

Abstract
Elucidation of mechanisms that regulate hematopoietic stem cell self-renewal and differentiation would be facilitated by the identification of defined culture conditions that allow these cells to be amplified. We now demonstrate a significant net increase (3-fold, P < 0.001) in vitro of cells that are individually able to permanently and competitively reconstitute the lymphoid and myeloid systems of syngeneic recipient mice when Sca-1+lin− adult marrow cells are incubated for 10 days in serum-free medium with interleukin 11, flt3-ligand, and Steel factor. Moreover, the culture-derived repopulating cells continued to expand their numbers in the primary hosts at the same rate seen in recipients of noncultured stem cells. In the expansion cultures, long-term culture-initiating cells increased 7- ± 2-fold, myeloid colony-forming cells increased 140- ± 36-fold, and total nucleated cells increased 230- ± 62-fold. Twenty-seven of 100 cultures initiated with 15 Sca-1+lin− marrow cells were found to contain transplantable stem cells 10 days later. This frequency of positive cultures is the same as the frequency of transplantable stem cells in the original input suspension, suggesting that most had undergone at least one self-renewal division in vitro. No expansion of stem cells was seen when Sca-1+TER119− CD34+ day 14.5 fetal liver cells were cultured under the same conditions. These findings set the stage for further investigations of the mechanisms by which cytokine stimulation may elicit different outcomes in mitotically activated hematopoietic stem cells during ontogeny and in the adult.
Keywords: interleukin 11, Flk-2, Flt3-ligand, Steel factor, ex vivo expansion
A cardinal property of hematopoietic stem cells is their ability to divide without significant alteration of their proliferative potential or differentiation state. The first demonstration that such stem cell self-renewal divisions can occur in vitro (albeit without a net increase in their numbers) made use of retroviral marking to identify the clonal progeny in multiple recipients of individual hematopoietic stem cells that had been amplified in 4-week-old long-term cultures (LTC) (1). Subsequent attempts to improve the output of stem cells in these cultures by using various cell lines as feeders have allowed input numbers to be maintained, but a net expansion has not been achieved (2–4). At the same time, attention has focused increasingly on studies of the responses of highly purified stem cell-containing populations to defined cytokines. This latter approach allows cellular sources of negative regulators to be minimized and different cytokines to be evaluated for their ability, either alone or in combination, to replace the stimulating function of stromal cells. This has made it possible to demonstrate the production of very large numbers of differentiated progeny without significant loss of stem cell numbers or activity, but a net increase in these cells has not been documented (5–9).
Many cytokines either alone or in various combinations have been found to stimulate the proliferation in semisolid medium of primitive hematopoietic cells capable of multilineage differentiation (10). However, several studies now indicate that the ability to proliferate in semisolid medium is an acquired property and may not be possessed by most of the in vivo reconstituting stem cells present in the marrow of normal adults (11–13). In addition, the cytokine responsiveness of hematopoietic cells is known to change both as a function of their ontological and their differentiation states (14–16). Thus, responsiveness to cytokines that are active in semi-solid cultures may not be predictive of cells with in vivo repopulating ability. Because of the important stimulatory role of stromal fibroblasts in the LTC system (17) and preliminary evidence of the potent and synergistic mitogenic activity of the stromal-derived cytokines, flt3-ligand (FL), Steel factor (SF), and interleukin 11 (IL-11) on primitive fetal liver cells (18), we chose to focus initially on these three cytokines. Recently, several groups have reported that IL-11 together with cytokine cocktails, including either FL or SF, can sustain hematopoietic stem cell activity in vitro (8, 9, 13, 19), and retroviral-mediated expression of IL-11 has been found to enhance hematopoietic stem cell self-renewal in vivo (20). Our results now show that the combination of IL-11 plus FL plus SF is sufficient to concomitantly obtain a severalfold expansion in vitro of cells identified as hematopoietic stem cells by the most stringent functional criteria available. These include their ability to clonally reconstitute both lymphoid and myeloid systems of primary recipients for periods of 1 year without any diminution of their ability to concomitantly expand their numbers in vivo. Interestingly, however, this cytokine mixture was not effective in eliciting a similar response from stem cells present in the day 14.5 fetal liver.
MATERIALS AND METHODS
Animals.
C57BL/6 (Ly5.2) and C57BL/6:Pep3b (Ly5.1) mice and C57BL/6-W41/W41 (Ly5.2) mice originally were obtained from commercial stocks and J. Barker, respectively, both at The Jackson Laboratory. These mice then were bred and maintained at the Joint Animal Facility of the British Columbia Cancer Research Centre in microisolator units provided with sterilized food, water, and bedding. Irradiated animals were additionally provided with acidified water (pH 3.0).
