhibition (4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione [TDZD-8]) or RNA interference–mediated knockdown of GSK3β promoted cell survival. Furthermore, RNA interference–mediated knockdown of Bax abrogated the cell death induced by constitutively active GSK3β. In a cell-free assay, TDZD-8 inhibited the phosphorylation of a peptide containing the Bax serine163 site targeted by stress-activated GSK3β. In rats, TDZD-8 inhibited ischemia-induced activation of GSK3β, Bax, and caspase 3; ameliorated tubular and epithelial cell damage; and significantly protected renal function. Taken together, GSK3β-mediated Bax activation induces apoptosis and tubular damage that contribute to acute ischemic kidney injury.
Acute ischemic renal failure remains a common cause of death that precipitates organ failure by causing apoptosis, necrosis, autophagy, and the desquamation of viable proximal tubule epithelial cells from the basement membrane.1–7 Although epithelial cell apoptosis has been detected in the intact kidney after ischemia8–10 and in cultured renal epithelial cells subjected to metabolic stress,11,12 it has been difficult to assess its contribution to human organ failure.2 More importantly, efforts to improve outcome in ischemic organ failure have been limited by an incomplete understanding of the signal events that regulate renal epithelial cell injury.
Glycogen synthase kinase-3β (GSK3β) is a 47-kD serine-threonine kinase that was first observed to phosphorylate and inactivate glycogen synthase, a distal enzyme in the glycogen synthesis pathway.13 GSK3β is an ideal “survival” enzyme, because it controls several extrametabolic processes that are perturbed by ischemia, including cytoskeletal dynamics, gene expression, proliferation, and apoptosis.14–22 Normally, GSK3β is suppressed by proliferative, prosurvival signals that increase serine9 phosphorylation, such as WNT ligands, EGF, IGF-I and -II, and fibroblast growth factor,16,19,23 as well as Akt.19 Conversely, GSK3β is activated by noxious stimuli, including serum starvation,23 hypertonic stress,24 potassium deprivation,25 hypoxia,26 endotoxin exposure,27 and tissue ischemia.21,28–31 In acute models of injury, GSK3β promotes the systemic inflammatory response, increases the proinflammatory release of cytokines, induces apoptosis, and alters cell proliferation.32,33
Although the mechanism by which GSK3β regulates epithelial cell survival after ischemia is poorly characterized, substantial evidence indicates that GSK3β inhibition preserves organ function after ischemia of the brain,34 heart,35 and gut.36 In the ischemic cell, GSK3β targets multiple proteins with the potential to regulate survival; however, most are transcription factors such as β-catenin/TCF-Lef genes, cAMP response element binding protein, heat shock factor-1, nuclear factor of activated T cells, and NF-κB as well as cyclin D117,18,26,37,38 that are unlikely to mediate acute cell survival during or immediately after transient ischemia.
In contrast to these transcription factors and cell-cycle regulators, immediate cell fate after de-energization or ischemia is regulated by BCL2 proteins that target mitochondria, resulting in the activation of both caspase-dependent and -independent cell death cascades.12,39–41 Intriguingly, GSK3β directly phosphorylates and activates Bax, a proapoptotic member of the BCL2 family.42 Both Bax and GSK3β are constitutively expressed and are regulated by phosphorylation at specific serine sites. Phosphorylation of serine9 inhibits GSK3β,19,43 whereas dephosphorylation of this residue activates GSK3β, thereby promoting downstream serine163 phosphorylation and activation of Bax.42 GSK3β has been linked to mitochondrial dysfunction after oxidative stress caused by renal ischemia or hypoxia-induced oxidative stress.44 In addition, GSK3β has been implicated in promoting mitochondrial permeabilization, a terminal event, by direct phosphorylation and destabilization of MCL-1, an antiapoptotic member of the BCL2 family.45 By directly phosphorylating and activating Bax, we propose that GSK3β mediates mitochondrial injury and determines early epithelial cell fate in vitro as well as the severity of tubular injury and organ dysfunction in vivo after an acute ischemic insult.
Using models of rotenone-induced ATP depletion in vitro and transient renal artery occlusion in rats in vivo, this study shows that direct manipulation of GSK3β regulates Bax activation, apoptosis, tubular injury, and organ dysfunction after metabolic stress or ischemia. Selective GSK3β upregulation (constitutively active GSK3β) or suppression (using RNA interference [RNAi] or 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione [TDZD-8], a selective GSK3β inhibitor), causes parallel changes in Bax and caspase 3 activation, apoptosis, and tubular injury, indicating that GSK3β mediates acute epithelial cell survival partly by disrupting the balance of pro- versus antiapoptotic BCL2 proteins in the renal epithelial cell. Specifically, GSK3β primarily operates via a Bax-dependent mechanism to regulate epithelial cell survival.
Results
Transfection of renal epithelial cells with constitutively active (S9A) or wild-type (WT) GSK3β resulted in an expected increase in total GSK3β content compared with empty vector (Figure 1A). In contrast, exposure of cells to one of three GSK3β RNAi directed against distinct GSK3β regions resulted in variable downregulation of GSK3β expression compared with either normal (nontransfected) or vector-exposed cells or cells incubated with nonspecific RNAi (Figure 1B, lanes 1, 2, and 7, respectively). Exposure to a mixture of three RNAis resulted in a marked reduction in immunoreactive GSK3β content compared with control (Figure 1B, lane 3).
Figure 1.
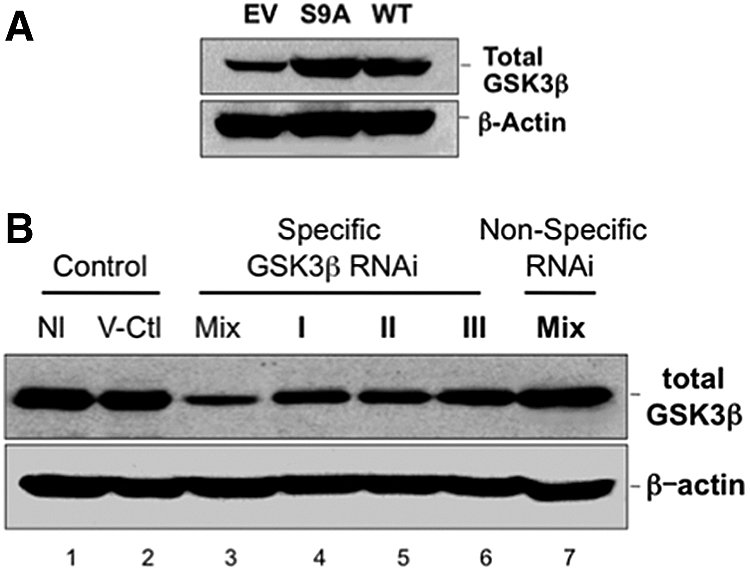
Effect of transfection and RNAi on GSK3β expression is shown. (A) Cell GSK3β content after transfection with empty vector (EV), constitutively active (S9A), or WT GSK3β (top); β-actin loading control (bottom). (B) Total GSK3β content in nontransfected cells (Nl), in cells transfected with vector only (V-Ctl), a mixture of three (Mix; top), single specific RNAi directed against GSK3β (I, II, or III) or a mixture of nonspecific RNAis (Mix-Non-Specific-RNAi); β-actin is loading control (bottom). Each lane contains 40 μg of total protein; results are representative of at least three separate studies.
