

Abstract
Instability of repetitive sequences is a hallmark of human cancer, and its enhancement has been linked to oxidative stress. Malondialdehyde is an endogenous product of oxidative stress that reacts with guanine to form the exocyclic adduct, pyrimido[1,2- α]purin-10(3H)-one (M1G). We used site-specifically modified single- and double-stranded vectors to investigate the mutagenic potential of M1G in bacteria and mammalian cells. M1G induced frameshift mutations (-1 and -2) when positioned in a reiterated (CpG)4 sequence but not when positioned in a nonreiterated sequence in Escherichia coli and in COS-7 cells. The frequency of frameshift mutations was highest when M1G was placed at the third G in the sequence. M1G induced base pair substitutions at comparable frequencies in both sequence contexts in COS-7 cells. These studies indicate that M1G, an endogenously generated product of oxidative stress, induces sequence-dependent frameshift mutations and base pair substitutions in bacteria and in mammalian cells. This finding suggests a potential role for the M1G lesion in the induction of mutations commonly associated with human diseases.
Many acquired and inherited human diseases are caused by alterations in DNA structure. For example, cancers are characterized by genomic instability that results in the accumulation of mutations (1). Microsatellites are short repetitive sequences that normally maintain a stable number of repeat units, but occasionally suffer frameshift mutations that lead to additions or deletions in the repeat units. Instability of microsatellites is linked to several cancer types and is generally attributed to mutations in mismatch repair proteins (2). However, several sporadic cancers exhibit microsatellite instability in the apparent absence of mismatch repair mutations, suggesting that other mechanisms may contribute to its evolution (2). Increasing evidence suggests that endogenous DNA damaging agents play a role in the induction of mutations responsible for human disease (3). In particular, oxidative damage that results from chronic inflammation has been implicated in the development of cancers, although the specific mechanisms by which inflammation contributes to carcinogenesis are unclear (4).
Oxidative stress decreases faithful maintenance of microsatellites in Escherichia coli and in several human cancer cell lines (5–7). Reactive oxidants can induce DNA base modifications and are a potential source of endogenous mutations (8). Although many of these oxidized bases have been examined for their mutagenic potential, relatively few induce frameshift mutations (9). In addition to base damage, oxidative stress induces indirect DNA damage by generating reactive aldehydes and epoxides from polyunsaturated fatty acid residues of membranes (10). One of these aldehydes, malondialdehyde (MDA), has been shown to revert the frameshift-sensitive Salmonella typhimurium tester strain, hisD3052 (11). This result is surprising because MDA is structurally distinct from typical frameshift mutagens that contain bulky, aromatic moieties and because the related bifunctional aldehydes, acrolein and methylglyoxal, do not induce frameshifts in Salmonella (12). MDA generates a variety of nucleic acid adducts upon reaction with DNA, and the major adduct isolated is an exocyclic adduct to guanine, pyrimido[1,2-α]purin-10(3H)-one (M1G) (Fig. 1) (13). M1G has been detected in several healthy human tissues including normal human colorectal mucosa and in human colorectal adenomas (14–18).
Fig. 1.

Endogenous generation of the M1G adduct. M1G exists in equilibrium with its ring-opened form, N2-(3-oxo-1-propenyl)-G. Ring-opening occurs at basic pH, and spontaneous conversion has been observed when M1G is opposite dC in DNA.
It is possible that M1G is the lesion responsible for MDA-induced frameshift mutations in Salmonella and that it contributes to frameshift mutations induced by oxidative stress. No studies have been performed to test this hypothesis either in bacterial or mammalian cells. Therefore, we constructed recombinant duplex M13 vectors containing single M1G residues and determined the replicative fate of the adducted genomes in WT E. coli strains or strains deficient in nucleotide excision repair (NER). To assess the mutagenic potential of M1G in mammalian cells, we inserted oligodeoxynucleotides containing a single M1G into a single-stranded shuttle vector that was replicated in simian kidney cells (COS-7). M1G was incorporated into reiterated CG-rich sequences and a nonreiterated sequence. Although it induced base pair substitutions in both sequence contexts, M1G induced frameshift mutations only in reiterated sequences. These studies not only establish that the MDA-derived adduct, M1G, is mutagenic in mammalian cells and E. coli, but also that it induces frameshift mutations in reiterated sequences that are models for microsatellites.
Materials and Methods
Materials. T4 polynucleotide kinase, T4 DNA polymerase, T4 DNA ligase, Exonuclease I, dNTP, M13K07 helper phage, BglII, BssHII, DpnI, HincII, and SphI were purchased from New England Biolabs. KspI was from Roche Molecular Biochemicals. Formamide was from Aldrich Chem (Metuchen, NJ). GELase was from Epicentre Technologies (Madison, WI). Tris·HCl, EDTA, Mops, calf thymus DNA, and lauryl sulfate were from Sigma. 5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-Gal) and isopropyl β-D-thiogalactoside (IPTG) were from Gold Biotechnology (St. Louis). Ultrafree-DA columns and Microcon-100 filters were from Amicon. CircleGrow media were purchased from Qbiogene (Carlsbad, CA). The Wizard Plus SV Miniprep system was from Promega. The QIAprep Spin Miniprep system was from Qiagen (Valencia, CA).
