

Abstract
In septic shock, reversible cardiac dysfunction starts within 24 h. Myocardial depressant factors are thought to cause myocyte dysfunction, resulting in alterations of intrinsic cardiac function. Nitric oxide is a myocardial depressant factor candidate. Here we identify endotoxin-induced myocarditis (EIM) a previously uncharacterized pathophysiological entity. Features of EIM include differential patterns of inducible NO synthase (NOS2) mRNA induction in the left (LV) and right (RV) ventricles during the systemic response inflammatory syndrome (SIRS) and the presence of myocarditis with focal areas of aseptic necrosis in the RV 24 h after SIRS induction. Even though clinical data lead to the presumption of myocardial injury in sepsis, the underlying pathophysiological mechanisms have not been previously elucidated. Gene expression profiling was used to test the hypothesis of differential LV and RV responses in EIM, and revealed novel patterns of qualitative and quantitative expansion of transcription. Those genes are novel targets for drug development in SIRS and sepsis. Our results demonstrate spatial and temporal heterogeneity of myocardial responses in EIM. These findings justify the design of treatments to ameliorate tissue injury in the RV. Because the complexity of the inflammatory response increases substantially as time elapses, we suggest a stepwise and multitarget therapeutic approach for SIRS and sepsis. Our findings can help identify innate immune pathways that could become targets for immunotherapy in the treatment of disease caused by potential bioterrorism agents.
The pathophysiology of the systemic inflammatory response syndrome (SIRS) and sepsis involves the CNS (1–3) and several peripheral systems that affect gastrointestinal, renal, and cardiovascular functions (4–6). In previous work we described temporal and spatial patterns of expression for genes encoding nitric oxide synthase type 2 (NOS2 or iNOS) and elements of the interleukin-1 system in the CNS (1, 2) during the course of lipopolysaccharide (LPS)-induced SIRS. Those data supported our hypothesis that a mechanism for immune system–brain communication could occur in brain vasculature through interleukin-1β–NOS2 interactions. To learn more about bidirectional brain–periphery immune system communications, we have studied the effects of systemic inflammation on peripheral tissues; here we focus on the heart.
Cardiac dysfunction generally starts during the first 24 h of the development of septic shock and it is reversible in survivors within 7–10 days (7, 8). In those individuals, there appear to be simultaneous right (RV) and left (LV) ventricle systolic and diastolic dysfunctions (9, 10). These changes reflect both intrinsic cardiac dysfunction and hemodynamic alterations that are characteristic of septic shock. Global myocardial ischemia has been refuted as an etiological hypotheses to explain intrinsic cardiac dysfunction during SIRS because myocardial lactate production is not increased (11), coronary blood flow is not decreased (12), and diminished oxygen extraction is more consistent with the notion of abnormal myocyte metabolism than with inadequate oxygen delivery. Currently, there is a consensus that myocardial dysfunction seems to result from myocardial depressant factors. Candidate mediators for myocardial depressant factor include NO as well as a series of other factors (such as endotoxin, cytokines, platelet-activating factor, arachidonic acid derivatives, kinins, complement components, endothelial factors, and reactive oxygen molecules) (10, 13, 14).
NO modulates both contraction and diastolic cardiac function in physiological states and in sepsis, and it may also limit the oxidative damage to cells by scavenging reactive molecules (15). Activation of constitutive NOS or NOS2 in the myocardium may play a role in the cardiac dysfunction complicating SIRS (16). We conducted studies to examine the effects of systemic inflammation in the heart, specifically to test the following three hypotheses: (i) NOS2 gene expression might be an element of the heart's response to systemic inflammation; (ii) myocardium tissue injury could be an element of the pathophysiology of SIRS; (iii) the extent of myocardium involvement could be assessed by characterization of differential patterns of gene expression.
Materials and Methods
Animals. Studies were carried out in accordance with protocols approved by the National Institutes of Health. Animals were treated as described before (1). Virus- and pathogen-free male Sprague–Dawley (200–250 g, Harlan, Indianapolis) rats were housed in a light- and temperature-controlled environment, with food and water ad libitum. Animals were treated with 5.0 mg of Escherichia coli LPS (055:B5, Sigma), prepared in 0.5 ml of saline and administered i.p. Groups of animals (n ≥ 6 per group) were studied 2, 6, or 24 h after a single injection of LPS or saline (control groups). None of the animals studied appeared to be moribund.
