

Abstract
Apoptosis is a highly regulated form of cell death, characterized by distinctive features such as cellular shrinkage and nuclear condensation. We demonstrate here that proteolytic activation of hPAK65, a p21-activated kinase, induces morphological changes and elicits apoptosis. hPAK65 is cleaved both in vitro and in vivo by caspases at a single site between the N-terminal regulatory p21-binding domain and the C-terminal kinase domain. The C-terminal cleavage product becomes activated, with a kinetic profile that parallels caspase activation during apoptosis. This C-terminal hPAK65 fragment also activates the c-Jun N-terminal kinase pathway in vivo. Microinjection or transfection of this truncated hPAK65 causes striking alterations in cellular and nuclear morphology, which subsequently promotes apoptosis in both CHO and Hela cells. Conversely, apoptosis is delayed in cells expressing a dominant-negative form of hPAK65. These findings provide a direct evidence that the activated form of hPAK65 generated by caspase cleavage is a proapoptotic effector that mediates morphological and biochemical changes seen in apoptosis.
In response to a variety of death stimuli such as activation of death receptors TNFR/Fas and UV irradiation, cells undergo apoptosis, an essential mechanism for maintaining normal tissue homeostasis. The hallmark events of apoptosis are morphological changes including nuclear condensation, cell shrinkage, membrane blebbing, and the formation of apoptotic bodies. These morphological changes are also accompanied by biochemical changes including DNA fragmentation and elevation of cytoplasmic Ca2+. The essential molecules that coordinate all these events are the caspase family of cysteinyl aspartate-directed proteases. Caspases exist as zymogens until proteolytically activated, either autocatalytically or by other caspases (reviewed in ref. 1). Among the caspase family, caspase-8/FLICE (2, 3), which can interact with death receptor-associated proteins such as FADD (4, 5), acts upstream of the proteolytic cascade. The ubiquitously expressed caspase-3 (CPP32/Yama/apopain; ref. 6), on the other hand, is implicated as a downstream effector protease of the proteolytic cascade (7). In a cell-free apoptosis system, naïve nuclei can be induced into apoptotic fragmentation when incubated with either lysates from apoptotic cells, or a combination of active caspase-3 and nonapoptotic lysates (8, 9). Therefore, the identification of substrates for caspase-3 is crucial for understanding signaling pathways leading apoptosis. By far, several molecules have been known to be cleaved by caspase-3 (reviewed in ref. 10); however, most substrates are destroyed by cleavage, and the biological function of the cleavage products in apoptotic cells remains unclear.
hPAK65 is a serine/threonine kinase (p21-activated kinase) that can be activated in vitro upon interaction with activated Rac1 and Cdc42, two p21-GTPases of the Rho subfamily and regulators of cell morphology and growth. Three mammalian PAKs have been identified with homologues in several species: human PAK1 (rat α-PAK/PAK65; refs. 11–13), human hPAK65/PAK2 (rat γ-PAK, rabbit PAKI; refs. 12, 14–17), and mouse mPAK3 (rat β-PAK; refs. 11 and 18). In contrast to the limited distribution of PAK1 and PAK3, hPAK65 is ubiquitously expressed in all tissues. The PAK proteins consist of an N-terminal p21-binding domain and a C-terminal kinase domain joined by a variable linker region. Treatment of hPAK65 with trypsin in vitro has been shown to cleave hPAK65 in the linker region, releasing a 40-kDa fragment containing the kinase domain, which subsequently becomes activated by autophosphorylation (17). The PAK proteins also can be activated in response to chemoattractants and coagulants (12, 15). However, the molecular mechanism by which PAK is regulated by these and other stimuli in vivo has not been shown.
PAKs may participate in the modulation of cytoskeleton. Ste20p has been shown to bind an actin-associated protein Bem1p (19). Additionally, PAK1 has be shown to phosphorylate and activate the myosins (20, 21), and the expression of a constitutively active PAK1 results in actin reorganization (22, 23). Thus, hPAK65 represents candidate effector proteins that may mediate morphological changes during apoptosis.
In this study, we identify hPAK65 as a substrate of caspases during apoptosis, and we demonstrate that hPAK65 is enzymatically activated by this cleavage. Most important, we show that the active hPAK65 induces morphological changes characteristic of apoptosis and enhances apoptosis in epithelial cells. Additionally, dominant-negative hPAK65 delays apoptosis. Our results indicate that hPAK65 plays an important effector role in death signaling.
MATERIALS AND METHODS
Cell Culture and Transfection.
Jurkat and HL-60 cells were grown in RPMI 1640 medium. Rat embryo fibroblasts (REF52), COS-7, NIH 3T3, and Hela cells were grown in high glucose DMEM. CHO cells were grown in HAM F12 medium. All cultures were supplemented with 10% heat-inactivated fetal calf serum (20% for HL-60). The transfection procedure has been described (24): for each 10-cm plate of cells, 5 μg total DNA including 1 μg marker plasmid encoding green fluorescence protein (GFP) or CD20 was transfected with lipofectamine (for NIH 3T3 and CHO; GIBCO/BRL) or LT-1 (for Hela and COS-7; TransIT, Madison, WI) according to the manufacturer’s suggestion. Transfection for kinase assays was the same except that 24 hr after transfection, cells were starved in DMEM without serum for another 16 hr and then harvested.
Preparation of Jurkat Cytosol, Caspases, and Cell-Free Apoptosis Assay.