Oligonucleotides. The 8-mer and 19-mer oligonucleotides used in mutagenesis experiments were synthesized as described and were of the following sequences, respectively: 5′-GGTXTCCG-3′, where X = dG or M1G, and 5′-GGTGTCCG1CG2CG3-CG4GCATC-3′, where Gn = dG, 1, N2-propanodeoxyguanosine (PdG), or M1G (19). Oligonucleotide scaffolds were purchased from Integrated DNA Technologies (Coralville, IA).
The 13-mer probes used for differential hybridization were synthesized in the Vanderbilt University DNA Chemistry Core Laboratory and were of the following sequences: -1 probe, 5′-TGTCCGCGCCGGC-3′; -2 probe, 5′-GTGTCCGCGCGGC-3′.
Bacterial Strains, Cell Lines, and Vectors. Bacterial strains used for mutagenesis studies are derivatives of the E. coli strain, AB1157 [thr-1, ara-14, leuB6, Δ(gpt-proA)62, lacY1, tsx-33, supE44, falK2, λ-, rac-, hisG4, rfbD1, mgl-51, rpsL31, kdgK51, xyl-5, mtl-1, argE3, and thi-1]. The specific strains were LM102 (AB1157; [F′ traD36, proAB, LacIQZΔM15]) and LM103 (AB1157uvrA6; [F′ traD36, proAB, LacIQZΔM15]). LM102 and LM103 were constructed as described (20). The E. coli strain JM105 was used as the host bacterium for the replication of the M13MB102-1 genome on indicator plates.
COS-7 cells were obtained from the American Type Culture Collection and grown in DMEM (GIBCO/BRL) supplemented with 10% FBS under a 5% CO2 atmosphere. The E. coli strain used for amplification and analysis of the DNA recovered from COS-7 cells was XL-1 Blue (Stratagene).
The pS189 shuttle vector was a gift from Michael Seidman (National Institutes of Health, Bethesda) and Alain Sarasin (Centre National de la Recherche Scientifique, Institut Gustave-Roussy, Villejuif, France). It possesses replication origins for simian virus 40 and πAN7 for replication in mammalian and bacterial cells, respectively (21). The use of M13K07 helper phage allows production of single-stranded pS189.
Construction and Characterization of M13MB102-1 Vector. To create a sensitive assay for the specific detection of frameshift mutations in bacteria, we designed a modified M13 vector that permits white-to-blue screening for frameshift mutants. A lacZα-mutant vector was constructed by using the Sculptor in vitro Mutagenesis System (Amersham Biosciences). An oligonucleotide corresponding to bases 6272–6297 of the M13MB102 vector, but lacking a dG at position 6284, was used to introduce a 1-bp deletion at position 6284 in the 5′ region of the lacZα gene. The mutated vector, M13MB102-1, was screened for the loss of a SacI restriction site at the mutated site by restriction digestion and sequenced to confirm the deletion at position 6284. The selectable vector contains a 1-bp deletion in the lacZα gene, rendering lacZα out-of-frame. A two-base deletion or one-base addition in the lacZα gene during replication restores the reading frame and is detected as a blue plaque in a background of clear plaques on indicator plates containing X-Gal and IPTG.
Preparation of Site-Specifically Modified M13MB102-1 Vectors. Construction of gapped duplex M13MB102-1 and ligation of 19-mer oligonucleotides were performed as described with slight modifications (22). Double-stranded M13MB102-1 was linearized with KspI and SphI then dialyzed with 12-fold excess of single-stranded M13MB102-1 in decreasing concentrations of formamide. The resultant gapped-duplex DNA was isolated by a 0.8% low melting point agarose gel run in 40 mM Tris·acetate/1 mM EDTA buffer (pH 8). The gapped-duplex band was excised, and the DNA was recovered by using the enzyme GELase, according to the procedure provided by Epicentre Technologies. M1G-, PdG- and dG-containing 19-mers (100 pmol) were phosphorylated before ligation using ATP (1 mM) and T4 polynucleotide kinase in 50 mM Mops buffer (pH 7.2). For ligation, gapped-duplex DNA was added to each of the phosphorylated 19-mers along with 400 units of T4 DNA ligase and ATP (1 mM). The ligation reaction proceeded for 4 h at 16°C in 50 mM Mops buffer. The reaction mixtures were then brought up to a volume of 100 μl with water, and the DNA was purified by filtration through modified poly(vinylidene difluoride) membranes from Millipore. The ligation products were then resolved on a 40 mM Mops, 0.8% low melting point agarose gel and recovered by using GELase enzyme.
Transformation of E. coli Cells and Determination of Mutation Frequency. Cells were UV-irradiated and transformed by electroporation as described (23). In brief, bacteria in logarithmic-growth phase were SOS-induced with UV light before making them competent for transformation. The UV dose was determined by irradiating cells at increasing times from 0 to 3 min and then plating dilutions of the irradiated cells on Luria–Bertani plates. The optimal UV dose corresponded to roughly a 10% survival rate of the cells compared with no exposure. For transformation, 3 μl of DNA sample (25 ng/μl) was added to 20 μl of cells. The cell/DNA mixture was placed into a chilled GIBCO/BRL microelectroporation cuvette, and the electroporations were performed at 1.5 kV/cm by using a GIBCO/BRL Cell-Porator E. coli electroporation system. After electroporation, 1 ml of SOC medium (20 g/liter bacto-tryptone/5 g/liter bacto-yeast extract/20 mM glucose/2.5 mM KCl/10 mM MgCl2/9 mM NaCl) was added, and the bacteria were plated on Luria–Bertani plates in the presence of competent bacteria and IPTG and allowed to grow overnight.