In Situ Hybridization Histochemistry. Hearts were removed, frozen, and stored at -80°C. Transverse sections in a plane perpendicular to the apicobasal axis were collected when they displayed the LV and the RV. Remaining LV and RV tissues were left in the frozen tissue block and subsequently used for microarray studies and semiquantitative RT-PCR. Ribonucleotide probes, directed against the rat NOS2 sequence, were used. Tissue fixing, hybridization, autoradiography, anatomical localization of the probe at the cellular level, and quantification of mRNA levels were performed as described (1). Consecutive nonhybridized slides were stained with hematoxylin and eosin for optimal histological characterization. ANOVA and post hoc analysis with the Student–Newman–Keuls test were used to determine the statistical significance of in situ hybridization results.
Microarray Studies. Total RNA was extracted from each ventricle by using TRIzol reagent (Invitrogen), cleaned with RNeasy (Qiagen, Valencia, CA), and gel verified. Samples were pooled by using the same amount of total RNA from each sample. Pools of five animals each were made as following: (i) saline LV, (ii) LPS LV, (iii) saline RV, and (iv) LPS RV for samples collected 6 and 24 h after treatment. We used GeneChip Rat Genome U34A arrays (Affymetrix, Santa Clara, CA) and followed their recommended protocol. Two independent replications of microarray experiments were performed (17).
Statistical Analyses of Microarray Data. Microarray image files were loaded into the DCHIP (18) software package. We used the Li and Wong algorithm (difference model) and computed two summary measures for each gene, the model-based expression index and present/absent call. The data set had 8,799 genes and ESTs. We removed 3,229 genes that were absent in all 16 of the samples (2 time points × 2 sides × 2 treatments × 2 replicates) from the analysis. Then we constructed a set of ANOVA models within each time point (6 and 24 h). Each set of ANOVA models consisted of individual models for each of the remaining 5,570 genes. The ANOVA model used the model-based expression index as the outcome measure and estimated the two main effects of treatment and side, and interaction effect of treatment by side. We set a significance threshold of 0.01 for each of these effects; all fold changes calculations use the formula from Affymetrix Microarray Suite software.
Chi-square (χ2) analyses were performed to investigate the difference in the numbers of up-regulated genes as defined by treatment P value ≤0.01 and standardized LPS expression > standardized saline expression between time points and sides. Z test for proportions was used to compare proportions of genes with significant up-regulated fold changes as defined by treatment effect P value ≤0.01 and treatment fold change ≥2 between sides at each time point. The proportion test was also used to compare the proportion of genes with fold change ≥5 between sides at 6 and 24 h.
Hierarchical clustering was performed by using CLUSTER and TREEVIEW software (19). Genes and arrays were clustered by using Average Linkage Clustering. Expression values in the cluster diagrams are standardized by subtracting the mean expression and dividing by the standard deviation.
Semiquantitative RT-PCR. First-strand cDNAs were prepared with SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) with 1 μg of RNA and random hexamers primers. The cDNAs were diluted five times after synthesis, and 0.5 μl were used in each PCR reaction. We used Quantum mRNA 18S Internal Standards (Ambion, Austin, TX) to perform multiplex PCR with the following modifications to the manufacturer's protocol. For all of the genes tested, except for β-actin, a ratio of 0.5:9.5 for 18S primer:competimer was used. The optimal cycle number was determined to be the one in which reactions for the specific gene and 18S in saline- and LPS-treated samples were in the linear range. For a given gene tested, all samples were assayed concomitantly, using aliquots of the same PCR mixture and first-strand cDNA derived from individual samples; and PCR products were loaded in the same agarose gel; gel was SYBR-green stained and analyzed by using AlphaImager System (Alpha Innotech, San Leandro, CA). Ratio of signal intensities of the interested gene versus the internal control was calculated for every sample. We randomly selected 11 genes with high fold change for confirmation and used β-actin as an experimental control. Data analysis was done by fitting a Liner Mixed Effects model to each of these 12 genes. Ratio values were transformed by using log (Ratio value + 1) to stabilize the variance. The ANOVA model estimated the two main effects of treatment and side, and the interaction effect of treatment by side. We set the threshold of 0.05 for each of these effects.
Results
Our studies focused in the temporal and spatial pattern of NOS2 expression in the myocardium, because NOS2 is a strong myocardial depressant factor candidate (13). In situ hybridization studies revealed that in the LV NOS2 gene expression levels increased significantly at 2 h (Fig. 1 E and F) and transiently peaked at 6 h (Fig. 1 G and H) (ANOVA, P < 0.01), then returned to baseline levels 24 h after LPS administration (Fig. 1 I and J). In the LV, NOS2 mRNA was found primarily on vasculature and myocytes after LPS treatment. This distribution is similar to previous reports (20, 21). The temporal patterns of NOS2 gene induction were compatible with those described in brain, pituitary, and peripheral tissues (1, 22, 23). In those tissues as in the LV, only residual NOS2 mRNA levels were present 24 h after SIRS induction.