Preparation of Jurkat cytosol was based on ref. 8 with some modifications. Briefly, 8 liters of suspension Jurkat cells were concentrated and washed in cold RPMI 1640, resuspended in low salt buffer (10 mM Hepes, pH 7.4, with 50 mM NaCl, 5 mM EGTA, 2 mM MgCl2, 2 mM DTT, and 200 μM PMSF at 3 × 108 cells per ml) and subjected to four freeze–thaw cycles. Membrane debris were removed by 20,000 × g, followed by 100,000 × g centrifugation. The supernatant was fractionated on sequential preparative Q and S Sepharose HP resins (Pharmacia).
Caspase-3 and caspase-8 were expressed in Escherichia coli and were fully active when assayed on the fluorogenic substrate DEVD-AFC (Enzyme Systems Products, Livermore, CA; excitation, 405 nm; emission, 505 nm). The caspase-3 was more than 80% pure, consisting of 17- and 11-kDa subunits as assayed by SDS/PAGE. The cell-free apoptosis system was essentially as described (8). Recombinant caspase-3 was added to Jurkat cytosol or chromatography fraction aliquots supplemented with 2 mM DTT and an ATP regeneration system and was incubated 30 min at 25°C. Naive Jurkat nuclei were added, and the preparations were further incubated at 37°C for 1 hr. Apoptotic nuclei were identified microscopically using Hoechst stain 23259. Jurkat nuclei were prepared essentially as described (25), except that cytocalasin B was not included, and the lysed suspension was centrifuged 20,000 × g over a 2-M sucrose pad.
Preparation of Fas-Triggered Jurkat Cell Extracts.
Jurkat cells were starved in RPMI 1640 without serum for 2 hr, then resuspended at 1 × 107 cells per ml in RPMI 1640 containing 500 ng/ml Fas antibody (CH-11, Upstate Biotechnology, Lake Placid, NY). Aliquots were taken at times indicated, washed, and resuspended in PBS. One-fifth of the samples were microfuged and resuspended in a freeze–thaw lysis buffer (10 mM Hepes, pH 7.5, 50 mM NaCl, 2 mM MgCl2, 1 mM EGTA, 1 mM EDTA, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 0.5 mM PMSF, and 10 μg/ml aprotonin). After three freeze–thaw cycles, cells were centrifuged and measured for caspase-3 activity on 50 μM substrate DEVD-AFC. The rest of the cell suspension was resuspended in lysis buffer [20 mM Tris, 137 mM NaCl, 1% Triton X-100, 15% glycerol, 50 μM Z-DEVD-FMK (Enzyme Systems Products), 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM PMSF, 10 μg/ml aprotonin], microfuged, and prepared for immunoblot analysis and kinase assays.
In-Gel Kinase Assay and PAK Activity Assay.
The in-gel kinase assay was performed as described (26), except that the separating gel was polymerized with 160 μg/ml histone H4 (Boehringer Mannheim). Endogenous hPAK65 kinase activity was measured by immunoprecipitation with hPAK65-C antibody, followed by an in vitro kinase reaction in kinase buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1 mM MnCl2, 50 μM ATP, 5 μCi[γ-32P]ATP) containing substrate Histone H4 (60 μg/ml). To analyze the COS-expressed PAKs, myc-tagged proteins were immunoprecipitated with anti-myc antibody (9E10, Boehringer Mannheim) with or without incubation with caspase-3 for 30 min at 37°C. Kinase assays were performed as above.
Plasmids.
Full-length hPAK65 was amplified from a human heart cDNA library (GIBCO/BRL), subcloned into a pET-28a vector downstream of a T7 promoter followed by a His-tag, and fully sequenced. Truncated hPAK65, denoted hPAK65-ΔN212 and representing a deletion of the protein N-terminal to the caspase-3 cut site, was amplified from the full-length DNA. For eukaryotic cell expression, all constructs were subcloned in the pBJ-myc vector containing an N-terminal myc-tag. PAK1 was also subcloned into the same pET or pBJ-myc vector.
Antibody.
The hPAK65-C antibody was a rabbit polyclonal IgG made to a C-terminal, 15-residue peptide of the hPAK65 sequence. The antibody recognized both recombinant PAK1 and hPAK65 expressed from E. coli.
c-Jun N-terminal Kinase (JNK) Assay.
JNK kinase assay was performed as described (24). COS-7 cells were transfected with 1 μg of HA-tagged JNK and 4 μg of various constructs in pBJ-myc vector as indicated. Cell lysates were immunoprecipitated with anti-HA antibody (Boehringer Mannheim), and the kinase activity was assayed using glutathione S-transferase (GST)-Jun (60 μg/ml; Santa Cruz Biotechnology) as substrate. Equal levels of JNK from each sample were judged by immunoblotting using anti-HA antibody.
Stable CHO Cell Lines and Crosslinking of Chimeric Receptors.
Constructs encoding the extracellular and transmembrane domains of human CD4 fused to either the wild-type cytoplasmic domain of murine Fas (Fas-wt-4) or a point mutant version in which Ile-225 was substituted by Val (Fas-cg-7) were transfected into CHO-K1 cells. Cells stably expressing the chimeric receptors were selected in 400 μg/ml Geneticin (GIBCO/BRL), and individual clones were analyzed for chimeric receptor expression by FACS analysis using antibodies to CD4. To trigger apoptosis, Fas-wt-4 or Fas-cg-7 CHO cells were treated first with 500 ng/ml anti-CD4 (Becton Dickinson) followed by 5 μg/ml anti-mouse IgG+IgM (Jackson ImmunoResearch). Fas-wt-4 cells are competent to trigger apoptosis upon CD4 crosslinking, whereas FAS-cg-7 cells are not.