To determine mutation frequencies, phage were eluted from the primary transformation plates, diluted, and then replated with JM105 on X-Gal/IPTG indicator plates to give ≈800 plaques per plate. Frameshift mutations (-2/+1) induced by M1G were detected by phenotypic screening with X-Gal/IPTG during the secondary plating. The adduct site in M13MB102-1 is upstream of the out-of-frame lacZ coding region, so mutations that cause a shift in the reading frame are detected as blue plaques against a background of clear plaques. The plaques on the secondary plates were then lifted by using nylon membranes, and the DNA was immobilized by UV-crosslinking. DNA was probed for frameshift mutations at position 6256 by differential hybridization with 13-mer probes.
Construction of pS189 Vectors Containing a Unique M1G Adduct. For mammalian cell studies, single-stranded pS189 DNA was digested at a unique BglII restriction site after hybridization with a 30-mer oligonucleotide to create a short region of double-stranded DNA. The oligonucleotide scaffold was removed from the linearized DNA by heating at 90°C, followed by rapid cooling to 0°C and filtration through a Microcon-100 filter. The single-stranded DNA was recircularized in buffer (50 mM Mops, pH 7.2/10 mM MgCl2) with either a 49-mer oligonucleotide scaffold, leaving an 8-bp gap, or a 51-mer scaffold, leaving a 19-bp gap, into which a lesion-containing 8-mer or 19-mer oligonucleotide was ligated (Fig. 2). Ligation was performed with 400 units of T4 DNA ligase, 1 mM ATP, in buffer (50 mM Mops, pH 7.2/10 mM MgCl2/5 mM DTT) at 15°C overnight. The scaffold was removed by the addition of an excess of its complement, heating to 90°C followed by rapid cooling in the presence of 100 mM NaCl. Remaining double-stranded DNA was removed by digestion with HincII. The constructs were ethanol-precipitated before transfection.
Fig. 2.

Scheme for site-specific incorporation of the M1G lesion into single-stranded pS189 shuttle vector. Single-stranded pS189 shuttle vector was annealed to a complementary 30-mer oligonucleotide scaffold to generate a unique BglII restriction site, digested with BglII, and hybridized to an oligonucleotide scaffold to create either an 8- or 19-bp gap. An unmodified or M1G-modified oligonucleotide was annealed to the gap, then ligated to regenerate the single-stranded vector.
Replication of pS189 in Mammalian Cells, Collection of Plasmid, and Shuttle into E. coli. COS-7 cells were transfected with the various constructs by using Lipofectamine reagent (GIBCO/BRL). Hirt lysate was made 48 h posttransfection (24), and the progeny phage were shuttled into the E. coli strain XL-1 Blue by electroporation. Transfectants were selected based on ampicillin resistance conferred by the plasmid. To select progeny plasmid containing the entire pS189 molecule (i.e., no large deletions or insertions), the XL-1 Blue transformation mixture was used to inoculate 2 ml of 2×YT (yeast/tryptone) medium supplemented with 100 μg/ml ampicillin. The culture was grown overnight, and plasmid DNA was isolated by using the Wizard Plus SV Miniprep DNA purification system (Promega). The plasmid DNA was separated on a 0.8% agarose gel, and DNA corresponding to the appropriately sized plasmid was excised and purified from the agarose by using Ultrafree-DA columns. The purified DNA was ethanol-precipitated.
Mutant Screening and Sequencing for pS189. The size-selected plasmid DNA recovered from COS-7 replication of vectors containing the 8-mer oligonucleotide sequence was used to directly transform XL-1 Blue E. coli. Transformants were selected on Luria–Bertani plates supplemented with ampicillin. Vector constructions using the 19-mer iterated sequence result in the placement of the DNA adduct in a unique BssHII restriction site. Size-selected plasmid DNA resulting from replication of 19-mer containing constructs was divided into two halves, and one half was digested with BssHII. The digested and nondigested samples were used to transform XL-1 Blue E. coli, and the ratio of colonies obtained with digested DNA to colonies obtained with undigested DNA was calculated. This value (defined as f1) was used to calculate mutation frequency, as described below.
For mutational analysis, 2 ml of CircleGrow medium supplemented with 100 μg/ml ampicillin was inoculated with isolated colonies and shaken overnight. Colonies transformed with 8-mer-containing inserts were isolated, as were colonies obtained with the BssHII-digested 19-mer-containing DNA. Plasmid DNA was extracted by using either the Wizard Plus SV Miniprep system (Promega) or the QIAprep Spin Miniprep system (Qiagen). The purified DNA was sequenced by the Vanderbilt University Sequencing Core Laboratory. Plasmids that contained a base pair substitution anywhere within the inserted oligonucleotide were scored as mutants. DNA sequences that did not contain intact flanking regions at the site of oligonucleotide insertion were discarded from the mutation analysis.
