

Abstract
To obtain comprehensive information on 17β-estradiol (E2) sensitivity of genes that are inducible or suppressible by this hormone, we designed a method that determines ligand sensitivities of large numbers of genes by using DNA microarray and a set of simple Perl computer scripts implementing the standard metric statistics. We used it to characterize effects of low (0–100 pM) concentrations of E2 on the transcriptome profile of MCF7/BUS human breast cancer cells, whose E2 dose-dependent growth curve saturated with 100 pM E2. Evaluation of changes in mRNA expression for all genes covered by the DNA microarray indicated that, at a very low concentration (10 pM), E2 suppressed ≈3–5 times larger numbers of genes than it induced, whereas at higher concentrations (30–100 pM) it induced ≈1.5–2 times more genes than it suppressed. Using clearly defined statistical criteria, E2-inducible genes were categorized into several classes based on their E2 sensitivities. This approach of hormone sensitivity analysis revealed that expression of two previously reported E2-inducible autocrine growth factors, transforming growth factor α and stromal cell-derived factor 1, was not affected by 100 pM and lower concentrations of E2 but strongly enhanced by 10 nM E2, which was far higher than the concentration that saturated the E2 dose-dependent growth curve of MCF7/BUS cells. These observations suggested that biological actions of E2 are derived from expression of multiple genes whose E2 sensitivities differ significantly and, hence, depend on the E2 concentration, especially when it is lower than the saturating level, emphasizing the importance of characterizing the ligand dosedependent aspects of E2 actions.
Estrogens bind to estrogen receptors (ERs), which belong to the steroid receptor family of transcription factors, and the liganded ERs activate or suppress transcription of genes by recruiting coactivator or corepressor proteins. ERs bind directly to genomic DNA sequences that are known as estrogen response elements, or they interact with genomic DNA indirectly through other DNA-binding proteins such as AP-1 or Sp1. ERs also interact with key components of other signal transduction pathways such as the Src tyrosine kinase or phosphatidyl inositol 3-kinase, affecting gene expression indirectly through these pathways (1). Moreover, changes in gene expression that occur as primary responses to estrogens may exert secondary influences on expression of other genes. Thus, the majority of the biological actions of estrogens are derived from, or at least associated with, induction or suppression of certain sets of genes, even when such actions are considered indirect or nongenomic.
To characterize estrogen effects on gene expression, several laboratories have used DNA microarrays and determined the transcriptome profiles of estrogen-dependent human breast cancer cells cultured in the presence or absence of 17β-estradiol (E2) (2–4). In these experiments, cells were subjected to hormone starvation for up to 5 days and then stimulated with 1–10 nM E2, which are high concentrations that usually saturate the E2 dose-dependent growth curves of ER-positive breast cancer cell cultures (5, 6). Therefore, the changes in transcriptome profiles reported in these previous studies, each of which handled only a single, high E2 concentration, were likely reflecting the maximum or near-maximum E2 effects. However, information on E2 dose-dependent effects at a lower, unsaturated range of concentrations would be critically important to understand estrogen actions in vivo, where cells are unlikely to be exposed to saturating concentrations of estrogens continuously.
The E2 sensitivity of a gene may be characterized by three aspects of the E2 dose-dependent profiles of the mRNA expression, namely, (i) the minimum E2 concentration required to induce or suppress expression of mRNA transcripts significantly, (ii) the E2 concentration that saturates E2 effects on mRNA expression, and (iii) the shape of the E2 dose–response curve for mRNA expression (i.e., linear, convex, or concave). The E2 sensitivity thus characterized may be determined by multiple factors: affinity of E2 to each of the two isoforms of ERs (i.e., ERα and ERβ), affinity of the liganded ERs to estrogen response element (EREs) (7, 8), location of the EREs in the transcriptional control sequences of genomic DNA (8), and availability of transcriptional coactivators and cosuppressors that are recruited to the transcriptional protein complexes formed around the liganded ERs (9). When the E2 sensitivity differs among genes, effects of a subsaturating concentration of E2, which would involve only a part of the whole repertoire of the E2-regulatable genes, may be different from the effect of a saturating concentration of estrogen not only quantitatively but also qualitatively. To examine such possibilities, it is necessary to develop a method that characterizes the quantitative aspects of E2 sensitivity of a large number of genes with a reasonable precision and a statistical basis.