Microinjections and Immunofluorescence Staining.
REF52 cells were microinjected with plasmids (0.5 μg/ml) as described (27). Sixteen hours after injection, cells were fixed with 4% paraformaldehyde, permeablized with 0.1% Triton X-100, and stained first with 20 μg/ml anti-myc antibody and then with the Texas Red-conjugated anti-mouse IgG (1/500; Amersham) and DAPI (0.5 μg/ml; Molecular Probes).
Flow Cytometry.
Transfected cells were collected and blocked with 5% normal mouse serum, followed by staining with phosphatidylethanolamine (PE)-conjugated CD20 (1/10; DAKO). Flow cytometry analysis was performed within 1 hr after cells were incubated with fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide (PI) as suggested by the manufacturer (R & D Systems). In each sample, 10,000 cells (CD20-positive, representing the transfected population) were analyzed.
RESULTS
Cleavage of hPAK65 by Caspases During Apoptosis.
To isolate substrates of caspases that may be actively involved in apoptosis, Jurkat cell cytosol was fractionated by sequential anion- and cation-exchange chromatography. Fraction aliquots were treated with recombinant caspase-3, added to naïve nuclei, and then assayed for apoptotic activity by quantitating the percentage of apoptotic nuclei. SDS/PAGE analysis followed by Coomassie staining was performed on column fractions incubated with and without added caspase-3 to identify candidate substrates that underwent a decrease in molecular weight following caspase treatment. A comparison of the apoptotic activity profile with the results of SDS/PAGE analysis revealed that in column fractions exhibiting peak apoptotic activity, a 65-kDa protein was cleaved into a 30- and a 36-kDa fragment, respectively, on the addition of caspase-3. Following preparative SDS/PAGE, the N terminus of the 36-kDa protein was sequenced and found to correspond to amino acids 213–242 of hPAK65/PAK2 (12, 14–17). The published hPAK65 sequence immediately upstream of the cleavage site is the pentapeptide DSHVD^, which is similar to the consensus recognition sequence for caspase-3 (28). The caspase-3 cleavage site (D212̂G213) is located in the linker region between the regulatory and kinase domain, 16 aa C-terminal to a known trypsin cleavage site (17). The hPAK65 homologues PAK1 and PAK-3 are most dissimilar to hPAK65 in this region and include insertions that interrupt the region susceptible to caspase-3 cleavage.
To examine whether the cleavage of endogenous hPAK65 occurs in cells undergoing apoptosis, we stimulated Jurkat cells with an agonistic anti-Fas antibody and compared the appearance of caspase activity with the cleavage of hPAK65 (Fig. 1A). Caspase activity, measured by cleavage of the fluorogenic peptide substrate DEVD-AFC, appeared by 1 hr and increased until ≈2 hr. At this time, apoptotic bodies were fully evident (data not shown). Western blotting using anti-peptide antibodies to the C terminus of hPAK65 showed that with similar kinetics to caspase activation, the 65-kDa full-length hPAK65 disappeared with the concomitant appearance of a 36-kDa species (Fig. 1B). The 68-kDa species, presumably PAK1, was not cleaved during the time studied (Fig. 1B).
Figure 1.
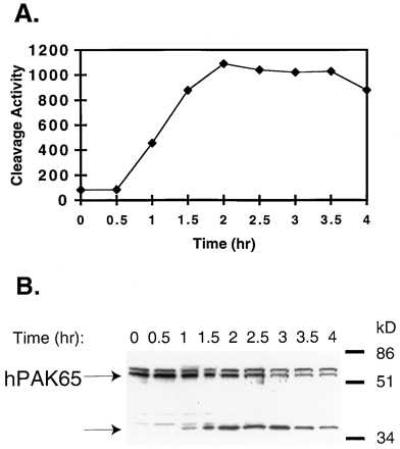
Endogenous hPAK65 cleavage during Fas-induced apoptosis. (A) Kinetics of caspase activation in Fas-stimulated Jurkat cells. Apoptosis was induced in Jurkat cells using anti-Fas antibody, and aliquots were taken at times indicated. Caspase-3 activity was measured on fluorogenic substrate DEVD-AFC. (B) Endogenous hPAK65 cleavage in Fas-stimulated Jurkat cells. The Jurkat cell extracts aliquoted from the same preparation were analyzed for hPAK65 cleavage by immunoblotting by using hPAK65-C antibody. The full-length and cleavage products of hPAK65 are indicated.
Caspase cleavage of hPAK65 also was observed in apoptotic CHO cells stably overexpressing a wild-type CD4-Fas chimeric receptor. Crosslinking of the extracellular domain of CD4 induced the CHO cells into apoptosis, resulting in the cleavage of hPAK65, whereas no hPAK65 cleavage occurred in crosslinked CHO cells expressing a mutant CD4-Fas chimera that is defective in signaling to apoptosis (data not shown). This indicates that endogenous hPAK65 cleavage occurs in both apoptotic epithelial CHO cells as well as in lymphocytes. Additionally, a similar cleavage pattern of hPAK65 was also observed in UV-irradiated HL-60 cells (data not shown), suggesting that the cleavage of hPAK65 is not limited to Fas-induced apoptosis.