Calculation of Mutation Frequency in the (CpG)4 Sequence After COS-7 Replication. Mutation frequency for the CpG repeat sequence was calculated as described by Kamiya and Kasai (25). Plasmids in the BssHII-resistant pool comprise plasmids with a mutation in either the target site or elsewhere in the enzyme site; undigested, nonmutant plasmids; and plasmids containing a random deletion from error-prone replication in mammalian cells. The plasmids in the first group were scored as mutants. The mutation frequency (MF) was calculated as follows:
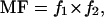 |
[1] |
where f1 = (number of colonies obtained with digested DNA)/(number of colonies obtained with undigested DNA) and f2 = (mutant colonies)/(total colonies sequenced). The plasmids from the fourth group appear to compose only a few percent of the total progeny plasmid.
Results
Frameshift Mutations in a (CpG)4 Sequence Replicated in E. coli. Oligonucleotides containing a single M1G adduct at each of four guanines in a 19-mer (CpG)4 repeat sequence (5′-GGTGTCCG1CG2CG3CG4GCATG-3′), were synthesized and purified as described. Modified duplex M13MB102-1 vectors were constructed by using the gapped-duplex method. Briefly, M1G-adducted oligonucleotides or unmodified oligonucleotides were ligated into gapped double-stranded M13MB102-1. After ligation, Form I0 DNA was isolated and transformed into SOS-induced repair-proficient or repair-deficient strains of E. coli. The frequency of frameshift mutations was analyzed by phenotypic screening of plaques after secondary plating on indicator plates containing X-Gal and IPTG. Frameshift mutations that restore the reading frame of lacZα (e.g., -2; +1) are detectable as blue plaques against a clear background.
Vectors containing the M1G adduct acquired frameshift mutations, as scored by the number of blue plaques. The mutation frequencies induced by M1G at various positions in the CpG repeat are presented in Table 1. M1G induced frameshift mutations at all four positions when transformed into the WT E. coli strain, LM102. The frequencies of reversion induced by M1G at the first, second, third, and fourth guanines in the CpG repeat sequence were 0.1 ± 0, 0.2 ± 0.2, 0.4 ± 0.3, and 0.2 ± 0.1, respectively. Sequence analysis of the bacteriophage DNA in the blue plaques revealed that the mutations were exclusively two-base deletions (ΔCG). Replication of M1G-adducted M13MB102-1 in NER-deficient cells (LM103) resulted in a 2- to 4-fold increase in frameshift mutations relative to replication in LM102 (Table 1). The highest mutation frequency was observed when M1G was at G3 in both WT and NER-deficient strains.
Table 1. Percentages of frameshift mutations detected in progeny of dG-, PdG, and M1G-adducted M13MB102–1 transformed into WT or repair-deficient E. coli strains.
| dG* | M1G-1 | M1G-2 | M1G-3 | M1G-4 | dG | PdG-1 | PdG-2 | PdG-3 | PdG-4 | |
|---|---|---|---|---|---|---|---|---|---|---|
| LM102† | <0.03 | 0.10 | 0.2 ± 0.2 | 0.4 ± 0.3 | 0.2 ± 0.1 | <0.05 | <0.03 | <0.03 | <0.03 | 0.04 ± 0.03 |
| LM103 (uvrA–) | 0.02 | 0.4 ± 0.3 | 0.4 ± 0.1 | 1.0 ± 0.3 | 0.5 ± 0.2 | <0.03 | 0.5 ± 0.3 | 0.10 ± 0.01 | 0.20 ± 0.06 | 0.30 ± 0.07 |
Sequence designations are as follows: 5′-GGTGTCCG1CG2CG3CG4GCATG-3′, where Gn = dG, M1G, or PdG
Results are expressed as mutation frequencies ± SD × 102 and were obtained by screening 1,000 plaques per experiment for each probe. Values are the average of three independent constructions, transformations, and hybridizations
M1G ring-opens to form N2-oxo-propenyl-G when placed opposite deoxycytosine in duplex DNA (Fig. 1) (26). Therefore, we used PdG as a model for the cyclic form of M1G because PdG is incapable of ring-opening in duplex DNA. PdG induced little or no frameshift mutations compared with unadducted vectors, when positioned at any of the four CpG repeat units in LM102 (Table 1). However, replication of PdG-modified M13MB102-1 vectors in NER-deficient LM103 did induce an increase in frameshift mutations (Table 1). The highest mutation frequency was observed with PdG modification at the first guanine of the CpG repeat, followed by adduction at the fourth and third positions.
Mutagenesis of M1G in Mammalian Cells. Exocyclic DNA adducts may exhibit different mutational specificities in mammalian cells than in E. coli (27). Therefore, we constructed site-specifically modified shuttle vectors for replication in mammalian cells. Single-stranded vectors are free from strand bias during replication and are poor substrates for repair enzymes. M1G was incorporated into a single-stranded shuttle vector, pS189, in two different sequence contexts (Fig. 2). In one vector, M1G was positioned at the third guanine in the frameshift-sensitive (CpG)4 repeat sequence. One key feature of this sequence is that M1G is embedded in a BssHII restriction site, which ultimately becomes a unique site in the construct. Base pair substitution or deletion mutations at or near the site of M1G render progeny plasmid resistant to cleavage by the BssHII enzyme. Thus, the mutant population may be enriched by treatment with restriction enzyme. Addition of GpC residues is not detectable via this assay because they do not eliminate cleavage by BssHII. Modified vectors were transfected into COS-7 cells. Forty-eight hours posttransfection, progeny plasmids were recovered and amplified in E. coli XL-1 Blue. Plasmid DNA from transformants was collected and sequenced to establish the mutational efficiency of the lesion, as described in Materials and Methods. Control experiments were performed with unmodified pS189 DNA.
