

Abstract
Because most potential molecular markers and targets are proteins, proteomic profiling is expected to yield more direct answers to functional and pharmacological questions than does transcriptional profiling. To aid in such studies, we have developed a protocol for making reverse-phase protein lysate microarrays with larger numbers of spots than previously feasible. Our first application of these arrays was to profiling of the 60 human cancer cell lines (NCI-60) used by the National Cancer Institute to screen compounds for anticancer activity. Each glass slide microarray included 648 lysate spots representing the NCI-60 cell lines plus controls, each at 10 two-fold serial dilutions to provide a wide dynamic range. Mouse monoclonal antibodies and the catalyzed signal amplification system were used for immunoquantitation. The signal levels from the >30,000 data points for our first 52 antibodies were analyzed by using p-scan and a quantitative dose interpolation method. Clustered image maps revealed biologically interpretable patterns of protein expression. Among the principal early findings from these arrays were two promising pathological markers for distinguishing colon from ovarian adenocarcinomas. When we compared the patterns of protein expression with those we had obtained for the same genes at the mRNA level by using both cDNA and oligonucleotide arrays, a striking regularity appeared: cell-structure-related proteins almost invariably showed a high correlation between mRNA and protein levels across the NCI-60 cell lines, whereas non-cell-structure-related proteins showed poor correlation.
High-throughput transcript profiling has generated large bodies of information on gene expression. However, proteomic profiling will yield more direct answers to our current biological and pharmacological questions, because the majority of known biological effector molecules, diagnostic markers, and pharmaceutical targets are proteins, not mRNA. The first broadly useful technology for proteomic profiling was two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) (1), which typically permits semiquantitation of the most abundant thousand or so spots, but poses the difficult problem of identifying the spots with particular proteins. More recently, microarray formats have been introduced for proteomic profiling, most of them based on robotic spotting of antibodies or other ligands that capture the protein molecules to be assessed (2, 3).
Reverse-phase protein lysate microarrays, recently reported by Paweletz et al. (4), are based on the opposite configuration. Samples to be assessed are robotically spotted, and an antibody is then used to measure the amount of a particular protein present in the sample. In contrast to 2D-PAGE and antibody arrays, the reverse-phase methodology assesses only one protein per slide, but it has the great advantage that all of the cell or tissue samples can be analyzed side by side in a single array. That is an advantage because, for functional studies, we are generally more interested in comparing protein levels across samples than in comparing samples across protein types (5, 6).
To date, however, the reverse-phase protein lysate microarray has been limited by technical considerations to relatively small numbers of spots. That became a serious problem when we wished to profile proteins in the 60 human cancer cell lines (NCI-60) used by the National Cancer Institute's Developmental Therapeutics Program since 1990 to screen >100,000 chemical compounds for anticancer activity (7–9). Hence, as will be described here, we developed, to our knowledge, new methods to obtain higher density, robotically spotted reverse-phase protein lysate microarrays with high precision of protein measurement. Our first application was to the NCI-60, but the technology is applicable much more broadly.
The NCI-60 set includes leukemias, lymphomas, and carcinomas of ovarian, renal, breast, prostate, colon, lung, and CNS origin. Because the screening data proved rich in information on the mechanisms of action of tested compounds (5, 7–10), our laboratory and many others have profiled the NCI-60 extensively at the DNA, RNA, protein, and functional levels for correlation with pharmacological sensitivities of the cells (5–13). Our laboratory's approach has been to profile the cells' characteristics in aggregate by using high-throughput, omic (14, 15) technologies. We began in the mid-1990's with 2D-PAGE (6), but were limited by the protein identification problem and switched focus to the mRNA level, applying cDNA microarrays (11, 12) and Affymetrix oligonucleotide chips (13). Studies at the DNA level are also in progress (16). Overall, the NCI-60 is the most extensively profiled set of cells anywhere, and the data sets on them have been widely used in the cancer research and bioinformatics communities. No cell lines in culture are fully representative of tumors in vivo, of course, but they have the advantages of reproducibility, availability in large numbers, and homogeneity in cell lineage (5).
Our reverse-phase protein lysate microarrays for the NCI-60 include 10 serial two-fold dilutions per cell sample (plus controls) and therefore permit measurements of considerable precision and wide dynamic range (4). The detection limit (defined as a signal two SD above background) for the reverse-phase arrays has been estimated at 2,000 molecules detected per spot for recombinant PSA. The functional sensitivity (defined as the lowest concentration measured with a coefficient of variation of 20% within an array) is ≈5,000 molecules per spot (4, 17). It is important to realize, however, that such a figure depends to a considerable extent on the physical characteristics of the particular target protein and on all aspects of the antigen–antibody interaction on the array.