Cell Purification.
The monoclonal antibodies (mAbs), staining, and analysis procedures have all been described in detail previously (6). Briefly, adult C57BL/6:Pep3b (Ly5.1) marrow cells suspended in Hanks’ balanced salt solution with 2% fetal bovine serum (FBS) (HF) were incubated sequentially at 5–8 × 106 cells per ml on ice with 6 μg/ml of 2.4G2 [anti-Fc receptor antibody (21)], then a mixture of biotinylated lineage-specific rat mAbs [anti-B220 (RA3–6B2), anti-GR-1 (RB6–8C5), anti-Ly-1 (53–7.3), anti-Mac-1 (M1/70)], and finally cyanine-5-succinimidyl ester (Cy5)-labeled anti-Sca-1 (anti-Ly6A/E; E13–161.7), fluorescein isothiocyanate (FITC)-labeled wheat germ agglutinin (where specified), and phycoerythrin (PE)-labeled streptavidin (SA-RPE; Southern Biotechnology Associates). Propidium iodide at 1 μg/ml (PI; Sigma) was added to the final wash. Alternatively, as specified, cells were incubated with biotinylated anti-CD34 (Ram34, PharMingen) and then FITC-labeled lineage-specific rat mAbs (anti-B220, anti-GR-1, anti-Ly1, anti-Mac-1, TER119), anti-Sca-1, and SA-RPE. Fetal liver cells were isolated from day 14.5 C57BL/6:Pep3b embryos as previously described (22) and incubated with 2.4G2 followed by staining with anti-CD34, anti-Sca-1, and TER119 mAbs. All cells were sorted on a FACStar+ (Becton Dickinson).
Serum-Free Suspension Cultures.
For most experiments, 1-ml cultures containing 200 purified cells suspended in Iscove’s medium supplemented with 1% (wt/vol) BSA, 200 μg/ml transferrin, 10 μg/ml insulin (BIT; StemCell Technologies, Vancouver, BC), 40 μg/ml low-density lipoproteins (Sigma), 10−4 M 2-mercaptoethanol, 2 mM glutamine, 50 μg/ml streptomycin, 50 units/ml penicillin, 100 ng/ml human IL-11 (Genetics Institute, Cambridge, MA), 100 ng/ml of human FL (Immunex), and 50 ng/ml murine SF (expressed in COS cells and purified in the Terry Fox Laboratory), unless specified otherwise, were incubated in 24-well plates (Nunc) at 33°C in 5% CO2 in air for up to 10 days with replacement of half of the medium (without removal of cells) every 3 days. Cells were harvested by removal of the suspended cells followed by rinsing each well with Iscove’s containing 2% FBS. When present, adherent cells were removed by incubation for 1–2 min at room temperature with 0.5 ml of trypsin-EDTA (GIBCO/BRL) followed by two rinses with medium. Cells pooled from at least four wells, or the equivalent of ≥10 competitive in vivo repopulating units (CRUs), were centrifuged at 350 g for 7 min, viable cell numbers were determined by eosin-dye exclusion, and assays were performed on appropriate aliquots. In the limiting dilution experiments, 15 Sca-1+lin− cells were cultured in 150-μl volumes in the individual wells of flat-bottomed 96-well culture plates (Nunc), and at the end of the 10-day culture protocol, the contents of each well were placed in a 1.5-ml microfuge tube after first adding 25 μl of Iscove’s medium plus 2% FBS containing 105 freshly isolated C57BL/6 marrow cells. Each well was then rinsed twice with 50 μl aliquots of Iscove’s medium with 2% FBS, and the combined harvest then was injected into individual irradiated C57BL/6 recipients (see below).
Assays for Colony-Forming Cells (CFC) and Long-Term Culture Initiating Cells (LTC-IC).