Progeny DNA were screened for base pair substitutions and frameshift mutations. Plasmid DNA was isolated from colonies resistant to BssHII, and the types of mutations were determined by sequencing. Of 106 BssHII-resistant clones analyzed, 83 exhibited targeted mutations. The frequency of targeted mutations was 2.0%. Eighty-two percent of the targeted mutations exhibited single mutations at the site of the adduct, and 18% exhibited mutations at the site of the adduct plus a nearby point mutation. The 23 remaining clones displayed nontargeted mutations. M1G principally induced G→A and G→T base substitutions and -2 deletions (Table 2). A few G→C and -1 deletions opposite the lesion were also observed (Fig. 3). Nontargeted mutations were observed in the vicinity of M1G, either singly or associated with a targeted mutation (Fig. 3). No mutations near the target site were observed when unmodified vectors were constructed and transformed.
Table 2. Targeted mutations detected in the BssHII site of a (CpG)4 repeat in progeny of dG- and M1G-adducted pS189 after replication in COS-7 cells.
| Number of targeted mutations*
|
Percent mutation frequency
|
|||||
|---|---|---|---|---|---|---|
| M1G→A | M1G→T | M1G→C | –2 | –1 | ||
| M1G-3:C | 27 | 19 | 12 | 20 | 5 | 2.0 |
| (33)† | (23) | (14) | (24) | (6) | ||
| dG:C | 0 | 0 | 0 | 0 | 0 | <0.09 |
Targeted mutations represent both single and multiple mutations found in the vectors described in Table 1. A total of 83 targeted mutations were detected
Values in parentheses are the percentages of mutations detected
Fig. 3.

Spectra of targeted, multiple mutations (depicted above 19-mer sequence) and untargeted mutations (depicted below 19-mer sequence) observed in (CpG)4 repeat sequence after shuttle vector replication in COS-7 cells. Mutants were selected as single colonies resistant to BssHII restriction digestion and identified by sequencing plasmid DNA. The Δ symbol represents deletion mutations. The numbers in parentheses refer to the number of times a mutation was observed. The TT mutation depicted in smaller font indicates a simultaneous base pair substitution and single-base insertion at the same base pair.
A second vector was constructed in which M1G was embedded in a nonreiterated sequence context (5′-GGTXTCCG-3′, where X = M1G), and replicated in COS-7 cells. This sequence was previously used in bacterial mutagenesis studies designed to probe for base pair substitutions (23). Because the nonreiterated sequence contains no unique restriction sites, we could not enrich the mutant population by restriction digestion. Therefore, replicated shuttle vector DNA was recovered and amplified in E. coli, and single colonies were selected and sequenced. Analysis of 600 sequences from unmodified samples and 660 sequences from adducted samples provided statistical evidence for a difference in mutation efficiency between these two groups. A total of 16 mutants were analyzed. The targeted mutation frequency was 2.0%. Small numbers of G→T transversions and G→A transitions were observed (Table 3). Three of the 13 targeted mutations observed occurred in tandem with nontargeted substitutions. Three individual nontargeted base pair substitutions were also observed, as seen with the CpG repeat sequence. None of the nontargeted mutations was observed in unmodified samples. This finding suggests that M1G is responsible for inducing not only mutations opposite the lesion but also at nearby sites. No deletion mutants were detected in this sequence.
Table 3. Percentages of targeted base pair substitutions detected in progeny of dG- and M1G-adducted TGT sequence in pS189 transfected into COS-7.
| M1G→G | M1G→A | M1G→T | M1G→C | Total mutation frequency* | |
|---|---|---|---|---|---|
| M1G:C | 98.0 | 0.5 | 1.5 | <0.15 | 2.0 |
| dG:C | 99.8 | <0.17 | 0.17 | <0.17 | 0.17 |
Targeted mutations include both single and multiple mutations detected in the DNA described in Table 2. The total number of targeted mutations detected was 13.
Assuming a 2.0% mutation frequency in M1G-modified samples and a 10-fold lower frequency for unmodified samples, 620 sequences were required for each group to afford 80% power to detect a significant difference (α = 0.05). A total of 630 M1G- and 620 dG-samples were sequenced to determine mutation frequencies. Results are expressed as mutation frequency × 102
Discussion
In this study, we compared the spectrum and frequency of mutations induced by a site-specific M1G adduct replicated in E. coli or COS-7 cells. The results of the bacterial studies showed differences in frameshift potential dependent on the position of M1G in a (CpG)4 repeat, with the adduct in the third CpG unit exhibiting the highest mutagenicity in both NER-deficient and NER-proficient strains of E. coli. The mutagenicity studies in COS-7 cells assayed for the induction of base pair substitutions and frameshifts by M1G within two sequence contexts. We found a targeted mutation frequency of 2.0% when M1G was positioned in either sequence. The most striking observation was the induction of frameshifts only in a (CpG)4 sequence, although both sequences acquired base pair substitutions.