Fig. 1.

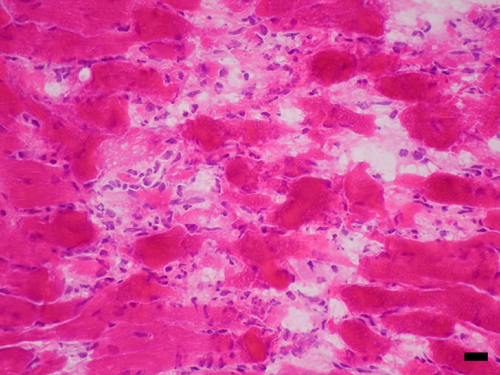
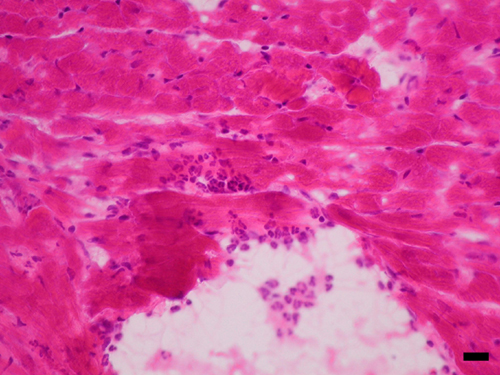
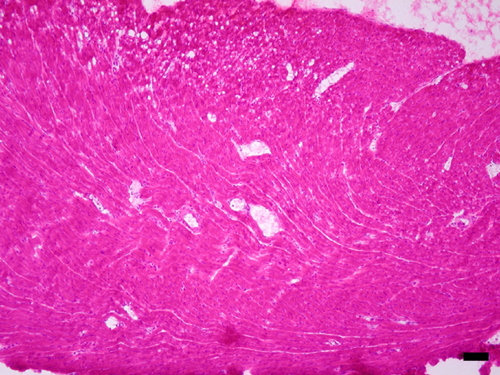
Induction of NOS2 mRNA in heart after LPS and identification of endotoxin-induced myocarditis in the RV. (A–D) A composite of film autoradiography showing the time course of NOS2 mRNA induction in the heart by in situ hybridization histochemistry. NOS2 mRNA induction starts at 2 h and progresses at 6 h after LPS administration. At 24 h, NOS2 levels return virtually to baseline in the LV, but are increased in the RV. (Scale bar = 2 mm.) (E–L)A composite of low-magnification (E, G, I, and K) and high-magnification (F, H, J, and L) images of NOS2 mRNA in heart ventricles. Shown are the LV at2h(E and F), 6 h (G and H), and 24 h (I and J) and the RV at 24 h (K and L). Arrows point to areas of increased NOS2 expression, which appear as white dots in dark-field (low magnification) and black dots in high-magnification images. Note high levels of NOS2 mRNA in the RV at 24 h (K and L). (Scale bar, 378 μm for E, G, I, and K and 30 μm for F, H, J, and L.) (M and N) Photomicrographs of hematoxylin/eosin-stained sections of heart ventricles 24 h after LPS administration. (M) In this section, the RV shows intense signs of inflammatory reaction. The myocardium is infiltrated by a moderate number of neutrophils that surround degenerate cardiomyocytes. Cardiomyocytes are fragmented, pale, and vacuolated, and in many areas the proteins are globular. Some globular proteins are surrounded by degenerate neutrophils characterized by fine nuclear debris and several neutrophils contain bright eosinophilic cytoplasmic material (phagocytosis). (N) Notice no signs of local inflammatory reaction in LV at 24 h after LPS administration. (Scale bar, 3.5 μm in M and 33.78 μm in N.)