In the present study, we applied the Affymetrix high-density oligonucleotide DNA microarray to determine the ligand sensitivity of E2-inducible and E2-suppressible genes in ER-positive MCF7/BUS human breast cancer cells. Using the Perl programming language, we developed a set of simple computer scripts that processed the DNA microarray data and characterized E2 sensitivity of each gene, implementing a standard test of metric statistics.
Materials and Methods
Cell Culture. MCF7/BUS cells (5, 10) were provided by A. M. Soto and C. Sonnenschein (Tufts University, Boston) and maintained in DMEM (4.5 mg/liter glucose) supplemented with 5% FCS that contained bioactive estrogens equivalent to ≈60 pM E2 (HyClone, defined grade) in xenoestrogen-free plastic ware (Corning). Their E2 dose-dependent growth curves were drawn following the e-screen protocol (5, 10). To determine E2 effects on the transcriptome, cells were washed three times with phenol red-free DMEM and cultured for 48 h in the presence of varying concentrations of E2; in these experiments, medium was supplemented with 5% charcoal/dextran-stripped FCS (HyClone) that did not contain significant amounts of steroid hormones. Throughout the experiments, all plastic ware was carefully selected to avoid xenoestrogen contamination.
DNA Microarray Experiment and RT-PCR. Total RNA samples were isolated from cell cultures by using a RNeasy mini-kit (Qiagen, Chatsworth, CA), and their amounts, purity, and integrity were evaluated by UV spectrophotometry and a RNA-nano Bioanalyzer (Agilent, Palo Alto, CA). Solid-phase archives of all RNA samples described in this study will be provided to investigators on request. Probe synthesis and hybridization of human U-133A GeneChip DNA microarrays (Affymetrix, Santa Clara, CA) were performed following the manufacturer's instructions. Gene expression data (CHP file of Affymetrix microarray suite 5.0 software) were normalized to a global target intensity of 500; by our hands, the background noise intensity was <50. RT-PCR primers and conditions for semiquantitative detection of E2-responsive genes are available on request.
Microarray Data Analysis. A master data set spreadsheet for E2 dose-dependent gene expression was generated from the CHP files by microarray suite 5.0 and converted into a tabdelimited text file. Each row of this text file represented a single gene and consisted of gene name, Affymetrix ID number, and normalized signal intensities of five repeated sets of E2 dose–response experiments. Each set of the E2 dose–response experiments consisted of data for 0, 10, 30, 60, and 100 pM E2. About 22,000 rows in this file represented all genes covered by the human U-133A chip.
When e(i, j) is a normalized signal intensity of a gene in experiment number i at E2 concentration of j [i = 1, 2, 3, 4, 5; j = 0, 10, 30, 60, 100 (pM)], distribution of E defined in the following equation approximates to the normal distribution (11, 12). This approximation, although not mathematically rigorous, is usually satisfactory for practical purposes (11, 12):
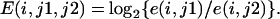 |
Because E is 0 when e(j1) = e(j2), and because E is > 0 when e(j1) is > e(j2), a single-tail Student's t test for a null hypothesis,
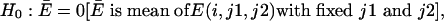 |
determines whether e(j1) is significantly greater than e(j2) or not.
Using the programming language Perl (13), simple computer scripts were developed to calculate E(i, j1, j2) for all combinations of (i, j1, j2) and to perform the above t test. Separate scripts were produced for each cutoff P value (P = 0.05, 0.01, 0.001) and each fold-increase cutoff number f. Thus, a script S(p, f) read a row from the master data set text file and copied it to another text file named (p, f, j1, j2) if e(j1) was significantly greater than e(j2) (P value < p) and e(j1) was more than f-fold greater than e(j2). Therefore, genes whose e(j1) was smaller than e(j2) were copied to file (p, f, j2, j1). The script then read the next row and repeated the process until it reached to the end of the master data set file, and text files (p, f, j1, j2) were generated for all possible combinations of the four parameters. Gene expression data for the hormone starvation experiments were processed similarly.