Compared with control, transfection with constitutively active GSK3β (GSK3β-S9A) increased total GSK3β content before, during, and after metabolic stress (Figure 2B), without marked differences in p-serine9 GSK3β content between the two groups (Figure 2A) as detected with a phospho-serine9–specific GSK3β antibody. The later observation is due to the fact that the constitutively active GSK3β-S9A mutant cannot be inactivated by phosphorylation at the serine9 position. Although GSK3β-S9A expression did not alter the content of active Bax at baseline (Figure 2C), this GSK3β mutant promoted Bax activation (detected by a 6A7 epitope–specific antibody) during and after stress in the absence of changes in total Bax content (Figure 2D). These results show that constitutively active GSK3β expression promotes stress-induced Bax activation.
Figure 2.
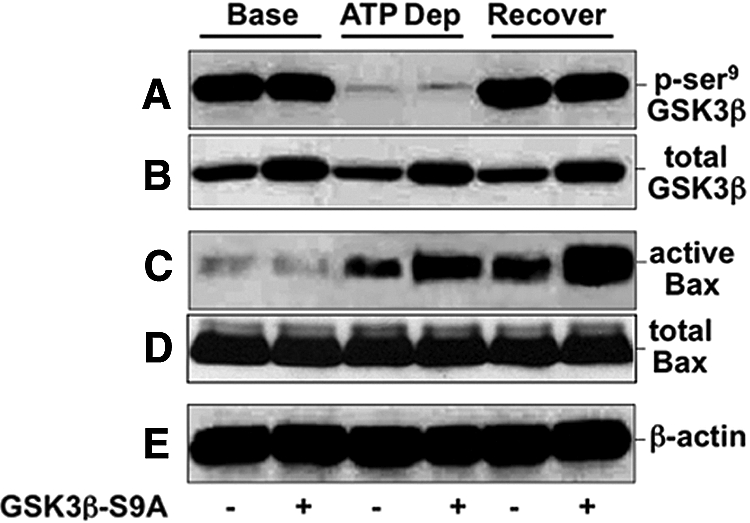
Effect of ATP depletion on GSK3β, active Bax, caspase 3, and apoptosis is shown. Content of total and p-serine9 GSK3β, active Bax (6A7 epitope), total Bax, and β-actin in cells at baseline (‘Base’); after 60 minutes of metabolic stress (ATP Dep); and after 60 minutes of recovery (Recover) in cells that express empty vector (GSK3β S9A −) or constitutively active GSK3β (GSK3β S9A +). Immunoblot results are representative of two to three independent studies; each lane contains 40 μg of total protein.
To confirm that GSK3β regulates Bax activation, we used both molecular and biochemical maneuvers to inhibit GSK3β. As expected, only specific RNAi downregulated total GSK3β expression at baseline (Figure 3A, lane 3), after 90 min stress (Figure 3A, lane 7), and during recovery (Figure 3A, lane 11). The decrement in total GSK3β caused by specific RNAi also decreased p-serine9 GSK3β content at these same time points (Figure 3B). Because phospho-GSK3β content was markedly reduced during ATP depletion (Figure 3B, lanes 5 through 8), the exposure time of the immunoblot was extended to increase sensitivity (Figure 3B, inset). With prolonged exposure time, only specific RNAi decreased p-serine9 GSK3β during ATP depletion. At baseline, no difference in the content of phosphoserine33,37 β-catenin, a substrate that is phosphorylated at these sites by GSK3β, was detected in the presence of nonspecific or specific RNAi (Figure 3C, lane 1 versus lanes 2 and 3). Likewise, TDZD-8, an agent that decreases kinase activity without affecting p-serine9 GSK3β content, did not change phosphoserine33,37 β-catenin (Figure 3C, lane 4). After stress, however, both specific RNAi and TDZD-8 markedly inhibited β-catenin serine33,37 phosphorylation (Figure 3C, lanes 7 and 8 and lanes 11 and 12). Furthermore, both specific RNAi and TDZD-8 diminished stress-induced Bax activation (Figure 3D, lanes 7 and 8 versus 5 and 6 and lanes 11 and 12 versus lanes 9 and 10).
Figure 3.

Effect of GSK3β regulation on active Bax after ATP depletion is shown. Content of p-serine9 GSK3β, total GSK3β, phospho33,37–β-catenin (a GSK3β substrate), active Bax (6A7 epitope), and β-actin (loading control) in cells at baseline, after 90 min metabolic stress (ATP Deplete), and after 60 min recovery (Recovery) in cells that were not exposed to RNAi (CTL) or were exposed to nonspecific RNAi (NS-RNA), anti-GSK3β RNAi (RNAi), or 10 μM TDZD-8 (TDZD), a GSK3β kinase inhibitor. Inset shows the indicated region of immunoblot in panel B subjected to a longer exposure time to permit bands to be visualized. Immunoblot results are representative of two to three independent studies; each lane contains 40 μg of total protein.
Expression of either WT GSK3β or constitutively active GSK3β significantly reduced cell survival after stress (Figure 4A). To limit cell death in control, we reduced the duration of ATP depletion to 60 min in this protocol. Interestingly, the lowest cell survival was observed in cells that express constitutively active GSK3β, a mutant that cannot be inactivated by phosphorylation (e.g., by Akt). In contrast, both specific RNAi directed against GSK3β or TDZD-8 significantly improved cell survival after stress (P < 0.05; Figure 4B). In fact, survival exceeded 90% during recovery from stress when GSK3β was inhibited with either of these molecular or pharmacologic manipulations; however, none of these maneuvers (GSK3β expression, specific or nonspecific RNAi or TDZD-8 exposure) caused significant changes in cell survival at baseline (data not shown).
Figure 4.

Effect of GSK3β expression on renal cell survival after ATP depletion is shown. (A) Cell survival [3-(4,5 dimethylthiazol)-2,5-diphenyl tetrazolium bromide (MTT) assay] after 60 minutes of stress with 6 hours of recovery in cells expressing empty vector (EV), constitutively active GSK3β (GSK3β-SA9), or WT GSK3β (GSK3β-WT) compared with control at baseline. *P = 005 versus EV by ANOVA; n = 5. (B) Cell survival after 90 minutes of stress with 6 hours of recovery in control (CTL) or cells exposed to either nonspecific (NS RNAi) or specific (GSK3β RNAi) RNAi directed against GSK3β or to TDZD-8. *P < 0.05 versus EV by ANOVA; n = 5. None of the maneuvers significantly altered cell survival at baseline (data not shown).
To confirm that reduced survival was attributable to apoptosis, we stained cells with Hoechst dye. At baseline, exposure to empty vector or vehicle (DMSO) did not alter cell morphology or increase nuclear staining with Hoechst dye (Figure 5A, left-hand panels). In contrast to normal-appearing, relatively large, faintly stained nuclei at baseline, ATP depletion caused marked changes in morphology and increased Hoechst staining in cells transfected with empty vector or exposed to either nonspecific RNAi or vehicle. In these groups, characteristic features of apoptosis were observed: many nuclei appeared smaller or rounded or were fragmented into apoptotic bodies that intensely stained with Hoechst (Figure 5A). Expression of constitutively active GSK3β dramatically increased ATP depletion–induced apoptosis and the appearance of apoptotic bodies and also decreased the number of adherent cells (Figure 5A, bottom). In contrast, specific GSK3β RNAi or TDZD-8 decreased the number of apoptotic cells and increased cell adherence after ATP depletion (Figure 5A, bottom).