We report here the results for an initial 52 proteins and use the data to address a question that has intrigued researchers for years: How similar are expression profiles at the RNA and protein levels? This correlation has previously been assessed across proteins within single cell types (18–20), but, because of the special characteristics of the reverse-phase format and the diversity of the NCI-60, we were able to assess the correlation in the more appropriate way, across disparate cell types for each protein. That, in turn permitted us to ask whether particular classes of proteins were better correlated than others with mRNA expression. The initial result was a striking difference between structural and nonstructural protein types.
Materials and Methods
Protein Lysate Preparation. The NCI-60 cell lines were cultured and protein prepared from them as described for our studies by using 2D gel electrophoresis (6). Briefly, cells were collected by scraping and washed three times with cold PBS. The resulting pellets were lysed in buffer containing 9 M urea (Sigma), 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS; Calbiochem), 2% pH 8.0–10.5 Pharmalyte (Amersham Pharmacia Biotech, Piscataway, NJ), and 65 mM DTT (Amersham Pharmacia Biotech) (21). After lysis, the samples were centrifuged briefly, and the supernatants were stored at –80°C. A reference pool was prepared by mixing equal volumes of all 60 cell lines.
Protein Lysate Array Design and Production. Arrays were prepared on nitrocellulose-coated glass slides (FAST Slides, Schleicher & Schuell) by using a pin-in-ring format GMS 417 arrayer (Affymetrix, Santa Clara, CA) with four 500-μm-diameter pins. Because the samples were viscous, we used the pin-in-ring format to avoid problems due to clogging of quills. Fig. 1A shows the design of the array. Ten two-fold serial dilutions were made from each lysate. Four 384-well microtiter plates (Genetix, New Milton, Hampshire, U.K.) were used to array 640 spots (plus eight spatial registration marks for use in image processing) on a 21 × 35-mm area of nitrocellulose membrane. The first dilution (four-fold) was made with buffer containing 5 M urea, 2% Pharmalyte, pH 8–10.5, and 65 mM DTT. The remaining dilutions were then made with buffer containing 6M urea, 1% CHAPS, 2% Pharmalyte, pH 8–10.5, and 65 mM DTT. Hence, only the lysate concentration changed along each dilution series. The urea concentration was thus kept at 6 M, and the CHAPS concentration at 2%, to keep proteins in their denatured forms. We could remove samples repeatedly from –80°C storage for use without heating. To avoid evaporation in the microtiter plate during spotting, we kept the humidity in the array chamber at 70–90% with a Vicks ultrasonic humidifier (Kaz, Hudson, NY). Arraying was completed for each microtiter plate within 70 min. Arrays were produced in batches of 20, and the occasional low-quality array (e.g., with many spot dropouts) was discarded.
Fig. 1.

NCI-60 reverse-phase protein lysate microarrays. (A) Staining with SYPRO ruby for total protein. Each row (see enlarged image at the left) consists of 10 two-fold dilutions of an NCI-60 cell line or the control pool. The pool was spotted at four locations to control for pin effects. Concentrated pool was spotted at the bottom right corner of each field to serve as a registration mark for scanning. (B) CSA staining for p300 expression. (C) Negative control. (D) Representative candidate antibodies prescreened for specificity by Western blotting (20 μg per lane) with NCI-60 pool. *, bands at the predicted molecular weight. Blots 1–7 (from the left) show a single predominant band at the expected molecular weight. Blots 8–10 represent antibodies rejected for the array application because (i) the target band is fainter than other bands (lane 8); (ii) the target band is approximately equal in intensity to other bands (lane 9); and (iii) the target band is dominant (lane 10), but the other bands persist when the lysate is diluted to a point that the target band is below saturation (data not shown).
Western Blotting. Murine monoclonal antibodies were screened for specificity by Western blotting with 20 μg of lysate protein per lane. The running buffer contained 62.5 mM Tris·HCl, pH 6.8, 2% SDS, 10% glycerol, and 2.5% 2-mercaptoethanol. We used a 4–15% SDS-polyacrylamide linear gradient gel (Tris·HCl Ready Gel, Bio-Rad), secondary alkaline phosphatase-conjugated goat anti-mouse antibody, and the chemiluminescent immunoblot detection system (Tropix, Bedford, MA). An antibody was accepted only if it produced a single predominant band at the expected molecular weight. Multiple types of information (including screening results) on the antibodies and their antigens were entered into a relational database, abminer, which can be accessed at http://discover.nci.nih.gov (S.M., S.N., R. Rowland, U. Shankavaram, F. Washburn, D. Asin, H.K.-M., and J.N.W., unpublished work).