Contrary to expectation, in the RV we found that NOS2 mRNA levels were remarkably elevated 24 h after LPS administration (Fig. 1 D, K, and L) (ANOVA, P < 0.01). NOS2 gene expression was also significantly increased at 2 and 6 h, and continued to be elevated at 24 h. Previous reports documented residual NOS2 mRNA induction at 24 h on homogenized cardiac tissue (21, 23). However, we identified a persistency of NOS2 gene induction and a novel pattern of distribution in the RV at 24 h, in which groups of cells expressed NOS2 mRNA in high levels. Those cells were observed in the free wall of the RV but spared the interventricular septum. Gene expression of NOS2 was found in cells located predominantly in the epicardium and endocardium/subendocardium, as well as in the myocardium layer. These cells were predominantly neutrophils, but endothelial cells and cardiomyocytes also expressed NOS2. At 24 h, the RV shows signs of intense inflammatory reaction, with significant neutrophil margination in vessels and invasion of myocardium (Fig. 1M, see also Figs. 6–8, which are published as supporting information on the PNAS web site), and areas of myocardium degeneration in which cardiomyocytes were fragmented, pale, and vacuolated and might contain globular proteins. Globular proteins were surrounded by degenerate leukocytes characterized by fine nuclear debris and the presence of bright eosinophilic cytoplasmic material (phagocytosis). This description is congruent with the diagnosis of acute myocarditis with areas of focal aseptic necrosis; therefore, the RV is susceptible to a process of endotoxin-induced myocarditis (EIM). No signs of local acute inflammatory reaction were found in the LV at 6 or 24 h, or RV at 6 h (Fig. 1N and Fig. 9, which is published as supporting information on the PNAS web site). These results indicate that patterns of gene expression and pathophysiological processes are different in the RV and the LV 24 h after SIRS induction.
To further characterize the transcriptional events related to EIM, we used DNA microarrays to study the RV and the LV at 6 and 24 h. Venn diagrams (Fig. 2) summarize our microarray results: 361 transcripts had at least one significant effect at 6 h (ANOVA, P ≤ 0.01) and 675 transcripts had at least one significant effect at 24 h (ANOVA, P ≤ 0.01). Genes that were differently expressed had predominantly an LPS (treatment) effect (306 and 592 transcripts for 6 and 24 h, respectively). It was unexpected to learn that more genes were significantly upregulated in the heart at 24 h than at 6 h (χ2 analysis, P = 0.0001, Table 1), because it is well documented that the expression of typical pro- and antiinflammatory genes return to levels close to baseline 24 h after SIRS induction in brain, pituitary, and peripheral tissues (1, 2, 24).
Fig. 2.

Summary of microarray results. In each Venn diagram, circles represent the main effects of our ANOVA analysis: LPS treatment (red circle), LPS treatment by side (yellow circle), and side (blue circle). Numbers inside each compartment represents the number of transcripts that are significant for that effect. The intersections of the sets represent genes with P < 0.01 for each of the effects involved in the intersection. Note the expansion of transcripts that are significant for treatment effect from 6 to 24 h.
Table 1. χ2 analyses of gene changes after LPS treatment.
| Up-regulated genes and time*
|
FC at 24 h in RV and LV*
|
FC at 6 h in RV and LV†
|
Large FC at 24 h in RV and LV*
|
Large FC at 6 h in RV and LV‡
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| With treatment effect | Entire data set | Treatment FC < ± 2 | Treatment FC ≥ ± 2 | Treatment FC < ± 2 | Treatment FC ≥ ± 2 | Treatment FC < ± 5 | Treatment FC ≥ ± 5 | Treatment FC < ± 5 | Treatment FC ≥ ± 5 | |
| 6 h | 139 | 3,903 | ||||||||
| 24 h | 423 | 3,758 | ||||||||
| Right | 375 | 174 | 183 | 76 | 505 | 44 | 246 | 13 | ||
| Left | 476 | 73 | 203 | 56 | 531 | 18 | 253 | 6 | ||
In this table, we report five separate χ2 analyses (first through fifth columns). These show associations between transcription changes and time or side of heart ventricle. The analyses revealed that significantly more genes are upregulated at 24 h than at 6 h after LPS treatment (first column). Significantly more genes had fold-change ≥ 2 in the RV at 24 h (second column) and at 6 h (third column). The proportion of genes with treatment fold changes ≥ 5 was significantly higher in the RV when compared to the LV at 24 h (fourth column), but not at 6 h (fifth column). FC, fold change.
P < 0.0001
P = 0.045
P = 0.101
More genes had fold changes 2 or higher in the RV when compared with the LV at 6 and 24 h (z test for proportions, P = 0.045 and P ≤ 0.0001, respectively; Table 1); this suggests that EIM is related to the exacerbated transcriptional response of the RV. Fig. 3 shows the cluster of genes with large transcription changes (5-fold or higher) in cardiac ventricles that have a significant treatment effect at 24 h. The number of those genes was significantly different in the RV and LV at 24 h but not at 6 h (χ2 analyses, P < 0.0001 and P < 0.101, respectively, Table 1). Semiquantitative RT-PCR confirmed the treatment effect for all 11 genes randomly selected (ANOVA, mixed effect model P < 0.05); we found no treatment effect for the β-actin gene (Fig. 4). In our studies, fold changes reflect the net change that occurs in many cell types; therefore, it is not surprising that most of our changes are relatively small, because large fold changes would reflect very large transcription increases in a significant proportion of cells.
Fig. 3.