The specific cleavage of hPAK65 was further characterized using in vitro-translated hPAK65 and PAK1. Jurkat cell lysates prepared at various times after Fas crosslinking were added to the translated products. hPAK65 was shown to be cleaved in the apoptotic lysate but not in the lysates from nonapoptotic cells (data not shown). Similarly, recombinant caspase-3 cleaved hPAK65 (Fig. 2, lane 6) but not PAK1 (lane 2). To determine whether the specific cleavage by caspase-3 occurs only at Asp-212, we introduced a point mutation by substituting an alanine for aspartate at position 212 (hPAK65-D212A). Caspase-3 did not cleave the in vitro-translated hPAK65-D212A, indicating that Asp-212 represents the only site within hPAK65 in which caspase-3 cleavage occurs (lane 4). We also showed that hPAK65 could be cleaved by activated recombinant caspase-8/FLICE (2, 3), an upstream “activator” caspase (lane 8). All of the observed cleavages could be inhibited by the addition of caspase inhibitor Z-DEVD-FMK (lanes 7 and 9).
Figure 2.
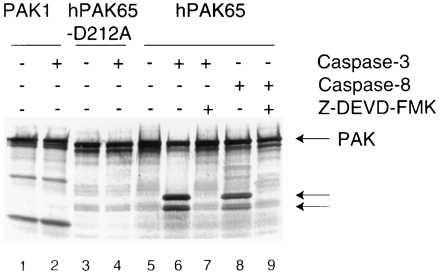
Specific cleavage of recombinant hPAK65 by caspases. hPAK65, cleavage-site mutant hPAK65-D212A, and PAK1 were in vitro translated and 35S-labeled, followed by incubation at 37°C for 30 min with: buffer, recombinant caspase-3, or caspase-8, +/− Z-DEVD-FMK (50 μM), as indicated. Products were separated by SDS/PAGE followed by autoradiography. Full-length hPAK65 and cleavage fragments are indicated by arrows.
Activation of hPAK65 by Caspase Cleavage.
Previously, proteolytic cleavage of hPAK65 by trypsin has been shown to cause hPAK65 autophosphorylation and activation (17). To test whether caspase cleavage of hPAK65 also results in kinase activation, we examined myc-tagged PAK proteins expressed in COS-7 cells. An N-terminal truncated form of hPAK65 corresponding to the active hPAK65 fragment generated during apoptosis (hPAK65-ΔN212) was found to be fully active whereas full-length hPAK65 was essentially inactive (Fig. 3A, lanes 1 and 2). Treatment of full-length hPAK65 with caspase-3, however, generated a fully active kinase, suggesting that proteolysis in the linker region releases the inhibitory effects of the N-terminal domain (lane 3). Treatment of a kinase-dead hPAK65 (hPAK65-K260R) with caspase-3 did not result in kinase activity (lane 4). To examine hPAK65 kinase activation in vivo, we performed an in-gel kinase assay on the Fas-activated Jurkat cell extracts from the experiment described in Fig. 1A by using histone H4 as a substrate. Fig. 3B shows the appearance of a single active kinase of 36 kDa that appeared with the same kinetics as that of hPAK65 cleavage during apoptosis. To further confirm that the 36-kDa kinase activity is derived from cleaved hPAK65, immunoprecipitation of hPAK65 from Jurkat apoptotic lysates over the same time course demonstrated that hPAK65 kinase activity increased during apoptosis and correlated directly with the occurrence of hPAK65 cleavage (Fig. 3C). These results suggest that, during apoptosis, the activation of hPAK65 is a consequence of caspase activation and may result in the phosphorylation of downstream targets.
Figure 3.
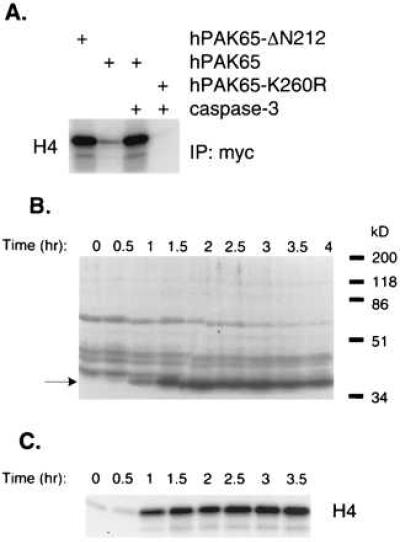
hPAK65 activation by caspase cleavage. (A) hPAK65 constitutively activated by N-terminal truncation. Various PAK constructs as indicated were expressed in COS-7 cells, immunoprecipitated with anti-myc antibody, and incubated with control buffer (lanes 1 and 2) or recombinant caspase-3 (lanes 3 and 4) at 37°C for 30 min. Kinase assays were performed using histone H4 as substrate. Equal amounts of protein in each reaction were confirmed by immunoblotting by using anti-myc antibody (data not shown). (B) A 36-kDa kinase induced during Fas-induced apoptosis. The same Jurkat cell extracts prepared as in Fig. 1 were subjected to an in-gel kinase assay. The induced 36-kDa kinase is indicated. (C) Endogenous hPAK65 kinase activity induced during Fas-induced apoptosis. The Jurkat cell extracts as in Fig. 1 were immunoprecipitated with hPAK65-C antibody, followed by in vitro kinase reactions in kinase buffer containing substrate histone H4. The reaction mixtures were then separated by SDS/PAGE.
JNK Activation by N-terminally Truncated hPAK65.