Detection of Specific and Total Protein on Microarrays. Each array was incubated with a specific primary antibody, which was detected by using the catalyzed signal amplification (CSA) system (DAKO). Briefly, each slide was washed manually with deionized water to remove urea. Then, in an Autostainer universal staining system (DAKO), it was blocked with I-block (Tropix) and incubated with primary and secondary antibodies. Also in the Autostainer, it was then incubated with streptavidin–biotin complex, biotinyl tyramide (for amplification) for 15 min, streptavidin-peroxidase for 15 min, and 3,3′-diaminobenzidine tetrahydrochloride chromogen for 5 min. Between steps, the slide was washed with CSA buffer. The signal was scanned with a Perfection 1200S scanner (Epson America, Long Beach, CA) with 256-shade gray scale at 600 dots per inch. For detection of total protein, arrays were stained with SYPRO ruby protein blot stain (Molecular Probes) and scanned with a FluorImager SI (Amersham Pharmacia Biotech) at 100-μm resolution. Spot images were converted to raw pixel values by a modified version of the p-scan (Peak quantification with Statistical Comparative Analysis) software (http://abs.cit.nih.gov/pscan) (22).
Dose Interpolation Data Analysis. Outliers traceable to defects in spotting were eliminated, and the data were then analyzed by using a dose interpolation (DI) algorithm developed for this study. Briefly, the maximum spot intensity (Imax) was defined heuristically to be the third highest value observed anywhere on the array, and the minimum intensity (Imin) was defined heuristically to be the mean of the tenth (i.e., last) dilution points over all cell types, Imin = Ī. The estimated dilution factor, 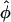 , for each cell type was then determined by interpolation in a monotonic linear spline fitted to the serial dilution curve. If the linear spline is represent by I = f, where φ is the true dilution factor, then
, for each cell type was then determined by interpolation in a monotonic linear spline fitted to the serial dilution curve. If the linear spline is represent by I = f, where φ is the true dilution factor, then
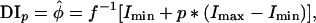 |
where p is the fraction of the way from minimum to maximum value of the intensity. On the basis of extensive optimization studies (L.Y., S.N., J.N.W., and P.J.M., unpublished work), we selected the P = 25% point because it optimally served two sometimes opposing purposes: (i) it minimized the measurement variance, and (ii) it yielded DIp values for as many as possible of the dose–response curves. These DI25 values were calculated for both target and total protein levels. To correct for any differences in protein content among the cell lysates, each experimental DI25 value was normalized by the mean over the 52 slides of the total protein DI25 values for the particular cell line. The final values formed a 52 × 64 matrix for 52 antibodies tested against the 60 cell lines plus four NCI-60 pools.
Results
Prescreening of Antibodies. Fig. 1D shows representative Western blot prescreening of 10 murine monoclonal antibodies against the NCI-60 pool. We tested >200 different antibodies, and ≈70% of them showed a single predominant band at the predicted molecular weight.
High-Density Reverse-Phase Protein Lysate Microarrays. Each array (Fig. 1 A) included serial dilutions of all 60 cell lines plus four pooled samples (640 spots in all). Fig. 1B shows, for illustration, an array incubated with mouse anti-human-p300 IgG. As a negative control, a duplicate slide was incubated with mouse anti-Aspergillus niger glucose oxidase IgG1 (Fig. 1C), which does not recognize any human antigen. Little nonspecific signal was seen, either with that control IgG or when primary antibody was omitted. The data for all 52 proteins can be accessed at http://discover.nci.nih.gov and in Table 1, which is published as supporting information on the PNAS web site.
Assessing Reproducibility. Fig. 2 shows the 64 dilution curves obtained for p300 expression and illustrates the DI25 calculation. To analyze experimental variability (see Fig. 5, which is published as supporting information on the PNAS web site), we measured p300 levels on arrays consisting of 24 repeats of the NCI-60 pool dilution series. This control study was repeated six times to investigate slide-to-slide, pin-to-pin, and row-to-row variation. We used a three-way random effects ANOVA to assess the components of variance, expressed as relative error (i.e., coefficient of variation) in the concentration estimate. The variation due to slide was 6.8%; due to pin, 6.6%; and due to row, 1.0%. The remaining variation (pure error) was 13.9%. The overall coefficient of variation was 17%. Additional indicators of reproducibility were obtained from the full arrays. The median Pearson correlation was +0.86 for all six possible pairwise combinations of the four cell pool replicates per slide across 52 antibodies. Because each pool on a slide was spotted with a different pin, any pin effects were included in this figure. The median Pearson correlation coefficient was +0.72 for the 15 pairwise comparisons of six replicate arrays across 60 cell lines (plus four cell pools) for p300 [Fig. 6, which is published as supporting information on the PNAS web site, shows the range (maximum minus minimum) and SD for each of the 52 proteins across the NCI-60 cell lines].