Figure 5.

Effect of ATP depletion on apoptosis and caspase 3 activity is shown. (A) Apoptosis assessed by staining cells with Hoechst dye before stress (Baseline) and 3 hours after 60 minutes of metabolic stress (ATP Depletion) in cells that express empty vector (Empty Vector), constitutively active GSK3β (GSK3β-S9A), nonspecific RNAi (NS-RNAi), or GSK3β-specific RNAi (GSK3β RNAi) or are exposed either to vehicle alone (DMSO) or to TDZD-8; apoptotic cells appear round and smaller and have more brightly stained nuclei than healthy cells stained with Hoechst dye. (B) Caspase 3 enzyme activity at baseline, immediately after stress (ATP deplete), and 15 or 60 minutes of recovery from stress (Rec15 and Rec60) in control (CTL) versus cells exposed to NS-RNAi, specific RNAi directed against GSK3β (GSK3β RNAi), or TDZD-8. Values are normalized to the baseline for each group. *P < 0.05 versus either control or NS-RNAi.
At baseline, caspase 3 enzyme activity, another surrogate measure of apoptosis, did not differ in control (nontransfected, non-RNAi exposed cells) versus cells exposed to nonspecific RNAi, GSK3β-specific RNAi, or TDZD-8 (Figure 5B). Compared with control or nonspecific RNAi, however, both specific GSK3β RNAi and TDZD-8 exposure significantly decreased stress-induced caspase 3 activation (P < 0.05).
To strengthen the causal relationship between the activation of GSK3β and Bax during ATP depletion, we devised a cell-free assay that measures the phosphorylation of Bax serine163, the residue targeted by GSK3β (see the Concise Methods section). In this assay, stress caused a six-fold increase in the phosphorylation of Bax serine163 (0.57 ± 0.03 versus 3.54 ± 0.19 pmol of phosphate incorporated into serine163 of the Bax peptide, respectively; P < 0.05; Figure 6A). In contrast, 10 μM TDZD-8 significantly reduced GSK3β-specific Bax serine163 phosphorylation during stress by >50% (3.54 ± 0.19 versus 1.72 ± 0.19 pmol of phosphate incorporated; P < 0.05 versus vehicle control). To support a pathogenic role of GSK3β in mediating Bax activation and apoptosis after stress, we assessed survival in cells in which Bax was suppressed before expressing constitutively active GSK3β (Figure 6B). Bax-specific RNAi (but not nonspecific RNAi) decreased Bax content (Figure 6B, immunoblot). Compared with control, nonspecific RNAi, Bax-specific RNAi, or GSK3β-S9A did not alter baseline cell survival (data not shown). GSK3β-S9A expression significantly decreased cell survival compared with control (44 ± 5 versus 69 ± 6%, respectively, Figure 6B). In contrast, suppressing Bax with specific RNAi resulted in almost 90% cell survival after stress. Interestingly, Bax suppression also significantly reduced cell death associated with GSK3β-S9A expression. In fact, Bax suppression almost completely prevented the loss of cell viability associated with overexpression of the constitutively active GSK3β mutant (81 ± 5 versus 44 ± 5% cell survival, respectively; P > 0.05).
Figure 6.

Effect of metabolic stress on Bax serine163 phosphorylation is shown. (A) 32[P] incorporation into a Bax-specific peptide containing serine163, the residue targeted by GSK3β in a cell-free system (see the Concise Methods section), at baseline, immediately after 60 minutes of ATP depletion, and in presence and absence of 10 μM TDZD-8. *P < 0.05 TDZD-8 versus vehicle; n = 5. (B) Survival (MTT assay) 8 hours after 60 minutes of ATP depletion in cells exposed to nonspecific RNAi plus empty vector adenovirus (NS-si+EV), constitutively active GSK3β (GSK3β-S9A), Bax-specific RNAi (Bax-siRNA), or constitutively active GSK3β plus Bax specific RNAi (GSK3β-S9A+Bax siRNA). *P < 0.05 versus no ATP depletion; †P < 0.05 versus ATP-depleted CTL, no GSK3β-S9A versus GSK3β-S9A; ‡P < 0.05 versus NS-si+EV or GSK3β-S9A; §P > 0.05 versus Bax siRNA alone; n = 5.
In renal cortical homogenates, transient renal ischemia in vivo caused virtually identical biochemical perturbations as were caused by metabolic stress in vitro. Specifically, ischemia activated GSK3β as indicated by a decrease in serine9 phosphorylation (Figure 7A, panel 1) but not in renal cortices harvested from either a sham-operated kidney with its renal artery encircled with a nonocclusive ligature or the nonischemic, contralateral kidney. Total GSK3β content did not change with any experimental maneuver (Figure 7, panel 2). Reperfusion for 30 minutes partially inactivated, whereas 60 minutes of reperfusion completely inactivated GSK3β (i.e., restored phospho-serine GSK3β content to the preischemic baseline level) in the sham kidney (data not shown). Administration of TDZD-8 (1 mg/kg intravenously) as a single dose 1 hour before ischemia blocked GSK3β kinase activity as indicated by a partial inhibition of stress-induced serine33,37 β-catenin phosphorylation (Figure 7, panel 3) but without altering total β-catenin content (Figure 7, panel 4). Ischemia also caused a dramatic increase in the accumulation of the 17-kD active caspase 3 cleavage product (Figure 7, panel 5) as well as marked Bax activation (Figure 7, panel 6). In contrast, TDZD-8 not only reduced Bax activation (Figure 7, panel 7) without altering total Bax content but also prevented the loss of immunoreactive Bcl2 after renal ischemia (Figure 7, panel 8). These results were confirmed by statistical analysis of densitometric data pooled from several immunoblot studies (Figure 7B).
Figure 7.

Effect of unilateral renal ischemia in the presence and absence of a GSK3β inhibitor on the apoptosis pathway is shown. (A) Effect of 30 minutes of renal ischemia on p-serine9 and total GSK3β content, p-serine33,37 and total β-catenin (a GSK3β substrate), active caspase 3, active and total Bax and Bcl-2; β-actin is a loading control (bottom). (B) Densitometric analysis of studies shown in A. *P < 0.05 versus non-uni-nephrectomy (Non-Unx); †P < 0.05 versus Unx control; n = 4.