Computer-Aided Knowledge Extraction. Medline citations that relate E2-responsive genes to estrogen, cancer, E2F1, or cell cycle were extracted from public and commercial literature databases by the opus knowledge extraction engine (X-Mine), which discovers meaningful relationships between gene names and the key words even when these do not appear in the title or abstract of a single publication. Extracted literature was verified manually to confirm the suggested relationships.
Results
Determination of Numbers of Genes Induced or Suppressed by Varying Low Concentrations of E2. The BUS subline of MCF7 human breast cancer cells has been used widely for quantitative detection of low concentrations of natural and xenobiotic estrogens (5, 10). Consistent with previous studies (5, 10), BUS cells showed a highly reproducible E2 dose-dependent growth curve that showed a significant response with as low as 2 pM E2 and saturated with 80–100 pM E2 (Fig. 1A). The growth-stimulating effect of 100 pM E2 was completely blocked by pure antiestrogen ICI182,780 (100 nM; data not shown), indicating that E2 binding to ERs is necessary for the BUS cell growth. Western blotting detected ERα protein, but not ERβ, in BUS cells and in an MCF7 cell stock obtained from the American Type Culture Collection (data not shown). In the present study, BUS cells were not subjected to hormone starvation before E2 treatment because, in a carefully controlled xenoestrogen-free cell culture environment, the viability of BUS cells was severely impaired after complete hormone starvation beyond 48 h.
Fig. 1.

Global effects of E2 on MCF7/BUS cell growth and transcriptome. (A) E2 dose-dependent growth profile of MCF7/BUS cells. Cells were cultured in the presence of indicated concentrations of E2 for 120 h, and cell numbers were determined. Each point represents mean ± SEM of five independent experiments. (B and C) Global profiles of sensitivities of E2-induced (B) and E2-suppressed (C) genes. BUS cells were exposed to varying concentrations of E2 for 48 h, and their transcriptome profiles were determined by DNA microarray. Columns indicate numbers of genes whose strength of mRNA expression was stronger (B) or weaker (C)[>2-fold difference and P < 0.01 (n = 5)] in the presence of E2 concentrations shown in the x axis than in the presence of the color-coded E2 concentrations.
To obtain comprehensive information on effects of subsaturating concentrations of E2 on gene expression, BUS cells were exposed to 0, 10, 30, 60, and 100 pM E2 for 48 h, and their transcriptome profiles were determined by DNA microarray. Data from five independent repeats of this experiment were compiled in a text file format, and the mean ± SEM of the signal intensity, which reflected the amount of the mRNA transcript, was calculated for each gene and each E2 concentration. The mean signal intensities were then compared between two E2 concentrations (denoted as j1 and j2), and genes whose strength of expression was significantly stronger when E2 concentration was j1 compared with j2 (cutoff P value ≤ p) with at least f-fold changes were copied from this text file to another text file labeled with the four parameters as (p, f, j1, j2) (P = 0.05, 0.01, or 0.001; f = 1, 2, or 4; j1, j2 = 0, 10, 30, 60, or 100 pM). Thus, a text file (p, f, j1, j2) lists E2-inducible genes when j1 > j2; on the contrary, it lists E2-suppressible genes when j1 < j2. A set of simple Perl scripts developed in our laboratory generated these gene lists for all possible combinations of the four parameters. Effects of hormone starvation on gene expression were analyzed similarly by using a DNA microarray data set from three independent repeats of hormone starvation experiments, and another class of gene lists (p, f, d1, d2), where d1 and d2 represented starvation periods (0, 1, or 2 days), were generated. Importantly, a gene list (p, f, j1, j2) is not identical to a list generated merely by subtracting one gene list (p, f, j2, 0 pM) from another gene list (p, f, j1, 0 pM). A gene list generated by subtraction between any two other gene lists will not be associated with meaningful parameters p or f.