Transcriptional patterns during EIM. Cluster analysis of genes and EST that have fold change 5 or higher at 24 h after a single LPS injection. High transcriptional fold changes accompany the histological image of acute myocarditis with focal areas of aseptic necrosis. In each treatment group, genes colored in red are up-regulated, and genes colored in green are downregulated when compared to the average gene expression across all conditions. Note that expression appears to be higher in the RV for most transcripts. The number of transcripts that have fold change 5 or higher is significantly larger in the RV than in the LV (Inset). Accession number denotes EST. 24SALR, saline treated RV at 24 h; 24SALL, saline treated LV at 24 h; 24 LPSR, LPS treated RV at 24 h; 24 LPSL, LPS treated LV at 24 h.
Fig. 4.

Semiquantitative RT-PCR of genes induced at 24 h after LPS. Our results confirm large fold changes for 11 genes randomly selected out of genes and EST with fold change 5 or higher at 24 h (ANOVA, P < 0.05). GluT, protein-glutamine γ-glutamyltransferase; Gro, growth-related oncogene; ICBP, intracellular calcium-binding protein (MRP14); IL-1b, interleukin-1 β; Lcn, lipocalin 2; MIP1a, macrophage inflammatory protein-1 α; MIP2, macrophage inflammatory protein-2 precursor; MT2, metallothionein-2; Pap, pancreatitis-associated protein precursor; Reg, regenerating gene; TIMP, tissue inhibitor of metalloproteinase-1.
In the heart, Toll-like receptor 4 and CD14 are critical in mediating the proinflammatory response induced by LPS (25). Excessive production of immunomediators such as NO and proinflammatory cytokines can be toxic, cause cardiac decompensation (7, 9), and contribute to the pathophysiological sequelae of infection, ischemia, and trauma, by causing tissue injury. Tissue injury could be critical in the heart because cardiomyocytes are unable to regenerate (26). It is therefore particularly noteworthy that we found large transcription inductions were present during the course of EIM. Based on the functions of genes that had fold changes 5 or higher (Fig. 3), we suggest that the following local processes are physiologically significant in EIM: increased leukocyte chemotaxis (macrophage inflammatory protein-1 α, macrophage inflammatory-2, and growth-related oncogene), enhanced tendency to thrombophilia (plasminogen activation inhibitor), protection from foreign antigen by removal of antigen–antibody complexes (increased Fc γ receptor), increased immunoactivation (downregulated by activation gene and leukotriene arachinodate 5-lipoxygenase-activating protein), increased cardiac remodeling, and cellular turnover [sialoprotein, lysyl oxidase, p21 protein cip (or cyclin-dependent kinase inhibitor-1A)]. Large induction of cyclohydrolase-1 suggests a novel role for this gene in the modulation of cardiac contractile function during SIRS; cyclohydrolase is an enzyme that modulates muscular tonus by catalyzing the reaction to produce tetrahydrobiopterin, an essential cofactor for phenylalanine, tyrosine, tryptophan hydroxylases, and NOS.
Interestingly, 33% of response at 6 h was replicated at 24 h (Fig. 10, which is published as supporting information on the PNAS web site). This pattern of activation could represent a stereotypical response of the innate immune system in the heart in the early phase of an acute inflammatory response. The identification of the critical elements of this standard response could therefore facilitate the management of the inflammatory process in this organ. A core of 101 genes maintained sustained activation, but as time elapsed, different genes became involved in the response with differential expression in RV and LV. Thus, analysis of gene expression patterns may help estimate the duration of such an acute inflammatory process. Several cardiac pathophysiological conditions have been associated with proinflammatory cytokine expression (27). Our microarray findings are consistent with numerous reports of transcriptional changes by LPS or cytokines and recent transcription studies (24, 28). In situ hybridization findings of NOS2 mRNA induction at 24 h were replicated in the microarray results (ANOVA, P < 0.05 for treatment, side, and treatment by side effects) and fold change of +2.3 and -1.3 for the RV and LV, respectively.