Compared with nonischemic control, transient ischemia caused marked tubular injury, loss of brush border, tubular dilation, and intratubular cast formation (Figure 8A). In contrast, a single dose of TDZD-8 preserved tubular morphology after ischemic stress. Higher power magnification confirmed that TDZD-8 exposure per se did not alter the appearance of the cortical tubules at baseline but improved tubular morphology after ischemia (Figure 8B). Blinded analysis of multiple sections of renal cortex revealed that inhibition of GSK3β resulted in a marked decrease in several parameters of proximal tubule injury, including dilation, vacuolization, brush border loss, epithelial cell detachment, and intratubular cast formation (Table 1). A modest reduction in leukocyte infiltration and capillary edema were also detected in TDZD-8–treated animals. Importantly, inhibition of the apoptosis cascade and improvement in tubular morphology after ischemia resulted in a significant decrement in serum creatinine on days 1 through 6 after injury compared with vehicle-treated control (P < 0.03; n = 6; Figure 9). Creatinine peaked at 3.9 mg/dl on day 2 in vehicle-exposed versus 2.6 mg/dl in TDZD-8–treated animals. On the final day of observation, creatinine was 60% higher in the vehicle-exposed group. All 12 animals survived renal ischemia.
Figure 8.

Effect of renal ischemia in the presence and absence of a GSK3β inhibitor on renal histology is shown. (A) Hematoxylin- and eosin-stained sections harvested from the renal cortices of rats at baseline or 1 hour after 30 minutes of renal ischemia in the presence of vehicle (Ischemia) or TDZD-8 (Ischemia + TDZD-8). (B) Hematoxylin- and eosin-stained sections harvested from the renal cortices of rats at baseline in the presence of vehicle (Baseline) or TDZD-8 (Baseline + TDZD-8) and 1 hour after 30 minutes of renal ischemia (Ischemia versus Ischemia + TDZD-8). Magnifications: ×20 in A; ×40 in B.
Table 1.
Histologic injury score in renal cortical tissue sections obtained before ischemia (baseline), immediately after 30 minutes of renal artery occlusion (ischemia), and after 30 minutes of recovery
| Parameter | TDZD-8 |
|||||
|---|---|---|---|---|---|---|
| Baseline |
Ischemia |
Recovery |
||||
| Sham − | Sham + | − | + | − | + | |
| Proximal tubule | ||||||
| dilation | 0 | 0 | 1 | 0 | 2 to 3 | 1 |
| vacuolar changes | 0 to 1 | 0 | 0 | 0 | 1 to 2 | 0 |
| brush border loss | 0 to 1 | 0 | 1 | 1 | 3 to 4 | 1 to 2 |
| cell detachment | 0 | 0 | 1 | 0 | 1 to 2 | 0 to 1 |
| nuclear condensation | 0 | 0 | 0 | 0 | 0 to 1 | 0 |
| Intratubular casts | 0 | 0 | 1 to 3 | 0 | 2 to 4 | 0 |
| Interstitial edema | 0 | 0 | 0 | 0 | 1 | 1 |
| Leukocyte infiltration | 0 | 0 | 0 | 0 | 1 to 2 | 0 |
| Capillary edema | 0 | 0 to 1 | 1 | 1 | 1 to 2 | 0 |
Injury was scored in 10 randomly selected fields in each tissue section; n = 6 animals.
Figure 9.
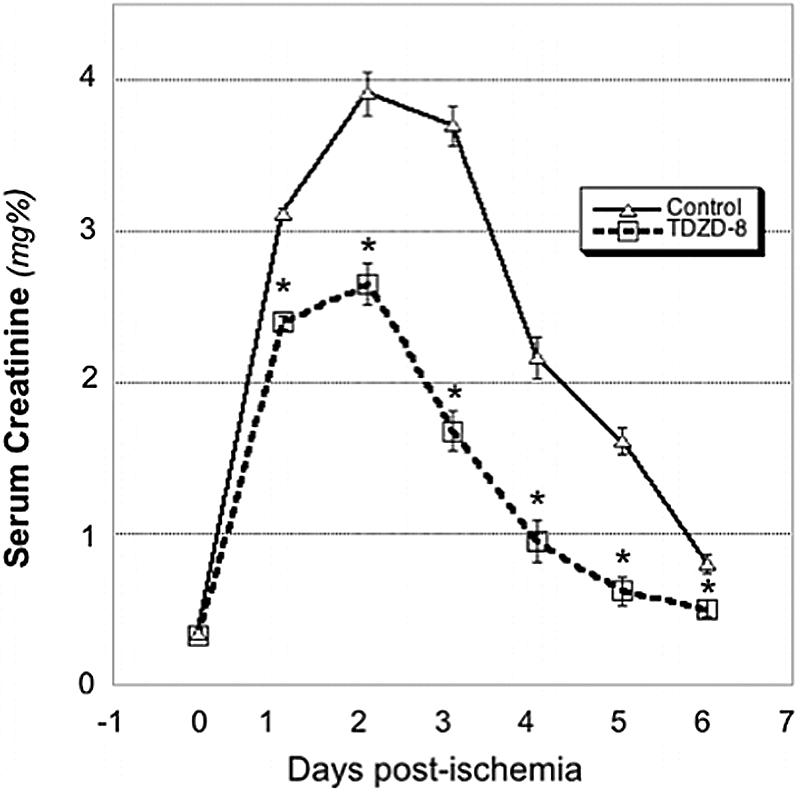
Effect of a GSK3β inhibitor on renal function after ischemia is shown. Serial serum creatinine levels in rats at baseline (Time 0) versus days 1 through 6 after 30 minutes of ischemia. *P < 0.03, vehicle versus TDZD-8; n = 6.
Discussion
A central feature of our in vitro and in vivo studies is the observation that stress transiently activates GSK3β, a multifunctional protein kinase that resides at the “biochemical intersection” between normal cell metabolism and cell survival. Although GSK3β affects inflammation and cell proliferation, it has recently been linked to apoptosis.27,33 Specifically, GSK3β affects BCL2 family proteins that regulate mitochondrial membrane permeabilization,45 a primary event in the apoptotic cell death pathway. In addition, Plotnikov et al.44 showed that renal ischemia or hypoxia increase reactive oxygen species formation, especially within renal tubules, reduced both mitochondrial membrane potential and respiration, and linked reactive oxygen species generation to GSK3β activation. This study extends those observations by identifying early, GSK3β-mediated events in the cell death pathway that link Bax-mediated mitochondrial injury to renal epithelial cell apoptosis and organ dysfunction.