To understand the global aspects of E2 effects on the transcriptome, we determined numbers of genes induced or suppressed significantly in the presence of one concentration of E2 (j1 pM) compared with another (j2 pM; j1 > j2). Fig. 1 B and C shows examples of such analyses (P = 0.01, f = 2; j1 is indicated by the x axis, and j2 is indicated by the color-coded inset). When j2 = 0 pM (red columns), numbers of both E2-inducible genes (Fig. 1B) and E2-suppressible genes (Fig. 1C) increased sharply along with the increasing j1, as expected. However, when j2 = 10 pM (purple columns), numbers of E2-inducible and suppressible genes were remarkably smaller than those with j2 = 0 pM, although they still showed steady increase along with increasing j1. When j2 = 30 pM (blue columns) or 60 pM (dark blue columns), numbers of E2-inducible or suppressible genes were trivial. Similar results were obtained with several different cutoff values for parameters p and f. These observations may imply that preexposing cells to very low concentrations of E2 (≤ 10 pM) might exert a significant influence on the efficiency of higher concentrations of E2 (such as 100 pM) to induce or suppress genes.
Fig. 1 B and C also shows that overall profiles of the numbers of E2-inducible and E2-suppressible genes were comparable, with the former being ≈1.5- to 2-fold greater than the latter. This feature was conserved with different cutoff P values (P = 0.05 or 0.001; data not shown), suggesting that E2 is as potent a gene suppressor as it is an inducer. However, the numbers of genes induced or suppressed with 10 pM E2 compared with no E2 were exceptions. When three different cutoff P values (P = 0.05, 0.01, and 0.001) were applied, the number of E2-inducible and E2-suppressible genes were 190 vs. 602, 38 vs. 122, and 3 vs. 16, respectively, thus consistently indicating that greater numbers of genes were suppressed than induced with this lowest-end E2 concentration. Although the present analysis does not provide information on the mechanistic aspects of gene induction and suppression by E2, it is tempting to speculate that these observations could imply that recruitment of transcriptional corepressors to liganded ERα might be more efficient than recruitment of coactivators when the E2 concentration is strictly limited.
Characterization of E2 Sensitivity of E2-Inducible and E2-Suppressible Genes. We next characterized the E2 sensitivity of each E2-inducible and E2-suppressible gene by using gene lists (P = 0.05, f = 4, j1, 0 pM) and (P = 0.05, f = 4, d1, 0 day) (j1 = 0, 10, 30, 60, 100 pM; d1 = 0, 1, 2 days). As shown in Fig. 2, mRNA expression of E2-inducible genes was enhanced with increasing E2 concentrations and, conversely, suppressed during hormone starvation. These genes were categorized into four classes of E2 sensitivity, namely, genes showing (i) significant (P < 0.01) and >2-fold induction with 10 pM E2 (class A); (ii) significant (P < 0.05) but <2-fold induction with 10 pM E2 (class B); (iii) significant (P < 0.05) induction with 30 pM E2 but not with 10 pM E2, although there were slight increases in amounts of mRNA transcripts with 10 pM E2 (class C); and (iv) no apparent changes in amounts of mRNA transcripts with 10 pM E2 (class D). Class E included several known E2 target genes that were not identified as E2-inducible genes with the strict criteria used to define other classes. Similarly, the mRNA expression profiles of E2-suppressible genes (class F) showed reduction along with increasing E2 concentration and enhancement during hormone starvation. Thus, Fig. 2 concisely shows the three attributes of E2 sensitivity, namely, the minimum E2 concentration required for significant gene induction or suppression, the E2 concentration that saturates transcriptional effects, and the shape of the E2 dose–response curve of gene expression. Moreover, computeraided knowledge extraction demonstrated that many E2-regulated genes identified in the present study have been reported as genes related to cancer and/or cell cycle regulation (Fig. 2, codes).
Fig. 2.

E2 sensitivity analysis. Color diagram shows induction (red) and suppression (green) of genes with E2 treatment and hormone starvation. Numbers in the diagram indicate statistical significance of induction or suppression determined by Student's t test; namely, 1, 2, and 3 indicate P < 0.05, P < 0.01, and P < 0.001, respectively. Each column represents one experimental condition, and each row represents a single gene, whose name and GenBank accession number are shown. The codes indicate known relationships of each gene with estrogen, cancer, cell cycle, and E2F family transcription factors retrieved from databasesby a computer-aided knowledge extraction engine and denoted by symbols Es, Ca, Cy, and E2F, respectively. (A–F) Classes A–F are defined based on the E2 sensitivity of genes, and details of the mRNA expression profiles of representative genes for each class are presented as paired line graphs. The left of each pair (filled symbols) shows the E2 dose–response profile (n = 5); right graphs (open symbols) shows profile of hormone starvation (n = 3). Each data point represents mean ± SEM. Single, double, and triple arrowheads indicate P ≤ 0.05, 0.01, and 0.001, respectively. Detailed mRNA expression profiles of all of the genes shown in the color diagram are in Fig. 4, which is published as supporting information on the PNAS web site.