NMR studies have shown M1G in duplex DNA to spontaneously ring-open to form N2-oxo-propenyl-G (28), whereas the exocyclic ring of M1G is stabilized opposite a two-base deletion in a CpG repeat (29). Both adduct forms distort the DNA helix, but there is no structural evidence to indicate which is present in the active site of a DNA polymerase. The exocyclic ring of PdG is unable to open and therefore is a model for ring-closed M1G. Pausing of DNA polymerases at oxidative DNA lesions may provide an opportunity for misalignment and has been proposed as a mechanism by which oxidative stress contributes to microsatellite instability (5). Surprisingly, PdG exhibited little frameshift potential in this study, although it is a strong replicative block. Previous work from our laboratory showed PdG to induce frameshifts at a frequency of 2.5% in the E. coli strain JM105 (31). In addition to the difference in the E. coli strains used for genome replication, differences in the lengths of the oligonucleotides used for vector construction and in the phenotypic assays used for mutant detection may contribute to the disparate results between the two studies. Changes in vector design and construction were adopted for the present study to minimize the possibility for genetic engineering artifacts.
An attractive hypothesis is that the ring-opened derivative of M1G, N2-oxo-propenyl-G, is the actual mutagenic lesion. The oxo-propenyl moiety may participate in base-pairing with a partner cytosine, possibly stabilizing a slipped mispairing intermediate of frameshift mutagenesis. However, M1G also induced frameshifts when introduced into single-stranded DNA, where it is expected to remain in the ring-closed form until a base (mainly cytosine) is incorporated. It will be interesting to investigate the possibility that M1G undergoes some degree of ring-opening during replicative bypass that might account for the induction of frameshift mutations in single-stranded vectors.
The primary base pair substitutions observed after replication of M1G in COS-7 cells are M1G→T transversions and M1G→A transitions, followed by M1G→C transversions. Fink et al. (23) observed a similar trend with site-specifically modified M13 phage genomes replicated in E. coli when M1G was positioned in the TGT sequence. The similarities in mutation frequency and spectrum between E. coli and mammalian cells are noteworthy. There have been several reports of differences in mutation frequency between mammalian cells and bacteria. Levine et al. (32) observed that 1, N6-ethenodeoxyadenosine strongly miscodes in mammalian cells, but does not miscode in E. coli. In contrast, the mutation frequency of PdG decreased from ≈70–100% in E. coli to 8% in COS cells (33). However, there are differences between the designs of our bacterial and mammalian cell studies, which may mask variations in mutation frequency. The use of single-stranded vectors enabled us to examine the ability of M1G to induce mutations in the absence of repair or replication strand bias in COS-7 cells, whereas double-stranded vectors were subject to both events in E. coli. Additionally, the distance of the DNA adduct in relation to the origin of replication may affect mutation rates (34).
Because of rigorous screening of progeny vector DNA by sequencing, we found that replication bypass of M1G is associated with a high incidence of multiple and untargeted mutations (Fig. 3). The cause of these untargeted mutations is unclear, although the ability of one DNA adduct to cause more than one mutation, even several bases away, has been observed by others (35–38). M1G may introduce an area of instability in DNA that could interfere with faithful replication beyond the adduct, causing multiple mistakes to be made by DNA polymerases. Recently identified error-prone, lesion-replicating polymerases are thought to be recruited to sites of DNA damage and displace a blocked replicative polymerase (39). These polymerases may incorporate beyond the damage site, inducing nearby mutations.
The mutation spectra observed in our study reflect those reported for lacZα mutations induced by treatment of M13 phage with MDA (20). In that study, 84% of mutations detected at G were base pair substitutions, with the remaining mutants comprising +G and ΔCG mutations. However, it is likely that not all of the frameshift mutations observed after MDA treatment can be attributed to M1G. One base pair additions in runs of deoxycytidine were also detected after MDA treatment, thereby suggesting a role for the MDA-deoxycytidine adduct M1C in frameshift generation. Other MDA–DNA adducts, such as the deoxyadenosine adduct, M1A, may also play an important role in MDA-induced mutagenesis. M1A and M1C do not undergo ring-closure, and the adducted moiety remains in the oxopropenylated form. It is likely that if the ring-opened form of M1G is responsible for the mutagenicity observed in our study, this may indicate a mutagenic potential for M1C and M1A. It will be interesting to evaluate the mutagenicity of these adducts in reiterated sequences known to be mutated in colorectal cancer [e.g., the A10 sequence in the TGF-βRII gene (40)].
M1G is produced endogenously by products of lipid peroxidation, prostaglandin biosynthesis, and DNA oxidation (Fig. 1) (41, 42). It has been detected in normal and malignant tissue (17, 18). Our findings demonstrate that M1G is a premutagenic lesion in mammalian cells and induces frameshift and base pair substitution mutations in both E. coli and COS-7 cells, demonstrating that M1G is likely to be responsible for some of the frameshift potential of MDA. The mechanism by which oxidative stress contributes to microsatellite instability is incompletely understood; however, oxidative DNA damage is likely to be a contributing factor. Surprisingly, although the mutagenicity of 19 DNA adducts derived from direct oxidation or lipid peroxidation products has been studied by using site-specific methodology, only a few have been shown to induce frameshift mutations, and those in very low frequency [i.e., 8-oxo-deoxyguanosine, 2-hydroxy-deoxyadenosine, 5-formyldeoxyuracil, and 3,N4-etheno-2-deoxycytidine (25, 43–46)]. Frameshift mutations that occur within the coding region of genes are often deleterious because these typically abrogate protein function. Instability of microsatellite sequences in genes involved in tumorigenesis has been linked to development of several types of sporadic cancers. For example, frameshifts are observed in reiterated deoxyguanosine tracts of Bax, APC, TGF-βRII, and IGFIIR (47–49). Although not demonstrated for all cancers that accumulate these frameshift mutations, microsatellite instability is often attributed to deficiencies in mismatch repair. Interestingly, PdG and, by analogy M1G, are recognized and can be repaired by the mismatch repair system (30). Thus, the production of MDA-derived adducts in reiterated base sequences may significantly increase sensitivity to frameshift mutation in mismatch-repair deficient individuals. Our findings suggest that the ability of MDA and its derived adducts to induce frameshift and base pair substitution mutations may provide a direct link between oxidative stress and human disease.