How does GSK3β induce mitochondrial injury and apoptosis? Evidence shows that GSK3β directly phosphorylates and activates Bax. Using a site-directed mutation of Bax serine163, Linseman et al.42 showed that this residue is critical for activating Bax and for translocating it to mitochondria. In this study, both metabolic stress and pharmacologic GSK3β inhibition altered the ability of GSK3β to phosphorylate Bax at the serine163 site in our cell-free assay (Figure 4), suggesting that GSK3β and Bax activation are causally linked. Interestingly, AKT has been shown to phosphorylate serine184, inactivating Bax.46 By simultaneously inactivating AKT and activating GSK3β, both ATP depletion and ischemia induce a perfect ménage a trios relationship that involves both of these kinases and Bax in a manner that induces mitochondrial membrane injury and apoptosis. That expression of constitutively active GSK3β alone and in the absence of metabolic stress does not activate Bax (Figure 2) or alter cell survival suggests that maximal Bax activation requires additional events. This observation is consistent with reports that distinct Bax domains (and likely Bax phosphorylation events47) regulate the signals that retain Bax in the cytosol or mediate its translocation to mitochondria.48,49
The first committed step in the Bax activation pathway is conformational change, followed by Bax oligomerization and translocation to mitochondria.50 Each of these three steps was recently confirmed in epithelial cells subjected to metabolic stress.51 After translocation, Bax permeabilizes the outer mitochondrial membrane by either forming de novo pores or opening existing ones.52,53 Considerable controversy surrounds the mechanism by which “Bax attack” alters membrane permeability. Similarly, the specific membrane site targeted by Bax is debated.53,54 Interaction between Bax and the voltage-dependent anion channel (VDAC) has received considerable attention, because VDAC is a key component of the membrane pore transition (MPT) complex responsible for releasing both cytochrome c and apoptosis-inducing factor (and others) after an apoptogenic stress.55 Although the role of VDAC in MPT has been questioned using knockout of VDAC 1 and 3 and knockdown of VDAC 2,56 substantial evidence suggests that GSK3β regulates MPT. By phosphorylating VDAC and displacing hexokinase, a second MPT complex component, GSK3β, could “prepare” the VDAC-binding site for attack by Bax.26,57
Regardless of the mechanism, activated Bax is clearly mitotoxic, particularly when the balance between pro- and antiapoptotic proteins is perturbed.39,53 The degradation of Bcl2 during metabolic stress in vitro12 or renal ischemia in vivo (Figure 7) by caspase 3 not only eliminates an anti-Bax protein58 but also generates a potent “Bax-like” fragment.59,60 In the renal cortex, ischemia induced a >20-fold increase in the active Bax-Bcl2 ratio (Figure 7), a critical determinant of the “apoptotic set point.”39 GSK3β inhibition in vivo partially restores the Bax-Bcl2 ratio by inhibiting Bax activation as well as by preserving Bcl2.
Although renal failure after ischemia has traditionally been attributed to acute tubular necrosis, it is now clear that necrosis fails to account for the severity of organ impairment, especially in humans.2,3,61 This information increases the likelihood that other forms of cell injury mediate organ dysfunction.2,5 The contribution of apoptosis to acute organ injury is confounded by the evanescent nature of apoptotic cells and the stochastic nature of the apoptotic process. Despite these limitations, caspase inhibitors reduce renal impairment62 and selective caspase knockout improves organ function in the postischemic mouse,63 indicating that apoptosis contributes to ischemic renal failure. In this study, ischemia caused tubular dilation, accumulation of detached epithelial cells into the lumen, intratubular cast formation, and mild inflammation in the relative absence of frank tubular necrosis (Figure 8). As well, a GSK3β inhibitor afforded a greater reduction in the indices of tubular injury than it did to reduce inflammation (Table 1). The modest reduction in the inflammatory response observed in this study may be because GSK3β activation promotes the systemic inflammatory response, increases cytokine release, and alters cell proliferation required for recovery after diverse renal insults.27,32,33,64 In a murine model of LPS-induced acute renal injury, GSK3β activation stimulated NF-κB followed by chemokine (C-C motif) ligand 5 known as CCL5 or RANTES expression, inflammation, and epithelial cell apoptosis.27 Their report suggested that GSK3β exerts potent proinflammatory effects that contribute to organ injury. In addition, caspase activation itself promotes inflammation.62
Given that GSK3β manipulation regulates apoptosis in renal epithelial cells in vitro, we propose that GSK3β-mediated apoptosis is an important cause of renal tubular injury and organ dysfunction after an ischemic insult (Figure 9). The observation that TDZD-8 inhibits ischemia-induced GSK3β kinase activity and Bax and caspase 3 activation, reduces tissue injury, and improves organ function is consistent with this interpretation. Furthermore, a single dose of TDZD-8, with a half-life of only a few hours,65 not only inhibited premitochondrial events in the apoptosis cascade but also preserved organ function during prolonged recovery, suggesting that these events are intimately linked. However, we cannot exclude the possibility that TDZD-8 affords protection by means other than its effect on GSK3β or that it alters survival in other cell types within the kidney. Importantly, we show that GSK3β is a readily modifiable cell survival signal that determines renal function after an acute ischemic insult.
Concise Methods
Reagents
All reagents were purchased from Sigma (St. Louis, MO) unless otherwise indicated.
Antibodies
Rabbit polyclonal antibodies directed against phospho-serine9 GSK3β (inactive), total GSK3β, and active caspase-3 (Cell Signaling Technology, Beverly, MA), as well as Bcl2 (Calbiochem, San Diego, CA) were obtained. MAbs directed against fibrillarin (Abcam, Cambridge, MA) and β-actin were also obtained. Both total (5B7) and active (6A7) Bax antibodies were used (Trevingen, Gaithersburg, MD). Secondary antibodies conjugated to horseradish peroxidase (Jackson Immunoresearch Laboratories, West Grove, PA) were used in combination with an enhanced chemiluminescence detection method (Pierce, Rockford, IL).
Cell Culture
A conditionally immortalized renal epithelial cell line derived from the mouse proximal tubule (BUMPT) was cultured as described previously.66 These cells were maintained for up to 40 passages in DMEM with high-glucose medium (Life Technologies BRL, Carlsbad, CA) containing 10% bovine serum and 10% penicillin-streptomycin at 37°C in an incubator containing 5% CO2.
GSK3β Overexpression
A WT and an established, constitutively active GSK3β construct (GSK3β-S9A) with a serine residue at position 9 was mutated to alanine21,42 and introduced into BUMPT cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The expression level of each mutant in cell lysates was confirmed by immunoblot analysis.
Inhibition of GSK3β
Both molecular and pharmacologic maneuvers were used to inhibit GSK3β. RNAi was achieved with short hairpin RNAs (shRNAs) expressed from the feline immunodeficiency virus (FIV)-based single-promoter pSIF1-H1-copGFP vector that produces shRNAs under control of H1 promoter and to express copGFP under the control of hCMV promoter (System Biosciences, Mountain View, CA). Each shRNA duplex was designed with a CTTCCTGTCAGA loop. Three GSK3β shRNA constructs were targeted to the following sequences: (1) 5′-GCTAGACCACTGTAACATAGT-3′, (2) 5′-GCTGTGTGTTGGCTGAATTGT-3′, and (3) 5′-GCGGGACCCAAATGTCAAACT-3′. Control shRNAs included the missense sequence, the nonspecific sequence, and the pSIF1-H1-copGFP vector. All constructs were sequenced before FIV packaging. HEK293TN cells were co-transfected with the lentiviral FIV-GSK3β-shRNA constructs and packaging plasmids using Lipofectamine Plus (Invitrogen). Forty-eight hours after transfection, culture supernatant containing viral particles was harvested, clarified by filtration through a 0.45-μm membrane filter (Nalge Nunc, Rochester, NY), and then concentrated by PEG precipitation. Fifteen hours after infection, the IU of each shRNA virus was estimated from the number of copGFP-expressing BUMPT cells. CopGFP-positive cells were then visualized and counted in 10 randomly selected fields using fluorescence microscopy. Cells were infected with virus (5 IU/cell) for 4 hours in the presence of 5 μg/ml polybrene, and the medium containing virus particles was then exchanged for fresh medium. For infection with multiple viruses, cells were sequentially infected with a single virus for 4 hours followed by 4 hours of recovery, and the cycle was then repeated. Infected cells were cultured for 48 hours, and >90% of the population were GFP expression as assessed by direct visualization. The effect of each construct on total cell GSK3β content was assessed by immunoblot analysis. TDZD-8, with an IC50 of 4 μM, inhibits GSK3β by binding to its active site without altering the degree of serine9 phosphorylation.33,65 TDZD-8 dissolved in DMSO has been used to adequately and selectively inhibit GSK3β in in vitro (10 μM) as well and in vivo (1 mg/kg body wt, intravenously) studies.