Burst-forming unit-erythroid (BFU-E), colony-forming unit (CFU)-granulocyte/macrophage (GM), and CFU-granulocyte/erythroid/megakaryocyte/macrophage (GEMM) progenitor numbers were determined by plating suitable aliquots of test cells in methylcellulose medium supplemented with 10 ng/ml murine IL-3, 10 ng/ml human IL-6, 50 ng/ml murine SF, and 3 units/ml human erythropoietin (HCC-3434; StemCell Technologies) and scoring the corresponding types of colonies (containing ≥30 cells each) present after 12 days of incubation at 37°C (23). The LTC-IC content of cell suspensions was determined by limiting dilution analysis as previously described (24). Briefly, purified or cultured cells were washed and resuspended in myeloid long-term culture medium (StemCell Technologies) to which freshly dissolved 10−6 M hydrocortisone sodium hemisuccinate (Sigma) was added. Varying cell numbers were then plated on 12 to 16 replicate wells of 96-well culture plates (Nunc) containing irradiated preestablished mouse marrow LTC feeder layers or confluent layers of S17 cells (25) and maintained for 4 weeks at 33°C with weekly removal of half of the nonadherent cells and replacement of half of the medium, and then both the adherent and nonadherent cells removed from each well and assayed for myeloid CFC. LTC-IC frequencies were calculated by the method of maximum likelihood from the proportion of negative wells (wells containing no detectable CFC) measured for each input dilution of cells tested (26).
Transplantation Procedures and Quantitation of CRUs.
These procedures have been described in detail previously (6, 27). Briefly, C57BL/6 (Ly5.2) recipients were irradiated with a single dose of 900 cGy (95 cGy/min) from a 137Cs γ-ray source and then injected intravenously with 15–100 freshly purified cells, or the number of 10-day progeny obtained from 5–125 purified cells, plus 105 freshly isolated C57BL/6 marrow cells (4–10 recipients per cell dose per experiment). C57BL/6-W41/W41 (Ly5.2) recipients were similarly irradiated but with a lower dose (400 cGy) and were injected with test cells only. Aliquots of peripheral blood (PB) were taken from these recipients at intervals between 5 weeks and 1 year later and assessed for the presence of Ly5.1 (test cell-derived) lymphoid and myeloid cells. Red blood cells were lysed with NH4Cl, and the nucleated cells were stained with biotinylated anti-GR-1 mAb for 30 min on ice, followed by a single wash in HF and a further incubation of the cells for 30 min with FITC-conjugated anti-Ly5.1 antibody (A20–1.7) and SA-RPE. The cells were then washed twice in HF with 1 μg/ml PI included in the final wash and analyzed on a FACSort (Becton Dickinson). Animals were considered to be reconstituted (positive) only if their PB contained both ≥1% test cell-derived (Ly5.1+) myeloid (GR-1+) cells and lymphoid cells (defined by their low orthogonal light-scattering characteristics). CRU frequencies were calculated by the method of maximum likelihood from the proportions of animals that were negative (6, 26, 27) when these were assessed 4–12 months posttransplant using a generalized linear model with binomial errors and a complementary log–log link function. Differences in CRU frequencies were tested by adding the appropriate interaction term in the model and assessing significance by the likelihood ratio test.
In the two secondary transplant experiments, groups of primary W41/W41 recipients were killed 18 or 20 weeks posttransplant and their marrow cells then transplanted into new groups of irradiated W41/W41 mice. In one experiment, the marrow cells from the primary recipients were first stained and Ly5.1+ (test cell-derived) and Ly5.1− (recipient-derived) cell populations were isolated using the FACStar+ to allow these two genotypes to be assayed separately for their contents of CFC, LTC-IC, and CRU.
RESULTS
Expansion of Adult Marrow Progenitors in Serum-Free Cultures Containing IL-11 plus FL and SF.
Table 1 shows the frequencies of different types of progenitors (CFC, LTC-IC, and CRU) measured in each of the various populations of Sca-1+lin− normal adult mouse marrow cells that were used to initiate serum-free suspension cultures containing 100 ng/ml IL-11, 100 ng/ml FL, and 50 ng/ml SF as described in Methods. Stem cells (CRU) were assessed for their ability to individually and competitively repopulate both lymphoid and myeloid compartments for at least 4 months after transplantation into either lethally irradiated C57BL/6 mice given a radioprotective dose of 105 syngeneic normal marrow cells or sublethally irradiated congenic W41/W41 recipients. The use of sublethally irradiated W41/W41 recipients simplifies the CRU assay and has been found to detect both unseparated and Sca-1+lin− CRU with the same sensitivity and specificity as the standard CRU assay (ref. 13 and unpublished findings). In one experiment, Sca-1+lin− cells that bind wheat germ agglutinin were isolated, and in three of the six experiments, the CD34− fraction of Sca-1+lin− cells (28) was isolated. Evidence of an amplification in the input CRU population after these had been in culture for 9 to 10 days was obtained in five of six experiments (Table 1). In a single experiment (part of experiment 1 in Table 1, data not shown), the combination of IL-11 plus FL at the same concentrations was found to maintain (but not expand) CRU numbers, whereas FL plus SF without IL-11 resulted in a 4-fold decrease in CRU. Coculture of the same input cells on S17 feeders, even with the addition of any of three combinations of IL-11, FL, and SF (including all three together), resulted in a 5-fold decrease in CRU numbers (data not shown). Interestingly, addition of 1,000 units/ml of human thrombopoietin (Zymogenetics, Seattle, WA) to the IL-11 plus FL and SF cytokine combination (experiment 6, Table 1) abrogated the increase in CRU numbers stimulated by IL-11 plus FL and SF alone and resulted in maintenance only of CRU (data not shown).