To examine the ability of the activated PAK to stimulate JNK activation, a series of PAK constructs was cotransfected with HA-tagged JNK1 into COS-7 cells. As shown in Fig. 4, hPAK65-ΔN212 stimulated JNK1 activity similar to the constitutively active PAK1 (PAK1-L107F; ref. 13), at least twofold over the vector control. In contrast, full-length PAK1, hPAK65, and hPAK65-K260R were unable to activate JNK1. JNK1 activation by active PAK was significant, albeit weaker, than that seen by constitutively active Cdc42 (Cdc42-V12; Fig. 4) and consistent with several previous reports (13, 18, 29–31). Cdc42 lies upstream of hPAK65 and may stimulate multiple effectors (32, 33), resulting in a higher level of JNK activity than PAK alone. Consistently, the moderate dominant-negative effect of hPAK65-K260R on Cdc42-induced JNK1 activation (Fig. 4) supports the redundancy of multiple mediators of Cdc42 in JNK1 activation.
Figure 4.
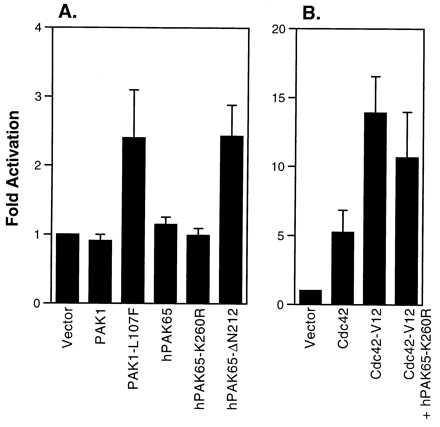
JNK activation by N-terminally truncated hPAK65. HA-tagged JNK was cotransfected into COS-7 cells with various myc-tagged constructs as indicated. Cell lysates were immunoprecipitated with anti-HA antibody, and the kinase activity was assayed using GST-Jun as substrate. Equal amounts of JNK were loaded on the gel as shown by immunoblotting by using anti-HA antibody (data not shown). Data are presented as fold activation over vector control and represent the means ± SE of three individual experiments.
Morphological Changes Induced by Active hPAK65.
To understand the consequence of hPAK65 activation during apoptosis, we introduced the active N-terminally truncated hPAK65 fragment into a variety of cell lines, including REF52, COS-7, NIH 3T3, Hela, or CHO. Sixteen hours after transfection or microinjection with myc-tagged PAKs and marker GFP, cells expressing the active fragment of hPAK65, hPAK65-ΔN212, but not other inactive PAK proteins markedly shrank and rounded up. Fig. 5 shows that COS-7 cells transfected with vector (Fig. 5A), kinase-dead hPAK65-ΔN212-K260R (Fig. 5B), full-length hPAK65, PAK1, or C-terminally truncated hPAK65 (hPAK65-ΔC212; data not shown) exhibited no changes in cell morphology. However, cells expressing either hPAK65-ΔN212 (Fig. 5C) or constitutively active PAK1 (PAK1-L107F; data not shown) showed dramatic morphological changes. The nucleus of shrunken cells also appeared condensed. Fig. 6 shows REF52 cells microinjected with full-length hPAK65 (Fig. 6 A and B) or hPAK65-ΔN212 (Fig. 6 C and D). As shown by DAPI staining, the shrunken nuclei were observed only in cells expressing active hPAK65. At this time, the rounded cells did not exhibit other apoptotic characteristics, such as formation of apoptotic bodies, DNA fragmentation as determined using TdT-mediated dUTP nick end-labeling (TUNEL) assay, or phosphatidylserine externalization as determined by annexin V binding. Furthermore, the morphological changes of the cells and nuclei were unaffected by preincubation of the cells with Z-VAD-FMK, a specific caspase inhibitor (data not shown), indicating that the morphogenic effect of active PAK expression was independent of caspase activation. As a positive control, cells injected with FADD exhibited changes characteristic of apoptosis within 4 hr, and this process was blocked by Z-VAD-FMK (data not shown).
Figure 5.
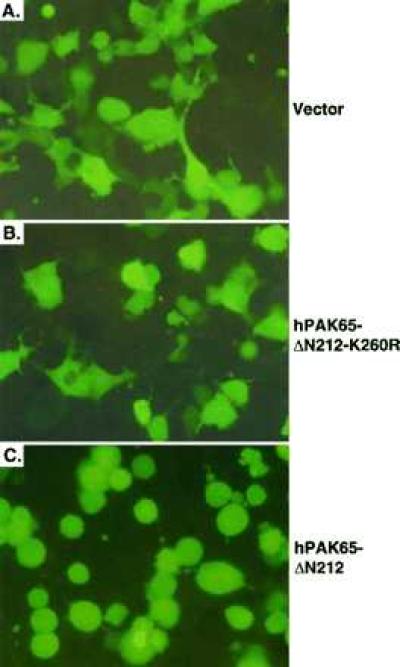
Morphological changes of cells transfected with active hPAK65. COS-7 cells were transfected with plasmids encoding GFP and myc-tagged vector (A); hPAK65-ΔN212-K260R (B); and hPAK65-ΔN212 (C). Sixteen hours after transfection, cells were directly photographed through fluorescence optics without fixing. Pictures were representative of three independent experiments.
Figure 6.