Acknowledgments
We are grateful to C. A. Rouzer for a critical reading. This work was supported by National Institutes of Health Grants CA87819, ES00267, and CA68484. L.A.V. was supported by National Institutes of Health Training Grant T32ES07028.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MDA, malondialdehyde; M1G, pyrimido[1,2-α]purin-10(3H)-one; PdG, 1,N2-propanodeoxyguanosine; NER, nucleotide excision repair; X-Gal, 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside; IPTG, isopropyl β-D-thiogalactoside.
References
- 1.Lengauer, C., Kinzler, K. W. & Vogelstein, B. (1998) Nature 396, 643-649. [DOI] [PubMed] [Google Scholar]
- 2.Jackson, A. L. & Loeb, L. A. (1998) Genetics 148, 1483-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marnett, L. J. & Plastaras, J. P. (2001) Trends Genet. 17, 214-221. [DOI] [PubMed] [Google Scholar]
- 4.Jackson, A. L. & Loeb, L. A. (2001) Mutat. Res. 477, 7-21. [DOI] [PubMed] [Google Scholar]
- 5.Jackson, A. L., Chen, R. & Loeb, L. A. (1998) Proc. Natl. Acad. Sci. USA 95, 12468-12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zienolddiny, S., Ryberg, D. & Haugen, A. (2000) Carcinogenesis 21, 1521-1526. [PubMed] [Google Scholar]
- 7.Gasche, C., Chang, C. L., Rhees, J., Goel, A. & Boland, C. R. (2001) Cancer Res. 61, 7444-7448. [PubMed] [Google Scholar]
- 8.Cooke, M. S., Evans, M. D., Dizdaroglu, M. & Lunec, J. (2003) FASEB J. 17, 1195-1214. [DOI] [PubMed] [Google Scholar]
- 9.Kamiya, H. (2003) Nucleic Acids Res. 31, 517-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marnett, L. J., Riggins, J. N. & West, J. D. (2003) J. Clin. Invest. 111, 583-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukai, F. H. & Goldstein, B. D. (1976) Science 191, 868-869. [DOI] [PubMed] [Google Scholar]
- 12.Marnett, L. J., Hurd, H. K., Hollstein, M. C., Levin, D. E., Esterbauer, H. & Ames, B. N. (1985) Mutat. Res. 148, 25-34. [DOI] [PubMed] [Google Scholar]
- 13.Seto, H., Okuda, T., Takesue, T. & Ikemura, T. (1983) Bull. Chem. Soc. Jpn. 56, 1799-1802. [Google Scholar]
- 14.Chaudhary, A. K., Nokubo, M., Reddy, G. R., Yeola, S. N., Morrow, J. D., Blair, I. A. & Marnett, L. J. (1994) Science 265, 1580-1582. [DOI] [PubMed] [Google Scholar]
- 15.Vaca, C. E., Fang, J.-L., Mutanen, M. & Valsta, L. (1995) Carcinogenesis 16, 1847-1851. [DOI] [PubMed] [Google Scholar]
- 16.Rouzer, C. A., Chaudhary, A. K., Nokubo, M., Ferguson, D. M., Reddy, G. R., Blair, I. A. & Marnett, L. J. (1997) Chem. Res. Toxicol. 10, 181-188. [DOI] [PubMed] [Google Scholar]
- 17.Everett, S. M., Singh, R., Leuratti, C., White, K. L., Neville, P., Greenwood, D., Marnett, L. J., Schorah, C. J., Forman, D., Shuker, D. & Axon, A. T. (2001) Cancer Epidemiol. Biomarkers Prev. 10, 369-376. [PubMed] [Google Scholar]
- 18.Leuratti, C., Watson, M. A., Deag, E. J., Welch, A., Singh, R., Gottschalg, E., Marnett, L. J., Atkin, W., Day, N. E., Shuker, D. E. & Bingham, S. A. (2002) Cancer Epidemiol. Biomarkers Prev. 11, 267-273. [PubMed] [Google Scholar]
- 19.Schnetz-Boutaud, N., Mao, H., Stone, M. P. & Marnett, L. J. (2000) Chem. Res. Toxicol. 13, 90-95. [DOI] [PubMed] [Google Scholar]
- 20.Benamira, M., Johnson, K., Chaudhary, A., Bruner, K., Tibbetts, C. & Marnett, L. J. (1995) Carcinogenesis 16, 93-99. [DOI] [PubMed] [Google Scholar]
- 21.Seidman, M. (1989) Mutat. Res. 220, 55-60. [DOI] [PubMed] [Google Scholar]
- 22.Burcham, P. C. & Marnett, L. J. (1994) J. Biol. Chem. 269, 28844-28850. [PubMed] [Google Scholar]
- 23.Fink, S. P., Reddy, G. R. & Marnett, L. J. (1997) Proc. Natl. Acad. Sci. USA 94, 8652-8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirt, B. (1967) J. Mol. Biol. 26, 365-369. [DOI] [PubMed] [Google Scholar]