Fig. 2.

Analysis of p300 expression. (A) An array incubated with p300 primary antibody and stained by CSA. (B) Sixty four dilution curves in eight fields on the array. y axes, p-scan intensity of p300 signal; x axes, log2(dilution factor). Numbers after cell line names are DI25 values. The order of cell line listing corresponds to placement on the array. (C) DI25 algorithm calculations for field 7. Broken line, the 25% level (at 43 units).
Clustering of Cells and Proteins. To organize the cell lines and proteins on the basis of expression patterns, we used the Clustered Image Map program package, cimminer (5), which can be accessed at http://discover.nci.nih.gov. The results for average linkage clustering with a correlation coefficient metric are shown in Fig. 3. Cell clustering patterns generally resembled those obtained at the transcript level (11–13). For example, MDA-MB435 (derived from the pleural effusion of a patient who had previously had breast cancer), and its c-erbB2 transfectant, MDA-N, clustered with seven melanotic melanomas. We have seen that same association at the mRNA level and also when clustering the cells by drug sensitivity patterns (11). Also, as seen in the transcript and pharmacological data, MDA-MB-435 and MDA-N clustered together essentially as although they were replicates. Despite high expression of c-erbB2 protein in MDA-N, the Pearson correlation between the two cell lines in the present study was very strong, +0.91 (two-tailed bootstrap 95% confidence limits = +0.66 to +0.98). Without c-erbB2, the correlation coefficient was even higher (+0.98; confidence limits = +0.95 to + 0.99). This observation is reflected in more detail in Fig. 7, which is published as supporting information on the PNAS web site. Fig. 7 shows very small differences between MDA-MB435 and MDA-N across the entire dilution curve.
Fig. 3.

Clustered image map relating the expression levels of 52 proteins in the NCI-60 cell lines. Data were generated by using the DI25 algorithm and data mean centered across both cells and proteins. (Right Lower Inset) The difference in c-erbB2 level between MDA-MB435 cells (DI25 = –1.4) and MDA-N (DI25 = +0.5). KRT, cytokeratins.
In terms of the protein axis in Fig. 3, all replicates (of Stat3, moesin, MGMT, E-cadherin, and p300) clustered together, even though the replicates in some cases represented different batches of arrays. An epithelial cluster consisted of cytokeratins 8, 18, and 20, as well as villin, c-erbB2, and E-cadherin. Overall, the measured protein expression patterns appeared to be technically reliable and biologically reasonable.
Comparison of Transcript and Protein Expression Patterns. For the comparison, we had NCI-60 data from two independent transcriptional profiling platforms, cDNA arrays and oligo arrays. To match transcript and protein names, we used the matchminer program (http://discover.nci.nih.gov; ref. 23). matchminer leverages several major public databases (i) to translate among various gene and protein identifiers for lists of genes, or (ii) to find the intersection between two such lists. Thirty-one of the 52 could be matched and appeared on all three array platforms. Because there are known problems in identification with both the cDNA and oligo array platforms, however, we excluded from further analysis any transcripts that did not show reasonable concordance (correlation coefficient >0.30) between the two. That level of concordance for any given gene would be highly unlikely to occur by chance (24). The 19 remaining molecular species showed a high correlation of Pearson correlation coefficients between cDNA/protein and oligo/protein data (+0.92; two-tailed bootstrap 95% confidence limits = +0.74 to +0.96). The overall cDNA/protein and oligo/protein correlation coefficients for these 19 were +0.52 (range –0.10 to +0.87) and +0.40 (range –0.15 to +0.88), respectively. Fig. 4 shows the relationships among these three types of measurements for 19 genes represented in all three databases. In this scattergram, genes for which cDNA/protein and oligo/protein values were similar appear near the major 45° diagonal. Proteins that matched the transcript level closely appear toward the upper right.
Fig. 4.