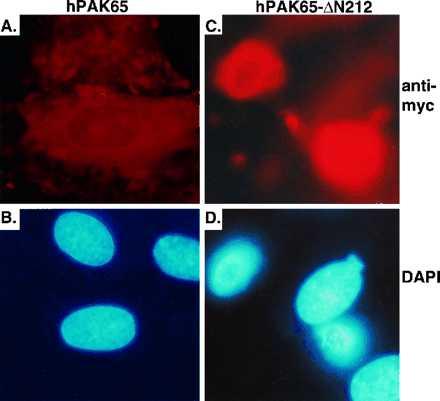
Cell and nuclear changes in cells microinjected with active hPAK65. REF52 cells were microinjected with plasmids encoding GFP and myc-tagged full-length hPAK65 (A and B), and hPAK65-ΔN212 (C and D). Sixteen hours after injection, cells were fixed and stained with immunofluorescent reagents as described in Methods. Pictures were taken from the same field by using different filters. A and C show PAK-expressing cells stained by anti-myc antibody. B and D show nuclei stained by DAPI. Nearly all cells injected with hPAK65-ΔN212 expressed the injected plasmids as evaluated by anti-myc staining (92/96), and greater than 80% of them rounded up (76/92). Injected plasmids were also expressed in most of the cells injected with full-length hPAK65 (114/118); whereas nearly no cell expressing inactive PAK showed any morphological changes (5/114). Similar results have been obtained from three independent experiments.
Apoptosis Induced by Active hPAK65 in Epithelial Cells.
We also noticed that many cells became detached after rounding up, and this detachment was most obvious in transfected epithelial Hela and CHO cells. To facilitate further characterization, Hela and CHO cells were individually transfected with various PAK plasmids and surface-expressed CD20. After 40 hr, the entire transfected pools, including detached cells, were stained with PI, FITC-conjugated annexin V, and PE-conjugated antibody to CD20 and analyzed by flow cytometry. The results showed that the number of apoptotic cells in the active hPAK65-expressing cell population increased 2- to 3-fold over that in the control cells (Fig. 7). Because the hPAK65-induced apoptosis was late compared with that in FADD-transfected cells, it may occur after cells have became detached from plates. It is known that cell detachment is able to induce apoptosis, termed “anoikis” (reviewed in ref. 34). Therefore, the morphological change induced by hPAK65 activation may enhance apoptotic process.
Figure 7.
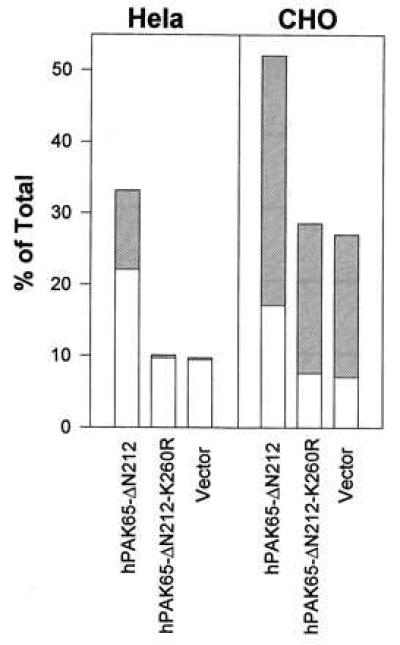
Induction of apoptosis by transfection of active hPAK65 in Hela and CHO cells. The indicated plasmids were cotransfected with surface-expressed CD20 into Hela and CHO cells. Forty hours after transfection, cells, including detached cells, were collected and stained with PE-conjugated CD20, FITC-conjugated annexin V, and PI. Open bars show the percentages of cells in early phase of apoptosis, which are annexin V-positive and PI-negative; hatched bars show the percentages of cells in late phase of apoptosis, which are annexin V- and PI-double-positive. Data represent means of three individual experiments. A similar level of protein expression among all constructs was confirmed by immunoblot analysis (data not shown).
Apoptosis Delayed by Dominant-Negative hPAK65.
To further address the proapoptotic effect of activated hPAK65 during apoptosis, the kinetics of apoptosis in CHO cells stably expressing a CD4-Fas chimera were measured as transfected with various hPAK65 constructs. As shown in Fig. 8, apoptosis was significantly delayed in cells expressing a dominant-negative form of hPAK65 (hPAK65-D208A, D212A, K260R) in which, in addition to the ATP-binding site, the caspase-recognition site was double-mutated to avoid competing for proteases. Similarly, a C-terminal fragment of hPAK65 with a mutation in its ATP-binding site (hPAK65-ΔN212-K260R) also delayed apoptosis. As a control, cells expressing only the N-terminal regulatory domain of hPAK65 showed no effect on apoptosis, further indicating that the interfering effect is a result of the kinase domain of hPAK65. Therefore, apoptosis is promoted by the kinase activation of hPAK65.
Figure 8.
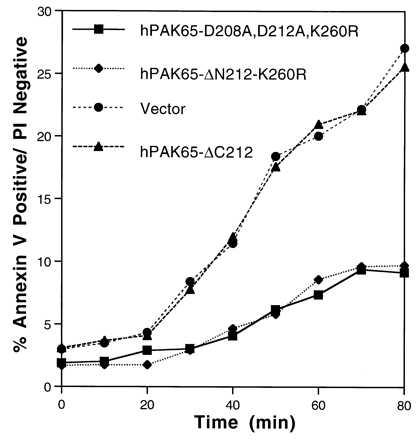
Apoptosis delayed by transfection of dominant-negative hPAK65 in CHO cells stably expressing a CD4-Fas chimera. Forty hours after transfection, cells were collected and stained with PE-conjugated CD20 and CD4. Apoptosis was initiated by adding anti-mouse IgG + IgM, and cells were incubated at 37°C. Aliquots were taken at times indicated, stained with FITC-conjugated annexin V and PI for 20 min at room temperature, and analyzed immediately by flow cytometry.