E2 Sensitivity Analysis Revealed Dissociation Between E2 Induction of Autocrine Growth Factors and E2 Dose-Dependent Growth Profile of MCF7/BUS Cells. Transforming growth factor-α (TGF-α) and stromal cell derived factor-1 (SDF-1) have been reported as E2-inducible autocrine growth factors that support E2-dependent growth of MCF7 cells (14, 15). Therefore, the apparent absence of induction of the mRNA transcripts for these genes with up to 100 pM E2 (Fig. 2, class E) seemed contradictory because MCF7/BUS cell growth was enhanced with 2–80 pM E2 (Fig. 1 A). Semiquantitative RT-PCR analysis, which was performed with limited cycles of amplification that did not reach to saturation, confirmed E2 dose-dependent induction and hormone starvation-dependent reduction of the mRNA transcripts for well-established E2 target genes CTSD/Cathepsin D and WISP2, whereas expression of the mRNA transcripts for control gene GAPDH was not affected by either E2 treatment or hormone starvation (Fig. 3A). When analyzed similarly, weak and consistent expression of the TGF-α mRNA transcript was observed with no evidence of influence of E2 treatment or hormone starvation (Fig. 3A), and this observation was persistent with several different numbers of PCR amplification cycles (data not shown). On the other hand, mRNA transcript for SDF-1 was not detected at all with varying conditions of RT-PCR (Fig. 3A). These results confirmed the DNA microarray data that had indicated that neither low concentrations of E2 (≤ 100 pM) nor hormone starvation exerted any significant influence on expression of the mRNA transcripts for TGF-α and SDF-1 genes.
Fig. 3.

mRNA expression profiles of E2-inducible genes with high or low E2 sensitivities: Semiquantitative RT-PCR. (A) mRNA expression in BUS cells exposed to the indicated concentrations of E2 for 48 h or hormone-starved (HS) for up to 2 days. (B) Expression of TGF-α and SDF-1 mRNA transcripts in BUS cells exposed to 10 or 100 nM E2 for 48 h. Primer pair 1 for SDF-1 detection was used for the experiment of A.
Because experiments described in previous reports that characterized TGF-α and SDF-1 as E2-inducible autocrine growth factors used 10 nM E2 [a concentration that was >100 times greater than that which saturated the E2 dose-dependent growth curves of MCF7 cells (5) and MCF7/BUS cells (Fig. 1 A)] we presumed that induction of the mRNA transcripts for these genes might require higher E2 concentrations than we had in the DNA microarray experiments. In fact, TGF-α mRNA transcripts were strongly induced with 10 nM E2 (Fig. 3B; the apparent absence of the RT-PCR product for the cells cultured in the absence of E2 was caused by the reduction of the PCR cycle number applied to avoid PCR saturation). The SDF-1 mRNA transcripts were also induced dramatically when cells were exposed to 10 nM E2 and 100 nM E2 (Fig. 3B). Specific detection of the SDF-1 mRNA was confirmed by using two different pairs of PCR primers. These results indicated that E2-dependent induction of the TGF-α and SDF-1 mRNA transcripts required an E2 concentration that was far higher than an E2 concentration that saturated the E2 dose-dependent growth curve of MCF7/BUS cells.