Table 1.
Progenitor activity in the Sca-1+lin− populations of adult mouse marrow cultured and assayed for evidence of CRU expansion
| Experiment no. | Input phenotype | Percent of original BM cells | Progenitors per 200 input cells
|
Expansion of CRU numbers after 9–10 days† | ||||
|---|---|---|---|---|---|---|---|---|
| BFU-E | CFU-GM | CFU-GEMM | LTC-IC | CRU* | ||||
| 1 | Sca-1+lin−WGA+ | 0.04 | ND | ND | ND | ND | 2 (12) | 5× (12) |
| 2 | Sca-1+lin−CD34− | 0.18 | ND | ND | ND | ND | 3 (8) | 4× (12) |
| 3a | Sca-1+lin−CD34− | 0.13 | 0.5 | 18 | 0.5 | 6 | 4 (8) | 4× (12) |
| 3b | 4 (8) | 4× (12) | ||||||
| 4 | Sca-1+lin−CD34− | 0.24 | 0.6 | 6 | 1.9 | 3 | 4 (6) | 10× (10) |
| 5 | Sca-1+lin− | 0.18 | 0.5 | 19 | 2.5 | 6 | 6 (8) | 1× (8) |
| 6 | Sca-1+lin− | 0.13 | 0.4 | 24 | 2.2 | 4 | 5 (18) | 4× (22) |
ND, not done.
Sublethally irradiated W41/W41 mice were used as recipients in CRU assays performed in experiments 1, 2, 3a, 4, and 6. Lethally irradiated C57BL/6 recipients were used as recipients in experiments 3b and 5. The number in each case is shown in parentheses.
Values shown are fold–expansion relative to input CRU numbers. The number of mice used to determine the frequency of CRU in the cultured populations is shown in parentheses.
Fig. 1 shows the pooled data for recipient mice injected with fresh or cultured cells from all of the six experiments presented in Table 1. The mean CRU frequencies (and 95% confidence intervals; n = the number of mice contributing to the estimate) calculated from these combined data sets are 1 CRU per 55 fresh cells (1/40–1/76, n = 68) and 1 CRU per 16 cultured cells expressed as input cell equivalents (1/12–1/23, n = 88). These values indicate a significant (P < 0.001) 3.5-fold net expansion in CRU numbers during the 9- to 10-day culture period. As shown in Fig. 2, this increase is higher than the 1.7-fold increase in CRU numbers seen after only 5 days of culture but lags slightly behind the rate of increase in LTC-IC (≈2.5-fold after 2 days and ≈7-fold after 10 days). In contrast, the total cell number and the number of cells detectable as CFC increased much more rapidly (>100-fold within 10 days). Interestingly, as can also be seen in Fig. 2, during the first 5 days in culture the CFC population expanded even faster than the total number of cells, indicating an imbalance in the initial rate of influx of cells into the CFC compartment as compared with their rate of loss from that compartment. Then, during the next 5 days, this situation was reversed.
Figure 1.
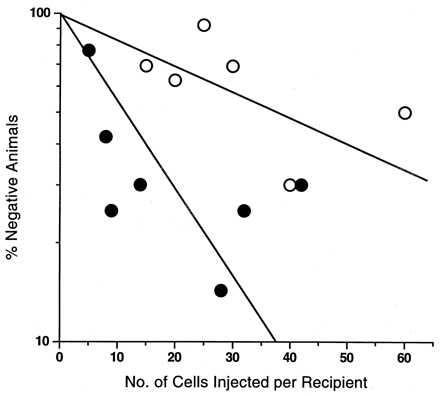
Proportion of negative mice (mice not showing repopulation of both lymphoid and myeloid compartments) 4–12 months after being transplanted with WGA+ Sca-1+lin− or CD34−Sca-1+lin− marrow cell populations (○) or the cells derived from them after 9–10 days incubation in 100 ng/ml IL-11, 100 ng/ml FL, and 50 ng/ml SF (•). The data for the cultured cells are expressed per freshly isolated purified cell equivalents to allow their increased absolute numbers to be seen directly on this graph. Data are pooled from the six experiments described in Table 1 (4–15 recipients per point). The lines shown connect the CRU frequency values determined from the two sets of pooled data by the method of maximum likelihood (26) (at 37% negative mice, see text) to the origin.