DISCUSSION
In this paper we demonstrate the activation of a kinase, hPAK65, during apoptosis as a result of a specific cleavage by caspases. The downstream effects of hPAK65 activation include the activation of JNK. The ectopically expressed active hPAK65 induces morphological changes, including cell and nucleus shrinkage, which are typically observed during the initial stages of apoptosis. This phenotypic change is independent of caspase activation because the presence of a caspase inhibitor does not block it, confirming that the effects of hPAK65 are downstream of the caspase cascade. Additionally, the morphological changes induced by hPAK65 activation result in apoptosis. In two epithelial cell lines, CHO and Hela cells, apoptosis is observed after the cells rounded up and detached. The proapoptotic effect of activated hPAK65 is further supported by the evidence that apoptosis is delayed in cells expressing the dominant-negative form of hPAK65.
We have observed the cleavage of hPAK65 during apoptosis in all of the cell lines that we have tested, including Jurkat, CHO, and HL-60. From our in-gel and immunoprecipitated kinase assays, we conclude that the cleavage product is an activated kinase with a molecular mass of 36 kDa (Fig. 3 B and C). It has also been reported that a 36-kDa kinase was activated in a variety of apoptotic systems, including UV-irradiated HL-60, tumor necrosis factor-treated tumor cell lines, and serum-starved NIH 3T3 cells (26). It is likely that the 36-kDa kinase reported is the caspase-activated hPAK65. Therefore, cleavage and activation of the ubiquitously expressed hPAK65 may be a common phenomenon during apoptotic signaling involving the activation of the caspase cascade.
Recently, Rudel and Bokoch (35) also reported the cleavage of hPAK65/PAK2 by caspases during apoptosis, yet the biological function of the activated hPAK65 was not directly addressed in that study. They showed that a stable Jurkat cell line constitutively expressing a dominant-negative form of PAK1 was resistant to Fas-induced formation of apoptotic bodies but surprisingly had an enhanced externalization of phosphatidylserine at the cell surface. In our study, the phenotype of the proteolytic product of hPAK65 was explored by expressing the carboxyl-terminal kinase domain in several cell types, including REF52, COS-7, NIH 3T3, Hela, or CHO cells. With no exception, all cells expressing active hPAK65 exhibit dramatically altered cell morphology. Furthermore, our results show that dominant-negative hPAK65 delays the process of phosphatidylserine externalization, supporting the proapoptotic effect of activated hPAK65 during apoptosis. Therefore, these observations provide unambiguous evidence that the activated hPAK65 acts as a mediator of morphological changes during apoptosis. The discrepancy between Rudel and Bokoch’s results and ours may arise from the clonal differences that occurred during the selection of the stable cell line. Additionally, Rudel and Bokoch (35) used dominant-negative PAK1 as opposed to dominant-negative hPAK65, and the role of PAK1 in apoptosis is yet not clear. In fact, it seems unlikely that PAK1 participates in apoptosis because it lacks the caspase cleavage site that is found in hPAK65, and we have also shown that PAK1 is not cleaved by caspases (Fig. 2B).
The positive role of hPAK65 cleavage in promoting apoptosis is also supported by the finding that introduction of the activated hPAK65 fragment is sufficient to trigger apoptosis in CHO and Hela cells (Fig. 6). Because this apoptosis is late in comparison with that observed in FADD-transfected cells, it may be secondary to cell detachment (anoikis), a property exclusive to epithelial and endothelial cells (reviewed in ref. 34). It has been suggested that, in the absence of cell contact with extracellular matrix, death-inducing molecules become activated and survival factors become down-regulated (36, 37). Therefore, at least in epithelial cells, during apoptosis morphological changes induced by hPAK65 activation can accelerate the execution of the death signal.
The direct target of activated hPAK65 in apoptosis remains unknown. In addition to the phosphorylated myosin heavy chain mentioned above, PAK proteins are known to phosphorylate p47-phox, a component of NADPH oxidase, which is involved in the production of superoxide in neutrophils (12); however, the engagement of truncated hPAK65 in the production of superoxide awaits further investigation. The production of reactive oxygen species (ROS), including superoxide, has been implicated as a mediator of apoptosis (reviewed in ref. 38).
In summary, the present study not only indicates a signaling pathway of apoptosis via hPAK65, but also demonstrates a mechanism of direct activation of endogenous hPAK65 in vivo and presents a new function of hPAK65 as an effector of morphological changes during apoptosis.
Acknowledgments
We thank Anna Lisa Fear for the preparation of purified caspases, Dr. K. Koths for advice concerning production and use of caspase, and G. Wang for DNA sequencing. We also thank Drs. W. Fantl, M. Kavanaugh, A. Klippel, S. Kothakota, and G. Pronk for helpful comments on the manuscript. The caspase-8, caspase-3, and PAK1 clones were generous gifts from Drs. T.-h. T. Chen, V. Dixit (University of Michigan), and S. Coughlin (University of California at San Francisco), respectively.