Discussion
Information on ligand sensitivity of E2-inducible and E2-suppressible genes may provide important insights into not only the biological actions of E2 itself but also factors that influence E2 effects such as expression of ER coregulator proteins (9, 16), ER interactions with other signaling pathways (1), and the presence of other estrogenic agonists and antagonists such as selective ER modulators or xenobiotic estrogens. To obtain systematic data on E2 sensitivity of large numbers of genes, in the present study we designed a simple method for determining E2 sensitivity of gene expression by using Affymetrix DNA microarray and Perl scripts and applied it to characterize the E2 responses of ERα-positive MCF7/BUS human breast cancer cells. In this study, the E2 exposure time was fixed to 48 h, when expression of the mRNA transcripts for several representative E2 target genes (e.g., progesterone receptor or WISP2; see Fig. 2) reached a maximum level after E2 stimulation (time course data not shown). Characterization of the combined aspects of the ligand sensitivity and the time course of E2 effects on the transcriptome is an important issue and should be addressed in future studies.
The present E2 sensitivity analysis identified a number of known E2 target genes successfully as indicated with code Es in Fig. 2, supporting the validity of our approach and method. The list of E2-inducible genes also included many genes involved in cell cycle progression and cancer as indicated by codes Cy, E2F, and Ca in Fig. 2; induction of some of these genes may involve BRCA1 and/or E2F1, which are E2-inducible transcription factors and key regulators of cell cycle and carcinogenesis (see Fig. 2 and refs. 17 and 18). On the other hand, cyclin D1 and c-MYC, two representative E2-inducible genes in human breast cancer cells, were not picked, although cyclin D1 mRNA expression showed a moderate increase with increasing E2 concentration (Fig. 2, class E). This increase is likely caused by the transient nature of induction of their mRNA transcripts, which peaks at ≈8 h after E2 exposure and decreases thereafter (19, 20). We also attempted to identify genes whose E2 dosedependent curves of mRNA expression showed U-shape or inverted U-shape. However, analyses using several different criteria identified only false positives, whose nonstandard mRNA expression profiles were not confirmed by RT-PCR (data not shown).
A number of additional E2-inducible and E2-suppressible genes have been identified in the present study (Fig. 2), although some of them may not be direct targets of ERs. Elucidation of mechanisms of the E2 responsiveness of their mRNA expression and the biological significance of such regulation awaits further studies. Among these genes, the FHL2/four-and-a-half LIM domains 2 (Fig. 2, class B) is especially interesting because this gene encodes a transcriptional coactivator for AP-1 (21) and β-catenin (22). The FHL2 induction with E2 may imply possible interactions between the E2 actions and the mitogen-activated protein kinase signaling pathway (which involves formation of the AP-1 transcription factor) and/or Wnt signaling pathway (which involves stabilization of β-catenin transcriptional coactivator). Another interesting gene was ribonucleotide reductase M2 (RRM2), which encodes the rate-limiting enzyme of deoxyribonucleotide production during DNA synthesis. It has been reported that RRM2 mRNA transcripts are inducible by E2F proteins (23) and BRCA1 (24). Moreover, a recent study identified RRM2 as a marker gene whose expression was stronger in invasive ductal carcinoma breast cancers than the ductal carcinoma in situ (25). These observations suggest a potential importance of RRM2 in growth and malignancy of breast cancer cells, and it would be interesting to determine whether the E2 sensitivity of RRM2 gene changes during the malignant progression of breast cancers.
Previous studies have reported that TGF-α and SDF-1 are E2-inducible autocrine growth factors for ERα-positive human breast cancer cells. The mRNA transcripts for both TGF-α (14) and SDF-1 (15) were strongly induced in MCF7 cells with 10 nM E2. Suppression of TGF-α production by small interfering RNA (14), and blocking the secreted SDF-1 peptide by a neutralizing antibody (15), resulted in significant inhibition of the E2-dependent growth of this cell line. However, although we were able to reproduce the induction of these mRNA transcripts in MCF7/BUS cells with 10–100 nM E2 (Fig. 3B), we also found that lower E2 concentrations that enhanced cell growth in a dose-dependent manner (i.e., 2–80 nM; Fig. 1 A) did not affect mRNA expression of these factors (Figs. 2E and 3A). Data shown in Fig. 3A rather suggested weak and consistent expression of TGF-α when cells were growing in the presence of subsaturating concentrations of E2. Interestingly, it has been proposed recently that TGF-α may enhance E2 sensitivity of MCF7 cells up to 10–100 times through activating the mitogen-activated protein kinase signaling pathway and, thereby, might facilitate E2-dependent cell growth (6). Moreover, we recently demonstrated that an ER coactivator CITED1 selectively enhanced the E2-dependent induction of the TGF-α mRNA transcript without affecting expression of pS2 mRNA transcript (E2 concentration was 10 nM) (26). Taken together, these observations may imply that induction of TGF-α might be regulated at the level of E2 sensitivity, which is determined by the availability of ER coactivators, and that TGF-α may function as an autocrine modifier of the growth-stimulating effects of E2 rather than as a classical autocrine growth factor. On the other hand, the inability to detect the SDF-1 mRNA in the presence of subsaturating concentrations of E2 (Fig. 3A) may suggest that this factor could function as an autocrine growth factor only when the cancer cells are exposed to saturating concentrations of E2, a situation that may occur in premenopausal women. Further studies are necessary to clarify the biological roles of these factors in relation to the E2 dose-dependent growth of breast cancer cells.