Metabolic Stress
ATP content was reduced by incubating BUMPT cells in glucose-free medium that contained rotenone (10 μM), a complex 1 electron transport inhibitor.67,68 This maneuver reduces ATP content to <2% of control within 10 minutes, a response that is comparable to that of cyanide66 or antimycin.11 For initiation of recovery, rotenone-free medium containing dextrose (5 mM) and heptanoic acid (2 mM) was added to bypass the rotenone-induced block of oxidative phosphorylation by supplying reducing equivalents to complex 2 in the electron transport chain.68 As recently reported by our laboratory, metabolic stress for 1 to 2 hours causes apoptosis without appreciable epithelial cell necrosis.51
Hoechst Assay
For quantification of apoptosis, cells were grown on coverslips and then stained with Hoechst dye 33342 for 10 minutes at 37°C and fixed with 2% paraformaldehyde for 30 minutes at 25°C.12 The cells were mounted and then evaluated by ultraviolet immunofluorescence microscopy using established criteria for apoptosis.12
Cell Viability
Cell viability was assayed using a modified colorimetric technique that is based on the ability of live cells to convert 3-(4,5 dimethylthiazol)-2,5-diphenyl tetrazolium bromide (Promega, Madison WI), a tetrazolium compound, into purple formazan crystals as previously reported.23 The number of surviving cells is expressed as a percentage of viable control cells detected at baseline. This assay accurately reflects cell survival in a model in which apoptosis is the primary cause of cell death.12,23,51,69
Caspase 3 Activity
Enzyme activity was measured in cell lysates with a commercial kit (Enzolyte AFC Capsase-3 Assay Kit 71114; San Jose, CA) according to the manufacturer's instructions.
Immunoblot Analysis
Cells were harvested on ice in a lysis buffer containing 20 mM Tris-HCl, 140 mM NaCl, 1 mM sodium orthovanadate, 1 mM NaF, 1 mM dithiothreitol, 1 mM PMSF, 1 mM Na4P2O7, 0.5% sodium deoxycholate; 0.4% NP-40, and a protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN) at pH 7.5. Cell lysates were incubated for 15 min on ice and then centrifuged at 14,000 × g for 15 minutes at 4°C for collection of the supernatant. The protein concentration of the supernatant was estimated with the Bio-Rad assay (Bio-Rad, Hercules, CA). Samples (20 to 50 μg) were boiled in 1× SDS sample buffer (Boston Bioproducts, Ashland, MA) and separated using SDS-PAGE. Total and phosphorylated and kinase content were assessed in the same samples using immunoblots performed in parallel. β-Actin, total GSK3β, β-catenin, and/or Bax was used as a loading control when appropriate. Data from each immunoblot were digitally acquired, and individual band densities were automatically reported.
Bax Knockdown
RNAi was used to silence Bax gene expression. Cells were plated in six- or 48-well culture plates with complete medium and allowed to grow for 24 hours to achieve 70% confluence. Cells were then transfected with an siRNA set composed of three target-specific 20- to 25-nt siRNAs that target Bax mRNA (Santa Cruz Biotechnology, Santa Cruz, CA). A nonspecific siRNA (Santa Cruz Biotechnology) that does not lead to the specific degradation of any known cellular mRNA served as a control. A mixture of OptiMEM and LipofectAMINE was incubated for 5 minutes at 25°C and was then incubated with the siRNA (100 pmol/well in six-well plates; 10 pmol/well in 48-well plates) for 20 minutes at 25°C. The siRNA mixture was then added to each well according to the manufacturer's protocol. The medium was changed 16 hours after transfection and incubated for 6 hours before infection with GSK3β-S9A lentivirus (as detailed already). Bax gene silencing was confirmed by immunoblot analysis.
GSK3β-Mediated Bax Serine163 Phosphorylation
GSK3 was immunoprecipitated from 600 μg of protein harvested from cell lysates as described already by constant mixing with 5 μg of anti-GSK3β mAb and protein A–Sepharose for 2 hours at 4°C. The resulting immunoprecipitates were washed three times in kinase assay wash buffer (20 mM HEPES, 20 mM β-glycerophosphate, and 1 mM EDTA at pH 7.5) and then resuspended in 35 μl of kinase assay mixture containing 4 mM MOPS, 2.5 mM β-glycerophosphate, 1 mM EGTA, 0.4 mM EDTA, 4 mM MgCl2, 0.05 mM dithiothreitol, 40 ng/μl BSA, 50 μM ATP, 2.5 μM cAMP-dependent protein kinase inhibitor peptide (IP20), and 100 μM [γ-32P]ATP (PerkinElmer Life Sciences, Boston, MA) at pH 7.2 and a synthetic Bax-specific peptide that flanks serine163 substrate WEGLLSYFGTP (400 ng/μl). For each assay, 30 μl of this mixture was used in the presence or absence of TDZD-8 (10 μM). The assay was terminated after 30 minutes of incubation at 30°C by spotting onto P81 ion-exchange paper. The paper was washed four times in 0.6% phosphoric acid, and bound radioactivity was quantified by scintillation counting. A commercially available GSK3β phosphorylation substrate (Upstate Biotech, Lake Placid, NY) was used to optimize the reaction conditions.
In Vivo Renal Ischemia
Male Sprague-Dawley rats that weighed 250 to 270 g each were anesthetized with thiopental sodium (55 mg/kg, intraperitoneally), a midline laparotomy was performed, and the left kidney was removed. A nontraumatic vascular clamp was placed on the right renal artery for 30 minutes, an insult that produces severe, reversible acute renal failure.70 Controls were subjected to either uni-nephrectomy (Unx or Unx CTL) or sham renal ischemia using a nonocclusive ligature without nephrectomy (Non-Unx). After transient ischemia, the clamp was removed and perfusion was confirmed by visual inspection. At serial time points after ischemia, the right kidney was harvested; the capsule was removed; the renal cortex was bluntly dissected into cortex and outer and inner medullas; and the tissues were prepared for immunoblot analysis to assess the content of Bcl2, active and total Bax, phospho- and total GSK3β, phospho and total β-catenin, and active caspase 3. Renal histology was examined in stained in 5-μm tissue sections. In a subsequent study, animals received either a single intravenous dose of 1 mg/kg TDZD-8 dissolved in DMSO 1 hour before ischemia32 or vehicle alone (control). These observations provided a physiologic basis for administering TDZD-8, a GSK3β inhibitor with an IC50 of 4 μM,33 as a single dose (1 mg/kg, intravenously) before ischemia, as reported by Dugo et al.32 in a rodent sepsis model. An observer who was blinded to the experimental conditions (R.B.) assessed the degree of tissue injury using a standardized, semiquantitative scoring scheme with four levels of severity for each of nine distinct categories on kidney sections stained with hematoxylin and eosin.71,72 Serum creatinine in tail-vein samples was measured at baseline and daily for 6 days after ischemia using a commercial kit (BioAssay Systems, Hayward, CA). All procedures were performed in adherence with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Statistical Analysis
Comparison of two groups was performed using a two-tailed t test. Differences among three or more groups were assessed using two-tailed ANOVA (Excel, Microsoft Corp., Redmond, WA). An experimental result was considered significant at P < 0.05.