Figure 2.
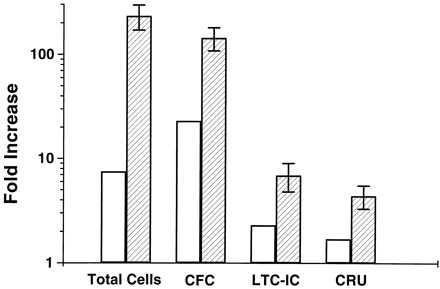
Expansion of total cells, CFC, LTC-IC, and CRU in cultures of Sca-1+lin− marrow cells stimulated in vitro by 100 ng/ml IL-11, 100 ng/ml FL, and 50 ng/ml SF for 5 (open bars, n = 2 experiments), or 9–10 days (hatched bars, n = 5–7 experiments). The fold increase (±1 SEM) was calculated by dividing the number of progenitors detected in the 5- or 9- to 10-day cultures by the corresponding number of progenitors calculated to be present in the cells used to initiate each culture. (Data include 1 experiment not described in Table 1.)
In Vivo Repopulating Cells Generated in Cultures of Purified Adult Marrow Cells Retain Their Self-Renewal Properties.
The extent and duration of lympho-myeloid reconstitution by fresh and cultured CRU was compared for periods of up to 1 year posttransplant. As illustrated by the results of a representative experiment shown in Fig. 3, there was no evidence that the durability of the lympho-myeloid reconstituting potential characteristic of freshly isolated CRU was compromised or diminished in the CRU that were generated in vitro, even in recipients of limiting numbers of CRU.
Figure 3.
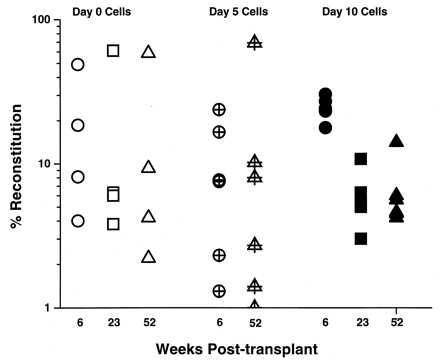
Levels of engraftment maintained in individual C57BL/6 mice injected with 40 Sca-1+lin−CD34− cells (day 0) or the cells generated from the equivalent of 30 or 32 of these cells after 5 (day 5) or 10 days (day 10) of culture in IL-11 plus FL and SF, respectively.
For a more stringent comparison of the self-renewal capacity of the CRU generated in the 10-day expansion cultures with those present in the normal marrow input populations, the number of CRU regenerated after 4 to 5 months in the primary recipients of both types of cells was assessed in two experiments. In these, all of the primary recipients (including both positive and negative animals) from groups that were transplanted with similar numbers of either fresh or cultured CRU were killed 18 or 20 weeks posttransplant. The pooled marrow cells from each group were then assayed separately in Ly5.2+ secondary recipients to determine the number of CRU present in the primary recipients whose origin could be traced to the initial transplant of fresh or cultured (Ly5.1+) cells. As shown in Table 2, the number of Ly5.1+ CRU regenerated in vivo from either fresh or cultured cells was similar. Hence, the calculated net expansion of these populations during the 18- to 20-week posttransplant period was also similar. There was also no difference in the average reconstitution of the lympho-myeloid compartments of the two groups of secondary recipients. Total CRU numbers (i.e., including those that might have been produced from the host cells and/or the originally coinjected 105 Ly5.2+ fresh marrow cells) were not determined but would have been expected to be subnormal (27, 29). The total (and Ly5.1+) cellularity, and CFC and LTC-IC content of the marrow obtained from primary recipients of fresh or cultured Ly5.1+ cells were also similar (Table 2) and indicated full recovery of the total cell and CFC populations, as expected. In contrast, the LTC-IC populations remained 2- to 3-fold below normal values, consistent with a close relationship of LTC-IC and CRU.
Table 2.