Transcript/protein correlation coefficients. The data were from two independent mRNA profiling platforms (cDNA and oligonucleotide arrays). Each point represents a single target protein. Molecular species were divided into two major categories according to SWISS-PROT and/or MIPS. Blue squares, cell-structure-related proteins; red squares, non-cell-structure-related proteins.
Discussion
We have developed a protocol for making reverse-phase protein lysate microarrays with larger numbers of spots than previously feasible and have applied those arrays to proteomic profiling of the NCI-60 human cancer cell lines. Previously, we had profiled the NCI-60 by 2D-PAGE (6), but the utility of those studies was limited by the difficulty of indexing protein spots across all of the gels and identifying the spots with particular proteins. The reverse-phase protein lysate microarray does not suffer those two problems, because all 60 cell types can be spotted on the same slide and because the proteins are detected with monoclonal antibodies of known (and Western blot-confirmed) specificity.
Unlike antibody arrays (2, 3) and similar alternative methods, the reverse-phase configuration does not require tagging of the lysate with a dye that could influence the binding (4), and it does not require careful matching of the antibodies' binding characteristics. Because standard enzyme-linked immunoassays would not have been sensitive enough for the array application, we used an amplified detection system (CSA). To assess the protein levels over a wide dynamic range, we spotted the lysate at 10 two-fold serial dilutions. That design, however, required larger reverse-phase microarrays than previously feasible. The protocol developed, a modification of that described by Paweletz et al. (4), has several useful features: (i) it permits use of protein lysate samples that have been collected in nonionic, urea-containing medium (e.g., for parallel assessment on 2D-PAGE gels); (ii) it keeps the proteins in a largely unfolded state by maintaining a high concentration of urea (6 M), plus CHAPS (2%) and DTT (65 mM), in the lysate throughout the serial dilutions; (iii) it uses a nonvolatile reducing agent (DTT); (iv) it avoids the necessity of boiling the lysate [particularly important if the original sample contained urea, because heated urea can covalently modify proteins (21)]; (v) it permits the microtiter plates (384-well) used in robotic spotting to be frozen and thawed between uses; (vi) it includes maintenance of a high relative humidity (≈80%) during spotting to prevent evaporation of liquid from the microtiter plates; and (vii) it includes staining for total protein (to normalize the data). We used this protocol to make arrays of 648 spots, incorporating 10 dilutions of the 60 cell lines, four control pools composed of all 60 lines, and eight spatial registration marks. With an overall coefficient of variation of 17%, this array methodology appears to be a useful tool for quantitative measurement.
The murine monoclonal antibodies used for detection were tested for specificity by Western blotting, and those that did not produce a single predominant band at the expected molecular weight were rejected. It should be understood, however, that lack of a significant second band with the pool could not rule out the possibility that a second band would be significant in one or two of the 60 lines, and it could not rule out crossreaction of the antibody with close family members that have similar molecular weight and bear the epitope. These potential problems are analogous to ones encountered with other expression platforms such as cDNA, oligo, and antibody microarrays. Further triage on the antibodies, for example by 2D Western blotting, would introduce additional complexities related to the distinctions among different posttranslational modifications.
The experiments produced 64 concentration-dilution curves for each slide. As illustrated in Fig. 2, we analyzed those curves using the program package p-scan (http://abs.cit.nih.gov/pscan), followed by our DI algorithm. That algorithm was the product of extensive sensitivity analyses to be presented elsewhere (L.Y., S.N., J.N.W., and P.J.M., unpublished work).
The results were analyzed statistically in a number of ways. The clustered image map in Fig. 3 shows cell lines and proteins clustered to bring like together with like and bring out patterns of coherence (5, 6). The cell clusters were similar to those obtained at the transcriptional level with cDNA microarrays (11, 12), as discussed more fully in Results.
Because it is technically easier, transcript profiling has developed more rapidly than has protein profiling, and the transcripts are often thought of, implicitly or explicitly, as surrogates for the proteins they code. However, protein levels cannot be inferred from transcript data. The two types of profiling give complementary information, and our mRNA and protein profiles for the NCI-60 provide an opportunity to compare the two across a wide variety of cell types. To our knowledge, that has not previously been done. Furthermore, the fact that we have profiled the transcript levels by two very different methods, cDNA arrays and Affymetrix oligonucleotide arrays, provides extra assurance of quality; we are able to focus the comparison on those genes for which the two gave concordant results. That assurance is important because of well known sources of error in both methods. Of the 52 proteins studied to date by reverse-phase protein lysate microarray, 31 can also be identified on both the cDNA and oligo arrays by using our matchminer program (23). Of those 31, 19 show a sufficient correlation (>0.30) between cDNA array and oligo array sets to indicate (at the two-tailed P = 0.03 level) that they do not represent different, unrelated transcripts. As shown in Fig. 4, the 19 show a high correlation coefficient (+0.92) between cDNA/protein and oligo/protein correlation coefficients (Fig. 4), providing a solid basis for analysis of the relationship between transcript and protein abundances.