Disclosures
None.
Acknowledgments
This work was supported by National Institutes of Health research grants (RO-1) DK-52898 (J.H.S.) and DK-53387 (S.C.B.) and a James A. Scherbenske Award from the American Society of Nephrology (S.C.B.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “GSK3β Plays Dirty in Acute Kidney Injury,” on pages 199–200.
References
- 1.Ueda N, Shah SV: Tubular cell damage in acute renal failure-apoptosis, necrosis, or both. Nephrol Dial Transplant 15: 318–323, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Bonegio R, Lieberthal W: Role of apoptosis in the pathogenesis of acute renal failure. Curr Opin Nephrol Hypertens 11: 301–308, 2002 [DOI] [PubMed] [Google Scholar]
- 3.Lieberthal W, Koh JS, Levine JS: Necrosis and apoptosis in acute renal failure. Semin Nephrol 18: 505–518, 1998 [PubMed] [Google Scholar]
- 4.Versteilen AM, Di Maggio F, Leemreis JR, Groeneveld AB, Musters RJ, Sipkema P: Molecular mechanisms of acute renal failure following ischemia/reperfusion. Int J Artif Organs 27: 1019–1029, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Ortiz A, Justo P, Catalan MP, Sanz AB, Lorz C, Egido J: Apoptotic cell death in renal injury: The rationale for intervention. Curr Drug Targets Immune Endocr Metabol Disord 2: 181–192, 2002 [PubMed] [Google Scholar]
- 6.Sheridan AM, Bonventre JV: Cell biology and molecular mechanisms of injury in ischemic acute renal failure. Curr Opin Nephrol Hypertens 9: 427–434, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Vincent JL, Bota DP, De Backer D: Epidemiology and outcome in renal failure. Int J Artif Organs 27: 1013–1018, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Basile DP, Liapis H, Hammerman MR: Expression of bcl-2 and bax in regenerating rat renal tubules following ischemic injury. Am J Physiol 272: F640–F647, 1997 [DOI] [PubMed] [Google Scholar]
- 9.Schumer M, Colombel MC, Sawczuk IS, Gobe G, Connor J, O'Toole KM, Olsson CA, Wise GJ, Buttyan R: Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol 140: 831–838, 1992 [PMC free article] [PubMed] [Google Scholar]
- 10.Gobe G, Zhang XJ, Willgoss DA, Schoch E, Hogg NA, Endre ZH: Relationship between expression of Bcl-2 genes and growth factors in ischemic acute renal failure in the rat. J Am Soc Nephrol 11: 454–467, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Lieberthal W, Menza SA, Levine JS: Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am J Physiol 274: F315–F327, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Knowlton AA, Christensen TG, Shih T, Borkan SC: Prior heat stress inhibits apoptosis in adenosine triphosphate-depleted renal tubular cells. Kidney Int 55: 2224–2235, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Frame S, Cohen P: GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson WJ, Nusse R: Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303: 1483–1487, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loberg RD, Vesely E, Brosius FC, 3rd: Enhanced glycogen synthase kinase-3beta activity mediates hypoxia-induced apoptosis of vascular smooth muscle cells and is prevented by glucose transport and metabolism. J Biol Chem 277: 41667–41673, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Ferkey DM, Kimelman D: GSK-3: New thoughts on an old enzyme. Dev Biol 225: 471–479, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Grimes CA, Jope RS: The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 65: 391–426, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Jope RS, Johnson GV: The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Doble BW, Woodgett JR: GSK-3: Tricks of the trade for a multi-tasking kinase. J Cell Sci 116: 1175–1186, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farooqui R, Zhu S, Fenteany G: Glycogen synthase kinase-3 acts upstream of ADP-ribosylation factor 6 and Rac1 to regulate epithelial cell migration. Exp Cell Res 312: 1514–1525, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Sanchez JF, Sniderhan LF, Williamson AL, Fan S, Chakraborty-Sett S, Maggirwar SB: Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol Cell Biol 23: 4649–4662, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novak A, Hsu SC, Leung-Hagesteijn C, Radeva G, Papkoff J, Montesano R, Roskelley C, Grosschedl R, Dedhar S: Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc Natl Acad Sci U S A 95: 4374–4379, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinha D, Wang Z, Ruchalski KL, Levine JS, Krishnan S, Lieberthal W, Schwartz JH, Borkan SC: Lithium activates the Wnt and phosphatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am J Physiol Renal Physiol 288: F703–F713, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Rao R, Hao CM, Breyer MD: Hypertonic stress activates glycogen synthase kinase 3beta-mediated apoptosis of renal medullary interstitial cells, suppressing an NFkappaB-driven cyclooxygenase-2-dependent survival pathway. J Biol Chem 279: 3949–3955, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Mora A, Sabio G, Risco AM, Cuenda A, Alonso JC, Soler G, Centeno F: Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cell Signal 14: 557–562, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ: Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113: 1535–1549, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Huang WC, Wang CY, Tsai CC, Chen CL, Chang YT, Kai JI, Lin CF: Inhibiting glycogen synthase kinase-3 reduces endotoxaemic acute renal failure by down-regulating inflammation and renal cell apoptosis. Br J Pharmacol 157: 1004–1013, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong H, Imahashi K, Steenbergen C, Murphy E: Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase–dependent pathway is cardioprotective. Circ Res 90: 377–379, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Kovacs K, Toth A, Deres P, Kalai T, Hideg K, Gallyas F, Jr, Sumegi B: Critical role of PI3-kinase/Akt activation in the PARP inhibitor induced heart function recovery during ischemia-reperfusion. Biochem Pharmacol 71: 441–452, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Park SS, Zhao H, Mueller RA, Xu Z: Bradykinin prevents reperfusion injury by targeting mitochondrial permeability transition pore through glycogen synthase kinase 3beta. J Mol Cell Cardiol 40: 708–716, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Li XL, Man K, Ng KT, Sun CK, Lo CM, Fan ST: The influence of phosphatidylinositol 3-kinase/Akt pathway on the ischemic injury during rat liver graft preservation. Am J Transplant 5: 1264–1275, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala EM, Louhelainen M, Foster SJ, Yaqoob MM, Thiemermann C: GSK-3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med 33: 1903–1912, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Obligado SH, Ibraghimov-Beskrovnaya O, Zuk A, Meijer L, Nelson PJ: CDK/GSK-3 inhibitors as therapeutic agents for parenchymal renal diseases. Kidney Int 73: 684–690, 2008 [DOI] [PubMed] [Google Scholar]
- 34.Collino M, Thiemermann C, Mastrocola R, Gallicchio M, Benetti E, Miglio G, Castiglia S, Danni O, Murch O, Dianzani C, Aragno M, Fantozzi R: Treatment with the glycogen synthase kinase-3beta inhibitor, TDZD-8, affects transient cerebral ischemia/reperfusion injury in the rat hippocampus. Shock 30: 299–307, 2008 [DOI] [PubMed] [Google Scholar]