Discussion
We document an expansion of the transcriptional response in the heart from 6 to 24 h after SIRS induction: a quantitative (number of transcripts) and a qualitative (magnitude of gene expression response) expansion occurred in both ventricles, but it was more intense in the RV, which is a site of EIM, representing an intense inflammatory response in vivo. In rodents, i.p. LPS that enters the portal vein can be eliminated entirely by the liver; thus, the lymphatic system is responsible for most of the LPS found in the systemic circulation. The peak plasma LPS level occurs 6 h after a single i.p. injection, and levels are ≈10 times lower at 24 h (29). There are interspecies variations in LPS organ uptake; in rats, [125I]LPS administered intravenously is recovered predominantly in liver and blood during the first 4 h (30). The lung seems to be a later route of LPS excretion; after remaining LPS-free for several hours, LPS-positive macrophage migration into the lungs starts and intensifies with time (31). Consequently, in our experiments, the concentrations of LPS that reach both ventricles are likely to be comparable because the lung is not a site of rapid LPS uptake (32). There is a striking correlation between the uptake of LPS and tissue injury, which is dose dependent (29, 33). In the heart, endotoxin-dependent signal transduction is activated in cardiomyocytes by LPS internalization (34). Therefore, differences in ventricular uptake of LPS could contribute to variation in the progression of the inflammatory response. Other factors that could contribute to this divergence in response include transcriptional differences that occur between 6 and 24 h, anatomical, histological, physiological, or functional ventricular differences. Our results clearly demonstrate that the myocardium is a heterogeneous tissue with marked differences between RV and LV responses.
The fact that intensive care patients without coronary artery disease have an unexpectedly high percentage of elevated levels of troponin T and I has lead to the suggestion of the existence of myocardial cell injury in sepsis and SIRS (35–37). Our findings that cardiac tissue is susceptible to endotoxin-induced cytotoxicity have provided a mechanism for those observations. RV myocardial cell injury has been unrecognized until now because the pathology of the RV has not been systematically examined. Human postmortem confirmation of our data may be confounded by the impact of prolonged and severe hypotension, resuscitation interventions including drug effects, bacterial endocarditis, or acute infarction in the myocardium.
A possible strategy to decrease tissue injury in the RV could involve the use of nonsteroid antiinflammatory agents to prevent the loss of terminally differentiated cardiomyocytes. In reality, the complexity of pro- and antiinflammatory processes could proscribe the use of systemic antiinflammatory drugs at specific phases of the inflammatory cascade response. Thus, the development of stepwise myocardium-specific, multitargeted strategies to prevent the amplification of the inflammatory reaction is justified, particularly in the light of evidence of compelling and alarming unrecognized myocardial injury in sepsis (35). Such strategies would require a close follow-up of myocardium status and would entail the development of a plan to curb local leukocyte recruitment and coagulation upon elevation of cardiac troponins.
The diagram in Fig. 5 illustrates the expansion in complexity and magnitude of transcriptional responses during the initial hours of SIRS induction in the heart. This cascade-like phenomenon in cardiac ventricles could result in EIM, which is characterized by a complex pattern of responses in the RV that differ from those in the LV. We also formulate a hypothetical influence of EIM in the outcome of sepsis (Fig. 5). The extent of this amplification process contrasts dramatically with immunotherapy strategies targeting one element of this cascade. In clinical cases, symptom presentation is generally temporally remote from the initial trigger event(s); consequently, strategies based on a single element of this cascade phenomenon are not likely to be powerful enough to significantly modify downstream events and change outcome. Recently, four clinical trials addressing inadequate host responses have reduced the mortality in sepsis (38), those studies have promoted aggressive clinical management to (i) improve tissue oxygenation; (ii) ameliorate hyperglycemia and insulin resistance; (iii) improve a relative adrenal insufficiency by using low-dose replacement steroids; and (iv) address a disordered coagulation/inflammation by replacing activated protein C, an anticoagulant that has substantial antiinflammatory properties. The identification of EIM as a distinct entity highlights that the complexity and heterogeneity of systemic inflammation may still reveal novel aspects of SIRS and sepsis. As the pathophysiology of SIRS and sepsis involves several peripheral systems and the CNS (2, 39), the determination of the number, impact, and identity of therapeutic candidates could benefit from increased knowledge of temporal patterns of gene expression in several key organs.
Fig. 5.

Diagram summarizing EIM data. LPS acts through toll-like receptor and initiates the heart's response to SIRS. Each spiral represents ≈50 genes or EST that are significantly changed in our microarray studies at P ≥ 0.01. Green spirals denote the number of genes that are common to the 6- and 24-h transcriptional response; they characterize a possible stereotypic response of the innate immune system in the heart. Red and black spirals designate genes that have a fold change of 2 or higher and 5 or higher, respectively. There is a small divergence in the response of the RV and LV at 6 h, which is intensified at 24 h. Note that the expansion of the transcriptional response in the heart at 24 h is more intense in the RV (it has more red and black spirals), the site for EIM. Hypothetically, the exacerbation of EIM would lead to death and the termination of the inflammatory process would result in recovery. R, right ventricle; L, left ventricle.