Equivalent expansion of CRU in vivo from transplants of fresh and cultured Sca-1+lin− mouse bone marrow cells
| Cells assessed | Fresh cell transplants (no./mouse)
|
Cultured cell transplants (no./mouse)
|
|||
|---|---|---|---|---|---|
| Experiment 2 | Experiment 3a | Experiment 2 | Experiment 3a | ||
| Primary (1°) recipients | |||||
| Initial transplant | Ly5.1+cells/1° recipient | 63 | 40 | 42* | 8* |
| Ly5.1+CRU/1° recipient | 1.2 | 0.8 | 3.2† | 0.6† | |
| No. of 1° recipients | 4 | 4 | 6 | 3 | |
| 18–20 weeks posttransplant | Total BM cells‡ (% Ly5.1+) | 19 × 107 (37%) | 19 × 107 (26%) | 17 × 107 (40%) | 17 × 107 (49%) |
| Total BM CFC (% Ly5.1+) | 33 × 104 (37%) | 35 × 104 (nd) | 32 × 104 (48%) | 40 × 104 | |
| Total BM LTC-IC (% Ly5.1+) | 5,300 (19%) | 4,900 (27%) | 4,400 (27%) | 2,500 (ND) | |
| Total Ly5.1+ CRU in BM§ | 24 | 27 | 24 | 18 | |
| Expansion of Ly5.1+ CRU | |||||
| in 1° recipient¶ | 20× | 34× | 7.5× | 30× | |
| Secondary (2°) recipients | |||||
| 16 weeks posttransplant | % Ly5.1+ cells in 2° recipient‖ | 22 ± 15% | 14 ± 6% | 28 ± 15% | 15 ± 10% |
| (n = 8/10) | (n = 7/8) | (n = 12/14) | (n = 4/8) | ||
For experimental details, see text. ND, not done. BM, bone marrow.
Expressed in terms of input (into the culture) cell equivalents.
Based on the number of CRU measured at the end of the 9- to 10-day cultures.
Calculated assuming that two femurs and two tibias together comprise 25% of the total marrow population of the mouse (41).
Determined by multiplying the frequency of Ly5.1+ CRU present in the 1° recipients’ BM (from assays in 2° mice) times the total number of cells in the BM of the 1° recipients.
Expansion = the number of Ly5.1+ CRU present 18 or 20 weeks posttransplant in the 1° recipients divided by the number of Ly5.1+ CRU present in the initial transplant, exclusive of potential losses due either to a seeding efficiency of <100% of the Ly5.1+ CRU injected into the 1° recipients and/or to subsequent CRU differentiation.
% repopulation of positive 2° mice; (n = number of positive/total).
Evidence that Most CRU in the Sca-1+lin− Population of Adult Mouse Marrow Generate Daughter CRU When Stimulated by IL-11 plus FL and SF in Vitro.
To obtain a first approximation of the frequency of cells able to generate CRU in the presence of IL-11, FL, and SF in vitro, mini-cultures were initiated with 15 Ly5.1+ Sca-1+lin− marrow cells each (i.e., ≈0.3 CRU per culture) so that the likelihood of a single culture containing >1 CRU was <0.05. Then, 10 days later, each well was assessed individually for the presence of CRU by injecting the entire culture together with 105 freshly isolated Ly5.2+ adult marrow cells into a single, irradiated Ly5.2+ (C57BL/6) recipient. A total of 100 mini-cultures were analyzed in two of these limiting dilution experiments. PB samples from 27 of the 100 test recipients contained detectable Ly5.1+ (culture-derived) lymphoid and myeloid cells for at least 4 months posttransplant, and in 6 of these, engraftment levels of 5, 6, 8, 21, 35, and 40% were measured. In the other 21 mice, the levels of engraftment were lower (mean ± SD = 1.5 ± 0.8%, n = 21). Thus, the proportion of cultures at the end of 10 days that contained at least 1 CRU was the same as that anticipated to have been seeded with 1 CRU (0.3 vs. 0.27), and hence, the calculated frequency of CRU “precursors” in the input innoculum (2%) was also the same as the anticipated original CRU concentration (1.5%).
Inability of IL-11 plus FL and SF to Expand Fetal Liver CRU Numbers.