- 25.Kamiya, H. & Kasai, H. (1997) Nucleic Acids Res. 25, 304-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao, H., Reddy, G. R., Marnett, L. J. & Stone, M. P. (1999) Biochemistry 38, 13491-13501. [DOI] [PubMed] [Google Scholar]
- 27.Pandya, G. A. & Moriya, M. (1996) Biochemistry 35, 11487-11492. [DOI] [PubMed] [Google Scholar]
- 28.Mao, H., Schnetz-Boutaud, N. C., Weisenseel, J. P., Marnett, L. J. & Stone, M. P. (1999) Proc. Natl. Acad. Sci. USA 96, 6615-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnetz-Boutaud, N. C., Saleh, S., Marnett, L. J. & Stone, M. P. (2001) Biochemistry 40, 15638-15649. [DOI] [PubMed] [Google Scholar]
- 30.Johnson, K. A., Mierzwa, M. L., Fink, S. P. & Marnett, L. J. (1999) J. Biol. Chem. 274, 27112-27118. [DOI] [PubMed] [Google Scholar]
- 31.Benamira, M., Singh, U. S. & Marnett, L. J. (1992) J. Biol. Chem. 267, 22392-22400. [PubMed] [Google Scholar]
- 32.Levine, R. L., Yang, I. Y., Hossain, M., Pandya, G. A., Grollman, A. P. & Moriya, M. (2000) Cancer Res. 60, 4098-4104. [PubMed] [Google Scholar]
- 33.Moriya, M., Zhang, W., Johnson, F. & Grollman, A. P. (1994) Proc. Natl. Acad. Sci. USA 91, 11899-11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pavlov, Y. I., Newlon, C. S. & Kunkel, T. A. (2002) Mol. Cell 10, 207-213. [DOI] [PubMed] [Google Scholar]
- 35.Kramata, P., Zajc, B., Sayer, J. M., Jerina, D. M. & Wei, C. S. (2003) J. Biol. Chem. 278, 14940-14948. [DOI] [PubMed] [Google Scholar]
- 36.Burnouf, D., Gauthier, C., Chottard, J. C. & Fuchs, R. P. P. (1990) Proc. Natl. Acad. Sci. USA 87, 6087-6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yarema, K. J., Lippard, S. J. & Essigmann, J. M. (1995) Nucleic Acids Res. 23, 4066-4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan, X., Suzuki, N., Johnson, F., Grollman, A. P. & Shibutani, S. (1999) Nucleic Acids Res. 27, 2310-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodman, M. F. (2002) Annu. Rev. Biochem. 71, 17-50. [DOI] [PubMed] [Google Scholar]
- 40.Lu, S. L., Akiyama, Y., Nagasaki, H., Saitoh, K. & Yuasa, Y. (1995) Biochem. Biophys. Res. Commun. 216, 452-457. [DOI] [PubMed] [Google Scholar]
- 41.Marnett, L. J. (1999) Mutat. Res. 424, 83-95. [DOI] [PubMed] [Google Scholar]
- 42.Dedon, P. C., Plastaras, J. P., Rouzer, C. A. & Marnett, L. J. (1998) Proc. Natl. Acad. Sci. USA 95, 11113-11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Page, F., Margot, A., Grollman, A. P., Sarasin, A. & Gentil, A. (1995) Carcinogenesis 16, 2779-2784. [DOI] [PubMed] [Google Scholar]
- 44.Kamiya, H. & Kasai, H. (1997) Biochemistry 36, 11125-11130. [DOI] [PubMed] [Google Scholar]
- 45.Miyabe, I., Zhang, Q. M., Sugiyama, H., Kino, K. & Yonei, S. (2001) Int. J. Radiat. Biol. 77, 53-58. [DOI] [PubMed] [Google Scholar]
- 46.Basu, A. K., Wood, M. L., Niedernhofer, L. J., Ramos, L. A. & Essigmann, J. M. (1993) Biochemistry 32, 12793-12801. [DOI] [PubMed] [Google Scholar]
- 47.Paoloni-Giacobino, A., Rey-Berthod, C., Couturier, A., Antonarakis, S. E. & Hutter, P. (2002) Hum. Genet. 111, 284-289. [DOI] [PubMed] [Google Scholar]
- 48.Huang, J., Papadopoulos, N., McKinley, A. J., Farrington, S. M., Curtis, L. J., Wyllie, A. H., Zheng, S., Willson, J. K., Markowitz, S. D., Morin, P., et al. (1996) Proc. Natl. Acad. Sci. USA 93, 9049-9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ouyang, H., Shiwaku, H. O., Hagiwara, H., Miura, K., Abe, T., Kato, Y., Ohtani, H., Shiiba, K., Souza, R. F., Meltzer, S. J. & Horii, A. (1997) Cancer Res. 57, 1851-1854. [PubMed] [Google Scholar]