In conclusion, EIM represents the heart's response to systemic inflammation. Future work should identify the mechanisms underlying the differential right and left ventricular activation in EIM. The genes we identify in this work are logical therapeutic targets that can lead to novel treatment strategies aimed at decreasing the morbidity and mortality of systemic inflammation and sepsis. The elements of the innate immune system identified here could also provide targets for immunotherapy in the treatment of disease caused by potential bioterrorism agents derived from bacterial toxins.
Supplementary Material
Acknowledgments
We thank the University of California, Los Angeles, Microarray Center and Dr. Stanley Nelson for their help and expertise in the conduction of array experiments. We thank Dr. D. Feinstein (University of Illinois, Chicago) for providing the rat NOS2 plasmid. This work was partially supported by the Intramural Research Program (Clinical Neuroendocrinology Branch, National Institute of Mental Health, Bethesda, MD). This work was also supported by National Institutes of Health Grants RR017611, RR016996, HL004526, DK058851, DK063240, HG002500, and GM061394 (to J.L.), MH062777 and RR017365 (to M.-L.W.), and RR00865. We are also supported by awards from National Alliance for Research on Schizophrenia and Depression (NARSAD) (M.-L.W.), Dana Foundation and Stanley Foundation (J.L.), and the Neuropsychiatric Institute (University of California, Los Angeles). J.H. was supported by a National Institutes of Health Postgraduate Training grant in Psychoneuroimmunology.
Abbreviations: SIRS, systemic inflammatory response syndrome; NOS2, inducible nitric oxide synthase; LPS, lipopolysaccharide; LV, left ventricle; RV, right ventricle; EIM, endotoxin-induced myocarditis.
References
- 1.Wong, M.-L., Rettori, V., Al-Shekhlee, A., Bongiorno, P. B., Canteros, G., McCann, S. M., Gold, P. W. & Licinio, J. (1996) Nat. Med. 2, 581-584. [DOI] [PubMed] [Google Scholar]
- 2.Wong, M.-L., Bongiorno, P. B., Rettori, V., McCann, S. M. & Licinio, J. (1997) Proc. Natl. Acad. Sci. USA 94, 227-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libert, C. (2003) Nature 421, 328-329. [DOI] [PubMed] [Google Scholar]
- 4.Bone, R. C. (1992) J. Am. Med. Assoc. 268, 3452-3455. [Google Scholar]
- 5.Dennhardt, R., Gramm, H. J., Meinhold, K. & Voigt, K. (1989) Prog. Clin. Biol. Res. 308, 751-756. [PubMed] [Google Scholar]
- 6.Dinarello, C. A. & Wolff, S. M. (1993) N. Engl. J. Med. 328, 106-113. [DOI] [PubMed] [Google Scholar]
- 7.Parker, M. M., Shelhamer, J. H., Bacharach, S. L., Green, M. V., Natanson, C., Frederick, T. M., Damske, B. A. & Parrillo, J. E. (1984) Ann. Intern. Med. 100, 483-490. [DOI] [PubMed] [Google Scholar]
- 8.Ellrodt, A. G., Riedinger, M. S., Kimchi, A., Berman, D. S., Maddahi, J., Swan, H. J. & Murata, G. H. (1985) Am. Heart J. 110, 402-409. [DOI] [PubMed] [Google Scholar]
- 9.Parker, M. M., McCarthy, K. E., Ognibene, F. P. & Parrillo, J. E. (1990) Chest 97, 126-131. [DOI] [PubMed] [Google Scholar]
- 10.Kumar, A., Haery, C. & Parrillo, J. E. (2000) Crit. Care Clin. 16, 251-287. [DOI] [PubMed] [Google Scholar]
- 11.Dhainaut, J. F., Huyghebaert, M. F., Monsallier, J. F., Lefevre, G., Dall'Ava-Santucci, J., Brunet, F., Villemant, D., Carli, A. & Raichvarg, D. (1987) Circulation 75, 533-541. [DOI] [PubMed] [Google Scholar]
- 12.Cunnion, R. E., Schaer, G. L., Parker, M. M., Natanson, C. & Parrillo, J. E. (1986) Circulation 73, 637-644. [DOI] [PubMed] [Google Scholar]