- 35.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K: Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol 43: 564–570, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Cuzzocrea S, Mazzon E, Esposito E, Muia C, Abdelrahman M, Di Paola R, Crisafulli C, Bramanti P, Thiemermann C: Glycogen synthase kinase-3beta inhibition attenuates the development of ischaemia/reperfusion injury of the gut. Intensive Care Med 33: 880–893, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Diehl JA, Cheng M, Roussel MF, Sherr CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12: 3499–3511, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maniatis T: A ubiquitin ligase complex essential for the NF-kappaB, Wnt/Wingless and Hedgehog signaling pathways. Genes Dev 13: 505–510, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN: Bcl-2/Bax: A rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol 4: 327–332, 1993 [PubMed] [Google Scholar]
- 40.Chiang-Ting C, Tzu-Ching C, Ching-Yi T, Song-Kuen S, Ming-Kuen L: Adenovirus-mediated bcl-2 gene transfer inhibits renal ischemia/reperfusion induced tubular oxidative stress and apoptosis. Am J Transplant 5: 1194–1203, 2005 [DOI] [PubMed] [Google Scholar]
- 41.Kelly KJ, Sutton TA, Weathered N, Ray N, Caldwell EJ, Plotkin Z, Dagher PC: Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am J Physiol Renal Physiol 287: F760–F766, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA: Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 24: 9993–10002, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jope RS: Lithium and GSK-3: One inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci 24: 441–443, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Plotnikov EY, Kazachenko AV, Vyssokikh MY, Vasileva AK, Tcvirkun DV, Isaev NK, Kirpatovsky VI, Zorov DB: The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int 72: 1493–1502, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR: Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21: 749–760, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM: Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 279: 21085–21095, 2004 [DOI] [PubMed] [Google Scholar]
- 47.del Mar Martinez-Senac M, Corbalan-Garcia S, Gomez-Fernandez JC: Conformation of the C-terminal domain of the pro-apoptotic protein Bax and mutants and its interaction with membranes. Biochemistry 40: 9983–9992, 2001 [DOI] [PubMed] [Google Scholar]
- 48.Suzuki M, Youle RJ, Tjandra N: Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell 103: 645–654, 2000 [DOI] [PubMed] [Google Scholar]
- 49.Cartron PF, Arokium H, Oliver L, Meflah K, Manon S, Vallette FM: Distinct domains control the addressing and the insertion of Bax into mitochondria. J Biol Chem 280: 10587–10598, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Hsu YT, Wolter KG, Youle RJ: Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A 94: 3668–3672, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Havasi A, Li Z, Wang Z, Martin JL, Botla V, Ruchalski K, Schwartz JH, Borkan SC: Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem 283: 12305–12313, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zamzami N, Kroemer G: Apoptosis: Mitochondrial membrane permeabilization: The (w)hole story? Curr Biol 13: R71–R73, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Chipuk JE, Bouchier-Hayes L, Green DR: Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ 13: 1396–1402, 2006 [DOI] [PubMed] [Google Scholar]
- 54.Bellot G, Cartron PF, Er E, Oliver L, Juin P, Armstrong LC, Bornstein P, Mihara K, Manon S, Vallette FM: TOM22, a core component of the mitochondria outer membrane protein translocation pore, is a mitochondrial receptor for the proapoptotic protein Bax. Cell Death Differ 14: 785–794, 2007 [DOI] [PubMed] [Google Scholar]
- 55.Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y: Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol 152: 237–250, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD: Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550–555, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pastorino JG, Hoek JB, Shulga N: Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res 65: 10545–10554, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Shimizu S, Konishi A, Kodama T, Tsujimoto Y: BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A 97: 3100–3105, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM: Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278: 1966–1968, 1997 [DOI] [PubMed] [Google Scholar]
- 60.Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM: Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem 274: 21155–21161, 1999 [DOI] [PubMed] [Google Scholar]
- 61.Oberbauer R, Schwarz C, Regele HM, Hansmann C, Meyer TW, Mayer G: Regulation of renal tubular cell apoptosis and proliferation after ischemic injury to a solitary kidney. J Lab Clin Med 138: 343–351, 2001 [DOI] [PubMed] [Google Scholar]
- 62.Melnikov VY, Faubel S, Siegmund B, Lucia MS, Ljubanovic D, Edelstein CL: Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J Clin Invest 110: 1083–1091, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA, Edelstein CL: Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int 66: 2202–2213, 2004 [DOI] [PubMed] [Google Scholar]
- 64.Dugo L, Collin M, Thiemermann C: Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock 27: 113–123, 2007 [DOI] [PubMed] [Google Scholar]
- 65.Evenson AR, Fareed MU, Menconi MJ, Mitchell JC, Hasselgren PO: GSK-3beta inhibitors reduce protein degradation in muscles from septic rats and in dexamethasone-treated myotubes. Int J Biochem Cell Biol 37: 2226–2238, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Sinha D, Wang Z, Price VR, Schwartz JH, Lieberthal W: Chemical anoxia of tubular cells induces activation of c-Src and its translocation to the zonula adherens. Am J Physiol Renal Physiol 284: F488–F497, 2003 [DOI] [PubMed] [Google Scholar]
- 67.Shang T, Joseph J, Hillard CJ, Kalyanaraman B: Death-associated protein kinase as a sensor of mitochondrial membrane potential: Role of lysosome in mitochondrial toxin-induced cell death. J Biol Chem 280: 34644–34653, 2005 [DOI] [PubMed] [Google Scholar]
- 68.Doctor RB, Bacallao R, Mandel LJ: Method for recovering ATP content and mitochondrial function after chemical anoxia in renal cell cultures. Am J Physiol 266: C1803–C1811, 1994 [DOI] [PubMed] [Google Scholar]
- 69.Ruchalski K, Mao H, Li Z, Wang Z, Gillers S, Wang Y, Mosser DD, Gabai V, Schwartz JH, Borkan SC: Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem 281: 7873–7880, 2006 [DOI] [PubMed] [Google Scholar]
- 70.Lieberthal W, Fuhro R, Andry CC, Rennke H, Abernathy VE, Koh JS, Valeri R, Levine JS: Rapamycin impairs recovery from acute renal failure: Role of cell-cycle arrest and apoptosis of tubular cells. Am J Physiol Renal Physiol 281: F693–F706, 2001 [DOI] [PubMed] [Google Scholar]
- 71.Goujon JM, Hauet T, Menet E, Levillain P, Babin P, Carretier M: Histological evaluation of proximal tubule cell injury in isolated perfused pig kidneys exposed to cold ischemia. J Surg Res 82: 228–233, 1999 [DOI] [PubMed] [Google Scholar]
- 72.Yin M, Zhong Z, Connor HD, Bunzendahl H, Finn WF, Rusyn I, Li X, Raleigh JA, Mason RP, Thurman RG: Protective effect of glycine on renal injury induced by ischemia-reperfusion in vivo. Am J Physiol Renal Physiol 282: F417–F423, 2002 [DOI] [PubMed] [Google Scholar]