DNA microarray data analysis is a growing field of science, and there are a number of software designed specifically for this application. However, it is not uncommon that specifications of such software do not exactly fit for the experimental design and objectives of a research project. In the present study, we used the Perl programming language to generate simple scripts to implement the algorithm of data processing. Recently, the use of Perl in biological research has been becoming increasingly popular (13), and the present study, together with some studies from other laboratories (27), has demonstrated the usefulness of the Perl scripting technique in DNA microarray data analysis. Although complicated methods of DNA microarray data analysis, such as unsupervised clustering, would require a complete software package, Perl scripting seems convenient and effective for the purpose of quickly implementing simple algorithms that are often sufficient to extract meaningful information from DNA microarray data.
In summary, we have designed a method for characterizing E2 sensitivity of large numbers of genes expressed in cell culture by using DNA microarray and Perl scripting. By applying this method to ERα-positive MCF7/BUS human breast cancer cells, we performed a comprehensive E2 sensitivity analysis and identified a number of E2-responsive genes. The importance of E2 sensitivity analysis was exemplified by the finding that E2 concentration required to induce the mRNA transcripts for TGF-α and SDF-1, two representative E2-inducible autocrine growth factors of MCF7 cells, was actually far greater than the E2 concentration that saturated the E2 dose-dependent MCF7/BUS growth curve.
Supplementary Material
Acknowledgments
We thank Ana Soto and Carlos Sonnenschein for providing MCF7/BUS cells and technical advice and Emmett Schmidt, Nick Dyson, Dennis Sgroi, and Myles Brown for helpful discussions and critiques. This work was supported by the Avon Breast Cancer Program and National Institutes of Health Grant R01-CA82230 (to T.S.).
Abbreviations: E2, 17β-estradiol; ER, estrogen receptor; RRM2, ribonucleotide reductase M2, TGF-α, transforming growth factor α; SDF-1, stromal cell-derived factor 1.
References
- 1.Pedram, A., Razandi, M., Aitkenhead, M., Hughes, C. C. & Levin, E. R. (2002) J. Biol. Chem. 277, 50768–50775. [DOI] [PubMed] [Google Scholar]
- 2.Levenson, A. S., Svoboda, K. M., Pease, K. M., Kaiser, S. A., Chen, B., Simons, L. A., Jovanovic, B. D., Dyck, P. A. & Jordan, V. C. (2002) Cancer Res. 62, 4419–4426. [PubMed] [Google Scholar]
- 3.Soulez, M. & Parker, M. G. (2001) J. Mol. Endocrinol. 27, 259–274. [DOI] [PubMed] [Google Scholar]
- 4.Inoue, A., Yoshida, N., Omoto, Y., Oguchi, S., Yamori, T., Kiyama, R. & Hayashi, S. (2002) J. Mol. Endocrinol. 29, 175–192. [DOI] [PubMed] [Google Scholar]
- 5.Villalobos, M., Olea, N., Brotons, J. A., Olea-Serrano, M. F., Ruiz de Almodovar, J. M. & Pedraza, V. (1995) Environ. Health Perspect. 103, 844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yue, W., Wang, J. P., Conaway, M., Masamura, S., Li, Y. & Santen, R. J. (2002) Endocrinology 143, 3221–3229. [DOI] [PubMed] [Google Scholar]
- 7.Nardulli, A. M., Romine, L. E., Carpo, C., Greene, G. L. & Rainish, B. (1996) Mol. Endocrinol. 10, 694–704. [DOI] [PubMed] [Google Scholar]
- 8.Klinge, C. M. (2001) Nucleic Acids Res. 29, 2905–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shang, Y. & Brown, M. (2002) Science 295, 2465–2468. [DOI] [PubMed] [Google Scholar]
- 10.Soto, A. M., Sonnenschein, C., Chung, K. L., Fernandez, M. F., Olea, N. & Serrano, F. O. (1995) Environ. Health Perspect. 103, Suppl. 7, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagota, A. (2001) Microarray Data Analysis and Visualization (Bioinformatics-By-the-Bay Press, Santa Cruz, CA).