To determine whether an expansion of the CRU population present in the fetal liver of 14.5-day-old mouse embryos could be obtained under culture conditions that were effective for stimulating adult marrow cells, Sca-1+TER119− CD34+ fetal liver cells were isolated and cultured for 10 days in IL-11, FL, and SF as described above. The selection of this phenotype was based on earlier experiments in which injection of 1,000 TER119−CD34+ cells (≈3.4% of the total day 14.5 fetal liver population) into 3 recipients each resulted in 73 ± 11% fetal liver cell-derived engraftment at 4 months posttransplant, whereas no engraftment was detectable at any time in recipients of 1,000 TER119− CD34− cells (≈10% of the fetal liver). Limiting dilution analysis of freshly isolated Sca-1+TER119− CD34+ cells (≈0.6% of the fetal liver) showed the frequency of CRU to be 1 per 50 (with a range defined by ± SE of 1/34 to 1/75). However, none of the recipients of cultured fetal liver cells showed any evidence of culture-derived lymphoid or myeloid cells in their peripheral blood at 7 or 19 weeks posttransplant (six and three mice injected with the cultured progeny of 50 and 14 Sca-1+ Ter119− CD34+ fetal liver cells, which would have contained 1 or 0.3 CRU, respectively).
DISCUSSION
In this study we show that stimulation of adult mouse marrow cells with a combination of IL-11, FL, and SF can lead to a significant net (3-fold) expansion of the hematopoietic stem cell population without diminution of the magnitude or permanence (up to 1 year) of their lympho-myeloid reconstituting ability or their ability to reamplify their numbers (up to 30-fold) subsequently in vivo. Two lines of evidence suggest that this expansion results from an ability of IL-11 in combination with FL and SF to stimulate hematopoietic stem cell self-renewal divisions in vitro. First, the expansion observed was obtained from populations known to be highly enriched (≈300-fold) in their content of cells with stem cell activity (6, 28). Second, analysis of cultures initiated with limiting numbers of CRU (0.3 per culture) indicated that the frequency of cells detectable as CRU in the original input suspension was the same as the frequency of cells able to generate (or maintain) detectable numbers of CRU under the culture conditions used. Although wide variation in the extent of repopulation obtained from the transplantation of individual cultures was noted, the design of the experiments used here would not allow discrimination between CRU numbers and activity as contributing to this variation. Even single CRU in normal marrow are known to exhibit large variations in their repopulating activity (1, 30, 31), and it might be expected that this would be exaggerated by the stochastic element that appears to characterize the self-renewal (vs. differentiation) behavior of many types of primitive hematopoietic cells [including CFU-GEMM, CFU-spleen (CFU-S), and LTC-IC] when these are stimulated to proliferate either in vitro (32–34) or in vivo (35). The use of suspensions that are more highly enriched in their CRU content should, however, allow these questions to be addressed using single cell cultures. The present findings thus open the door to future studies of critical issues in hematopoietic stem cell biology that concern the ability of different treatments to influence the subsequent functional repertoire of affected stem cells.
In recent studies of the properties of the clonal progeny of single CD34+CD38− cells originally isolated from normal adult human marrow and generated in vitro under similar conditions to those described here, we showed that the retention or loss of LTC-IC activity could be modulated independently of cell viability or cell cycle progression by exposure of the cells to different relative or absolute cytokine concentrations (34). These findings suggest that the types of receptors activated and the intensity and/or duration of that activation can influence the integrity of cellular properties that may be required for (or reflect the maintenance of) stem cell potential. Therefore, it is interesting to note that in the present studies, an increase in LTC-IC numbers closely paralleled increases in CRU, in contrast to the much more rapid expansions of CFC and total cell numbers. The LTC-IC and CRU assays are known to identify closely related populations in normal murine and human hematopoietic tissues of either adult (24) or fetal (36) origin, but increasing evidence indicates that these assays detect a spectrum of biologically distinct cell types that may vary in the extent to which they overlap under different circumstances. For example, we have shown that the rate of decline of cells detectable as LTC-IC in LTC of mouse bone marrow cells is slower than the rate at which the number of CRU decreases (37). Similarly, near-normal numbers of LTC-IC can be generated and detected in the marrow of adult mice when c-kit-mediated signaling is deficient, although a 15-fold reduction in CRU activity is exhibited by the same cell suspensions (23).
The present studies also bring into focus another emerging theme in hematopoietic stem cell biology—i.e., that ontogeny adds another dimension to the patterns of change that differentiating hematopoietic cells may undergo and, in particular, the ways these changes may be regulated. In the murine system, fetal liver stem cells have been shown to differ from their adult counterparts with respect to phenotypic markers, proliferative and self-renewal potential in vivo, dependence on SF in vivo for long-term maintenance of hematopoiesis, and cytokine responses in vitro (14, 16, 29, 36, 38–40). In this study, we demonstrate a difference in CD34 expression on fetal and adult hematopoietic stem cells (no CD34− CRU detectable in fetal liver in contrast to adult mouse marrow). In addition, we showed that exposure of fetal liver cells to a combination of IL-11, FL, and SF resulted in a loss of long-term repopulating ability in spite of their generation under these conditions of huge populations of differentiating lymphoid and myeloid cells (18). These findings predict that similar differences will characterize the responses of primitive human hematopoietic cells in adult marrow and cord blood, and further delineation of these differences may be crucial to the design of appropriate expansion or gene therapy protocols involving such ontologically distinct sources of cells.