ABBREVIATIONS
- PE
phosphatidylethanolamine
- PI
propidium iodide
- GFP
green fluorescence protein
- FITC
fluorescein isothiocyanate
- JNK
c-Jun N-terminal kinase
References
- 1.Nagata S. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 2.Muzio M, Chinnaiyan A M, Kischkel F C, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz J D, Zhang M, Gentz R, Mann M, Krammer P H, Peter M E, Dixit V M. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 3.Boldin M P, Goncharov T M, Goltsev Y V, Wallach D. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 4.Boldin M P, Varfolomeev E E, Pancer Z, Mett I L, Camonis J H, Wallach D. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 5.Chinnaiyan A M, O’Rourke K, Tewari M, Dixit V M. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 6.Fernandes-Alnemri T, Litwack G, Alnemri E S. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 7.Enari M, Talanian R V, Wong W W, Nagata S. Nature (London) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 8.Chow S C, Weis M, Kass G E, Holmstrom T H, Eriksson J E, Orrenius S. FEBS Lett. 1995;364:134–138. doi: 10.1016/0014-5793(95)00370-o. [DOI] [PubMed] [Google Scholar]
- 9.Enari M, Hase A, Nagata S. EMBO J. 1995;14:5201–5208. doi: 10.1002/j.1460-2075.1995.tb00204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter A G, Ng P, Janicke R U. BioEssays. 1997;19:501–507. doi: 10.1002/bies.950190609. [DOI] [PubMed] [Google Scholar]
- 11.Manser E, Chong C, Zhao Z S, Leung T, Michael G, Hall C, Lim L. J Biol Chem. 1995;270:25070–25078. doi: 10.1074/jbc.270.42.25070. [DOI] [PubMed] [Google Scholar]
- 12.Knaus U G, Morris S, Dong H J, Chernoff J, Bokoch G M. Science. 1995;269:221–223. doi: 10.1126/science.7618083. [DOI] [PubMed] [Google Scholar]
- 13.Brown J L, Stowers L, Baer M, Trejo J, Coughlin S, Chant J. Curr Biol. 1996;6:598–605. doi: 10.1016/s0960-9822(02)00546-8. [DOI] [PubMed] [Google Scholar]
- 14.Martin G A, Bollag G, McCormick F, Abo A. EMBO J. 1995;14:1970–1978. doi: 10.1002/j.1460-2075.1995.tb07189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teo M, Manser E, Lim L. J Biol Chem. 1995;270:26690–26697. doi: 10.1074/jbc.270.44.26690. [DOI] [PubMed] [Google Scholar]
- 16.Jakobi R, Chen C J, Tuazon P T, Traught J A. J Biol Chem. 1996;271:6206–6211. doi: 10.1074/jbc.271.11.6206. [DOI] [PubMed] [Google Scholar]
- 17.Benner G E, Dennis P B, Masaracchia R A. J Biol Chem. 1995;270:21121–21128. doi: 10.1074/jbc.270.36.21121. [DOI] [PubMed] [Google Scholar]
- 18.Bagrodia S, Taylor S J, Creasy C L, Chernoff J, Cerione R A. J Biol Chem. 1995;270:22731–22737. doi: 10.1074/jbc.270.39.22731. [DOI] [PubMed] [Google Scholar]
- 19.Leeuw T, Fourest-Lieuvin A, Wu C, Chenevert J, Clark K, Whiteway M, Thomas D Y, Leberer E. Science. 1995;270:1210–1213. doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- 20.Brzeska H, Knaus U G, Wang Z Y, Bokoch G M, Korn E D. Proc Natl Acad Sci USA. 1997;94:1092–1095. doi: 10.1073/pnas.94.4.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C, Lee S F, Furmaniak-Kazmierczak E, Cote G P, Thomas D Y, Leberer E. J Biol Chem. 1996;271:31787–31790. doi: 10.1074/jbc.271.50.31787. [DOI] [PubMed] [Google Scholar]
- 22.Manser E, Huang H Y, Loo T H, Chen X Q, Dong J M, Leung T, Lim L. Mol Cell Biol. 1997;17:1129–1143. doi: 10.1128/mcb.17.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sells M A, Knaus U G, Bagrodia S, Ambrose D M, Bokoch G M, Chernoff J. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- 24.Reinhard C, Shamoon B, Shyamala V, Williams L T. EMBO J. 1997;16:1080–1092. doi: 10.1093/emboj/16.5.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazebnik Y A, Cole S, Cooke C A, Nelson W G, Earnshaw W C. J Cell Biol. 1993;123:7–22. doi: 10.1083/jcb.123.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu M L, Sato M, Cao B, Richie J P. Proc Natl Acad Sci USA. 1996;93:8977–8982. doi: 10.1073/pnas.93.17.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reinhard C, Fernandez A, Lamb N J, Thomas G. EMBO J. 1994;13:1557–1565. doi: 10.1002/j.1460-2075.1994.tb06418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, Ding C K, Gallant M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, Munday N A, Raju S M, Smulson M E, Yamin T T, Tu V L, Miller D K. Nature (London) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 29.Frost J A, Xu S, Hutchison M R, Marcus S, Cobb M H. Mol Cell Biol. 1996;16:3707–3713. doi: 10.1128/mcb.16.7.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polverino A, Frost J, Yang P, Hutchison M, Neiman A M, Cobb M H, Marcus S. J Biol Chem. 1995;270:26067–26070. doi: 10.1074/jbc.270.44.26067. [DOI] [PubMed] [Google Scholar]
- 31.Zhang S, Han J, Sells M A, Chernoff J, Knaus U G, Ulevitch R J, Bokoch G M. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- 32.Teramoto H, Coso O A, Miyata H, Igishi T, Miki T, Gutkind J S. J Biol Chem. 1996;271:27225–27228. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- 33.Teramoto H, Crespo P, Coso O A, Igishi T, Xu N, Gutkind J S. J Biol Chem. 1996;271:25731–25734. doi: 10.1074/jbc.271.42.25731. [DOI] [PubMed] [Google Scholar]
- 34.Ruoslahti E, Reed J C. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 35.Rudel T, Bokoch G M. Science. 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- 36.Frisch S M, Vuori K, Kelaita D, Sicks S. J Cell Biol. 1996;135:1377–1382. doi: 10.1083/jcb.135.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne P H, Downward J. EMBO J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buttke T M, Sandstrom P A. Immunol Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]