The mean cDNA/protein and oligo/protein correlation coefficients for the 19 species across 60 cell lines are +0.52 and +0.40, respectively, but Fig. 4 clearly indicates a wide range of mRNA protein correlations for different molecular species. The highest individual values are +0.87 and +0.88 for cDNA/protein and oligo/protein array correlations of cytokeratin 8. The lowest are the analogous values of –0.10 and –0.15 for ISGF3G.
We next ask whether particular categories of molecules have high or low correlations. Visual inspection of the data points in Fig. 4 immediately leads to the hypothesis that proteins related to cell structure are more highly correlated with mRNA levels than are nonstructural proteins. To pursue that hypothesis more concretely, we use functional definitions obtained from SWISS-PROT (http://us.expasy.org) and/or MIPS (http://mips.gsf.de) through GeneCards (http://bioinfo.weizmann.ac.il) to classify the proteins as “cell-structure-related” or “non-cell-structure-related.” The structure-related proteins fell into three subcategories: type I membrane proteins (ERBB2, CDH1, CDH2, and MCP), actinbinding proteins (FN1, VIL1, MSN), and keratins (KRT8). The non-structure-related proteins also fell into three subcategories: nuclear proteins (PCNA, STAT3, ISGF3G, EP300, and MSH2), cell-cycle control proteins (CCNA2, CCNB1, MCM7, and MAPK1), and others (GSK3B and FADD).
As indicated in Fig. 4, the structure-related proteins are almost always better correlated with mRNA levels across the 60 cell lines. P = 0.0007 for the cDNA/protein correlation, and P = 0.004 for the oligo/protein correlation (two-tailed, nonpaired t test). For the corresponding nonparametric (Wilcoxon) tests of significance, P = 0.0005 and 0.005, respectively. These statistics do not imply a causal relationship, however; there could easily be a confounding factor.
Let us now consider more precisely why one might expect a high correlation between mRNA and protein expression levels. Assume the simplest possible kinetic model,
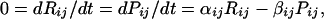 |
where i denotes the cell type (i = 1, 60), j the molecular species (j = 1,19), R the number of mRNA molecules per cell, P the number of protein molecules per cell, α the rate constant for producing protein from mRNA, and β the rate constant for degradation of protein. This equation holds for log-phase growth if one averages over all cell-cycle phases for an unsynchronized cell population. It holds regardless of whether an individual protein molecule has a defined lifetime or has an equal probability of being degraded at all points in time. Quite generally, for mathematical stationarity in cell properties, we have the proportionality
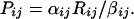 |
When we compare mRNA and protein levels across cell lines for a given molecule type, we are correlating Pj with Rj, where Pj is the vector (in this case of length 60) of levels of the jth protein across cell lines, and Rj is defined analogously. If, for a given j, αij/βij were the same for all i, the correlation would be perfect. A poor correlation between Pj and Rj implies that this ratio differs, for whatever reason, from cell type to cell type. Previous studies (18–20) of mRNA/protein correlation were addressing a different issue, the relationship between Pi and Ri (i.e., the correlation across molecular species for a given cell type). The former type of correlation is most naturally addressed by the reverse-phase format, because all cell types are represented on a single array for a given protein type. mRNA and antibody arrays have the opposite characteristic. Because of the various factors that influence antibody binding, neither lysate arrays nor antibody arrays give directly commensurate results from protein to protein.
Insofar as the correlation across cell types is high for structure-related species, we might be able to use mRNA profiling data to identify candidate molecular markers for use at the protein level by immunohistochemistry. We have, in fact, used this approach to identify villin and moesin as diagnostic markers for distinguishing colon from ovarian adenocarcinomas in the abdomen (25). That is an important differential diagnosis to make because it determines the type of chemotherapy given. We initially identified the markers by using cDNA and Affymetrix oligo microarrays. The reverse-phase protein lysate microarrays described here then showed them to be good markers at the protein level as well, and the findings were strikingly confirmed for clinical tumors in tissue array format. These markers are being followed up for possible use in clinical pathology. In accord with our overall findings, both villin and moesin were identified as cell structure-related proteins (26, 27), as indicated in Fig. 4. Also consistent with our findings here are published reports (although based on limited numbers of cell types) that the structural proteins cytokeratin 8, CDH1, villin, and moesin are transcriptionally regulated (20, 26–31).