- 13.Grocott-Mason, R. M. & Shah, A. M. (1998) Intensive Care Med. 24, 286-295. [DOI] [PubMed] [Google Scholar]
- 14.Meldrum, D. R. (1998) Am. J. Physiol. 274, R577-R595. [DOI] [PubMed] [Google Scholar]
- 15.McCann, S. M., Licinio, J., Wong, M. L., Yu, W. H., Karanth, S. & Rettori, V. (1998) Exp. Gerontol. 33, 813-826. [DOI] [PubMed] [Google Scholar]
- 16.Ungureanu-Longrois, D., Balligand, J. L., Okada, I., Simmons, W. W., Kobzik, L., Lowenstein, C. J., Kunkel, S. L., Michel, T., Kelly, R. A. & Smith, T. W. (1995) Circ. Res. 77, 486-493. [DOI] [PubMed] [Google Scholar]
- 17.Lockhart, D. J. & Barlow, C. (2001) Nat. Rev. Neurosci. 2, 63-68. [DOI] [PubMed] [Google Scholar]
- 18.Li, C. & Wong, W. H. (2001) Proc. Natl. Acad. Sci. USA 98, 31-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95, 14863-14968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buttery, L. D., Evans, T. J., Springall, D. R., Carpenter, A., Cohen, J. & Polak, J. M. (1994) Lab. Invest. 71, 755-764. [PubMed] [Google Scholar]
- 21.Luss, H., Watkins, S. C., Freeswick, P. D., Imro, A. K., Nussler, A. K., Billiar, T. R., Simmons, R. L., del Nido, P. J. & McGowan, F. X., Jr. (1995) J. Mol. Cell Cardiol. 27, 2015-2029. [DOI] [PubMed] [Google Scholar]
- 22.Ahn, K. Y., Mohaupt, M. G., Madsen, K. M. & Kone, B. C. (1994) Am. J. Physiol. 267, F748-F757. [DOI] [PubMed] [Google Scholar]
- 23.Liu, S. F., Barnes, P. J. & Evans, T. W. (1997) Crit. Care Med. 25, 512-518. [DOI] [PubMed] [Google Scholar]
- 24.Saban, M. R., Hellmich, H., Nguyen, N. B., Winston, J., Hammond, T. G. & Saban, R. (2001) Physiol. Genomics 5, 147-160. [DOI] [PubMed] [Google Scholar]
- 25.Knuefermann, P., Nemoto, S., Misra, A., Nozaki, N., Defreitas, G., Goyert, S. M., Carabello, B. A., Mann, D. L. & Vallejo, J. G. (2002) Circulation 106, 2608-2615. [DOI] [PubMed] [Google Scholar]
- 26.Rockman, H. A., Koch, W. J. & Lefkowitz, R. J. (2002) Nature 415, 206-212. [DOI] [PubMed] [Google Scholar]
- 27.Mann, D. L. (2003) Annu. Rev. Physiol. 65, 81-101. [DOI] [PubMed] [Google Scholar]
- 28.Zhou, A., Scoggin, S., Gaynor, R. B. & Williams, N. S. (2003) Oncogene 22, 2054-2064. [DOI] [PubMed] [Google Scholar]
- 29.Yasui, M., Nakao, A., Yuuki, T., Harada, A., Nonami, T. & Takagi, H. (1995) Hepatogastroenterology 42, 683-690. [PubMed] [Google Scholar]
- 30.Warner, A. E., DeCamp, M. M., Jr., Molina, R. M. & Brain, J. D. (1988) Lab. Invest. 59, 219-230. [PubMed] [Google Scholar]
- 31.Freudenberg, M. & Galanos, C. (1990) Adv. Exp. Med. Biol. 256, 499-509. [DOI] [PubMed] [Google Scholar]
- 32.Freudenberg, M. A., Freudenberg, N. & Galanos, C. (1982) Br. J. Exp. Pathol. 63, 56-65. [PMC free article] [PubMed] [Google Scholar]
- 33.Freudenberg, M. A. & Galanos, C. (1990) Int. Rev. Immunol. 6, 207-221. [DOI] [PubMed] [Google Scholar]
- 34.Cowan, D. B., Noria, S., Stamm, C., Garcia, L. M., Poutias, D. N., del Nido, P. J. & McGowan, F. X., Jr. (2001) Circ. Res. 88, 491-498. [DOI] [PubMed] [Google Scholar]
- 35.Guest, T. M., Ramanathan, A. V., Tuteur, P. G., Schechtman, K. B., Ladenson, J. H. & Jaffe, A. S. (1995) J. Am. Med. Assoc. 273, 1945-1949. [PubMed] [Google Scholar]
- 36.Turner, A., Tsamitros, M. & Bellomo, R. (1999) Crit. Care Med. 27, 1775-1780. [DOI] [PubMed] [Google Scholar]
- 37.Thiru, Y., Pathan, N., Bignall, S., Habibi, P. & Levin, M. (2000) Crit. Care Med. 28, 2979-2983. [DOI] [PubMed] [Google Scholar]
- 38.Cohen, J. (2002) Nature 420, 885-891. [DOI] [PubMed] [Google Scholar]
- 39.Bone, R. C. (1996) Ann. Intern. Med. 125, 680-687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}