- 12.Affymetrix (2003) GeneChip Expression Analysis (Affymetrix, Santa Clara, CA).
- 13.Stajich, J. E., Block, D., Boulez, K., Brenner, S. E., Chervitz, S. A., Dagdigian, C., Fuellen, G., Gilbert, J. G., Korf, I., Lapp, H., et al. (2002) Genome Res. 12, 1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenney, N. J., Saeki, T., Gottardis, M., Kim, N., Garcia-Morales, P., Martin, M. B., Normanno, N., Ciardiello, F., Day, A., Cutler, M. L., et al. (1993) J. Cell Physiol. 156, 497–514. [DOI] [PubMed] [Google Scholar]
- 15.Hall, J. M. & Korach, K. S. (2003) Mol. Endocrinol. 17, 792–803. [DOI] [PubMed] [Google Scholar]
- 16.Shang, Y., Hu, X., DiRenzo, J., Lazar, M. A. & Brown, M. (2000) Cell 103, 843–852. [DOI] [PubMed] [Google Scholar]
- 17.Spillman, M. A. & Bowcock, A. M. (1996) Oncogene 13, 1639–1645. [PubMed] [Google Scholar]
- 18.Wang, W., Dong, L., Saville, B. & Safe, S. (1999) Mol. Endocrinol. 13, 1373–1387. [DOI] [PubMed] [Google Scholar]
- 19.Prall, O. W., Sarcevic, B., Musgrove, E. A., Watts, C. K. & Sutherland, R. L. (1997) J. Biol. Chem. 272, 10882–10894. [DOI] [PubMed] [Google Scholar]
- 20.Jeng, M. H., Shupnik, M. A., Bender, T. P., Westin, E. H., Bandyopadhyay, D., Kumar, R., Masamura, S. & Santen, R. J. (1998) Endocrinology 139, 4164–4174. [DOI] [PubMed] [Google Scholar]
- 21.Morlon, A. & Sassone-Corsi, P. (2003) Proc. Natl. Acad. Sci. USA 100, 3977–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei, Y., Renard, C. A., Labalette, C., Wu, Y., Levy, L., Neuveut, C., Prieur, X., Flajolet, M., Prigent, S. & Buendia, M. A. (2003) J. Biol. Chem. 278, 5188–5194. [DOI] [PubMed] [Google Scholar]
- 23.Ishida, S., Huang, E., Zuzan, H., Spang, R., Leone, G., West, M. & Nevins, J. R. (2001) Mol. Cell. Biol. 21, 4684–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welcsh, P. L., Lee, M. K., Gonzalez-Hernandez, R. M., Black, D. J., Mahadevappa, M., Swisher, E. M., Warrington, J. A. & King, M. C. (2002) Proc. Natl. Acad. Sci. USA 99, 7560–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma, X. J., Salunga, R., Tuggle, J. T., Gaudet, J., Enright, E., McQuary, P., Payette, T., Pistone, M., Stecker, K., Zhang, B. M., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 5974–5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yahata, T., Shao, W., Endoh, H., Hur, J., Coser, K. R., Sun, H., Ueda, Y., Kato, S., Isselbacher, K. J., Brown, M. & Shioda, T. (2001) Genes Dev. 15, 2598–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudley, A. M., Aach, J., Steffen, M. A. & Church, G. M. (2002) Proc. Natl. Acad. Sci. USA 99, 7554–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}