In conclusion, we have developed reverse-phase protein lysate microarrays with larger numbers of spots than previously feasible and have used them for accurate measurements of the expression levels of 52 proteins to date across the NCI-60 cell lines. One significant finding was a class of molecular species (cell-structure-related) for which the mRNA/protein correlation was high. Exploration of the proteome is still in its early stages, and this technology can be expected to contribute significantly to our basic understanding, as well as to more practical endeavors like the identification of molecular markers and targets for therapy.
Supplementary Material
Acknowledgments
We thank E. A. Sausville, A. Monks, D. A. Scudiero, K. D. Paull, and others in the National Cancer Institute Developmental Therapeutics Program, whose work over the years and in collaboration with us has made these studies of the NCI-60 cell lines possible; D. A. Ross and others in the Brown/Botstein laboratory at Stanford University (Stanford, CA); J. Staunton and others in the Golub/Lander group at the Whitehead Institute (Cambridge, MA) for collaborations that generated the transcript profiles used for comparison with protein profiles in this work; and D. Beitner-Johnson for editing a draft of the manuscript.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NCI-60, the National Cancer Institute's Developmental Therapeutics Program to screen chemical compounds for anticancer activity; CSA, catalyzed signal amplification; DI, dose interpolation.
References
- 1.O'Farrell, P. H. (1975) J. Biol. Chem. 250, 4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 2.Haab, B. B., Dunham, M. J. & Brown, P. O. (2001) Genome Biol. 2, research0004.1–0004.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knezevic, V., Leethanakul, C., Bichsel, V. E., Worth, J. M., Prabhu, V. V., Gutkind, J. S., Liotta, L. A., Munson, P. J., Petricoin, E. F., III, & Krizman, D. B. (2001) Proteomics 1, 1271–1278. [DOI] [PubMed] [Google Scholar]
- 4.Paweletz, C. P., Charboneau, L., Bichsel, V. E., Simone, N. L., Chen, T., Gillespie, J. W., Emmert-Buck, M. R., Roth, M. J., Petricoin, E. F., III, & Liotta, L. A. (2001) Oncogene 20, 1981–1989. [DOI] [PubMed] [Google Scholar]
- 5.Weinstein, J. N., Myers, T. G., O'Connor, P. M., Friend, S. H., Fornace, A. J., Jr., Kohn, K. W., Fojo, T., Bates, S. E., Rubinstein, L. V., Anderson, N. L., et al. (1997) Science 275, 343–349. [DOI] [PubMed] [Google Scholar]
- 6.Myers, T. G., Waltham, M., Li, G., Buolamwini, J. K., Scudiero, D. A., Rubinstein, L. V., Paull, K. D., Sausville, E. A., Anderson, N. L. & Weinstein, J. N. (1997) Electrophoresis 18, 647–653. [DOI] [PubMed] [Google Scholar]
- 7.Paull, K. D., Shoemaker, R. H., Hodes, L., Monks, A., Scudiero, D. A., Rubinstein, L., Plowman, J. & Boyd, M. R. (1989) J. Natl. Cancer Inst. 81, 1088–1092. [DOI] [PubMed] [Google Scholar]
- 8.Monks, A., Scudiero, D., Skehan, P., Shoemaker, R., Paull, K., Vistica, D., Hose, C., Langely, J., Cronise, P., VAigro-Wolff, A., et al. (1991) J. Natl. Cancer Inst. 83, 757–766. [DOI] [PubMed] [Google Scholar]
- 9.Stinson, S. F., Alley, M. C., Kopp, W. C., Fiebig, H. H., Mullendore, L. A., Pittman, A. F., Kenney, S., Keller, J. & Boyd, M. R. (1992) Anticancer Res. 12, 1035–1053. [PubMed] [Google Scholar]
- 10.Alley, M. C., Scudiero, D. A., Monks, A., Hursey, M. L., Czerwinski, M. J., Fine, D. L., Abbot, B. J., Mayo, J. G., Shoemaker, R. H. & Boyd, M. R. (1988) Cancer Res. 48, 589–601. [PubMed] [Google Scholar]
- 11.Scherf, U., Ross, D. T., Waltham, M., Smith, L. H., Lee, J. K., Tanabe, L., Kohn, K. W., Reinhold, W. C., Myers, T. G., Andrews, D. T., et al. (2000) Nat. Genet. 24, 236–244. [DOI] [PubMed] [Google Scholar]
- 12.Ross, D. T., Scherf, U., Eisen, M. B., Perou, C. M., Rees, C., Spellman, P., Iyer, V., Jeffrey, S. S., Van de Rijn, M., Waltham, M., et al. (2000) Nat. Genet. 24, 227–235. [DOI] [PubMed] [Google Scholar]
- 13.Staunton, J. E., Slonim, D. K., Coller, H. A., Tamayo, P., Angelo, M. J., Park, J., Scherf, U., Lee, J. K., Weinstein, J. N., Mesirov, J. P., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 10787–10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinstein, J. N. (2002) Curr. Opin. Pharmacol. 2, 361–365. [DOI] [PubMed] [Google Scholar]
- 15.Weinstein, J. N., Scherf, U., Lee, J. K., Nishizuka, S., Gwadry, F., Bussey, A. K., Kim, S., Smith, L. H., Tanabe, L., Richman, S., et al. (2002) Cytometry 47, 46–49. [DOI] [PubMed] [Google Scholar]
- 16.Roschke, A. V., Tonon, G., Gehlhaus, K. S., McTyre, N., Bussey, K. J., Lababidi, S., Scudiero, D. A., Weinstein, J. N. & Kirsch, I. R. Cancer Res., in press. [PubMed]
- 17.Liotta, L. A., Espina, V., Mehta, A. I., Calvert, V., Rosenblatt, K., Geho, D., Munson, P. J., Young, L., Wulfkuhle, J. & Petricoin, E. F. (2003) Cancer Cell 3, 317–325. [DOI] [PubMed] [Google Scholar]
- 18.Anderson, L. & Seilhamer, J. (1997) Electrophoresis 18, 533–537. [DOI] [PubMed] [Google Scholar]
- 19.Gygi, S. P., Rochon, Y., Franza, B. R. & Aebersold, R. (1999) Mol. Cell. Biol. 19, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ideker, T., Thorsson, V., Ranish, J. A., Christmas, R., Buhler, J., Eng, J. K., Bumgarner, R., Goodlett, D. R., Aebersold, R. & Hood, L. (2001) Science 292, 929–934. [DOI] [PubMed] [Google Scholar]
- 21.Anderson, N. L., Esquer-Blasco, R., Hofmann, J.-P. & Anderson, N. G. (1991) Electrophoresis 12, 907–930. [DOI] [PubMed] [Google Scholar]
- 22.Carlisle, A. J., Prabhu, V. V., Elkahloun, A., Hudson, J., Trent, J. M., Linehan, W. M., Williams, E. D., Emmert-Buck, M. R., Liotta, L. A., Munson, P. J. & Krizman, D. B. (2000) Mol. Carcinog. 28, 12–22. [PubMed] [Google Scholar]
- 23.Bussey, K. J., Kane, D., Sunshine, M., Narasimhan, S., Nishizuka, S., Reinhold, W. C., Zeeberg, B., Ajay, W. & Weinstein, J. N. (2003) Genome Biol. 4, R27.1–R27.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee, J. K., Bussey, K. J., Gwadry, F. G., Reinhold, W. C., Riddick, G., Pelletier, S. L., Nishizuka, S., Szakacs, G., Annereau, J.-P., Shankavaram, U., et al. Genome Res., in press. [DOI] [PMC free article] [PubMed]
- 25.Nishizuka, S., Chen, S. T., Gwadry, F. G., Alexander, J., Major, S. M., Scherf, U., Reinhold, W. C., Waltham, M., Charboneau, L., Young, L., et al. (2003) Cancer Res. 63, 5243–5250. [PubMed] [Google Scholar]
- 26.Bretscher, A. & Weber, K. (1980) Cell 20, 839–847. [DOI] [PubMed] [Google Scholar]
- 27.Lankes, W. T. & Furthmayr, H. (1991) Proc. Natl. Acad. Sci. USA 88, 8297–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pringault, E., Arpin, M., Garcia, A., Finidori, J. & Louvard, D. (1986) EMBO J. 5, 3119–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calnek, D. & Quaroni, A. (1993) Differentiation (Berlin) 53, 95–104. [DOI] [PubMed] [Google Scholar]
- 30.Oda, T., Kanai, Y., Oyama, T., Yoshiura, K., Shimoyama, Y., Birchmeier, W., Sugimura, T. & Hirohashi, S. (1994) Proc. Natl. Acad. Sci. USA 91, 1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barilá, D., Murgia, C., Nobili, F. & Perozzi, G. (1995) Biochim. Biophys. Acta 1263, 133–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}