Acknowledgments
We thank Maya Sinclaire, Jessyca Maltman, Julie Parker, and Alex Hodge for expert technical assistance; Gayle Thornbury for FACS operation; Bernadine Fox for typing the manuscript; Dr. John Spinelli (University of British Columbia, Vancouver, BC) for statistical advice; and Dr. Peter Lansdorp (Terry Fox Laboratory), Stem Cell Technologies, Genetics Institute, Immunex, and Zymogenetics for gifts of antibodies, media, and cytokines. This work was supported by grants from the National Institutes of Health (HL 55435) and the National Cancer Institute of Canada with funds from the Terry Fox Run. C.J.E. is a Terry Fox Cancer Research Scientist of the National Cancer Institute of Canada.
ABBREVIATIONS
- FL
flt3-ligand
- SF
Steel factor
- FBS
fetal bovine serum
- CRU
competitive repopulating unit
- CFC
colony-forming cell(s)
- CFU
colony-forming unit
- LTC
long-term culture(s)
- LTC-IC
LTC-initiating cells
References
- 1.Fraser C C, Szilvassy S J, Eaves C J, Humphries R K. Proc Natl Acad Sci USA. 1992;89:1968–1972. doi: 10.1073/pnas.89.5.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kodama H, Nose M, Yamaguchi Y, Tsunoda J, Suda T, Nishikawa S. J Exp Med. 1992;176:351–361. doi: 10.1084/jem.176.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wineman J, Moore K, Lemischka I, Muller-Sieburg C. Blood. 1996;87:4082–4090. [PubMed] [Google Scholar]
- 4.Szilvassy S J, Weller K P, Lin W, Shrama A K, Ho A S Y, Tsukamoto A, Hoffman R, Leiby K R, Gearing D P. Blood. 1996;87:4618–4628. [PubMed] [Google Scholar]
- 5.Bodine D M, Crosier P S, Clark S C. Blood. 1991;78:914–920. [PubMed] [Google Scholar]
- 6.Rebel V I, Dragowska W, Eaves C J, Humphries R K, Lansdorp P M. Blood. 1994;83:128–136. [PubMed] [Google Scholar]
- 7.Traycoff C M, Cornetta K, Yoder M C, Davidson A, Srour E F. Exp Hematol. 1996;24:299–306. [PubMed] [Google Scholar]
- 8.Yonemura Y, Ku H, Hirayama F, Souza L M, Ogawa M. Proc Natl Acad Sci USA. 1996;93:4040–4044. doi: 10.1073/pnas.93.9.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yonemura Y, Ku H, Lyman S D, Ogawa M. Blood. 1997;89:1915–1921. [PubMed] [Google Scholar]
- 10.Ogawa M. Blood. 1993;81:2844–2853. [PubMed] [Google Scholar]
- 11.Petzer A L, Zandstra P W, Piret J M, Eaves C J. J Exp Med. 1996;183:2551–2558. doi: 10.1084/jem.183.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sitnicka E, Lin N, Priestley G V, Fox N, Broudy V C, Wolf N S, Kaushansky K. Blood. 1996;87:4998–5005. [PubMed] [Google Scholar]
- 13.Trevisan M, Yan X, Iscove N N. Blood. 1996;88:4149–4158. [PubMed] [Google Scholar]
- 14.Morrison S J, Hemmati H D, Wandycz A M, Weissman I L. Proc Natl Acad Sci USA. 1995;92:10302–10306. doi: 10.1073/pnas.92.22.10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakano T, Kodama H, Honjo T. Science. 1996;272:722–724. doi: 10.1126/science.272.5262.722. [DOI] [PubMed] [Google Scholar]
- 16.Rebel V I, Lansdorp P M. J Hematother. 1996;5:25–37. doi: 10.1089/scd.1.1996.5.25. [DOI] [PubMed] [Google Scholar]
- 17.Dexter T M, Spooncer E, Toksoz D, Lajtha L G. J Supramol Struct. 1980;13:513–524. doi: 10.1002/jss.400130410. [DOI] [PubMed] [Google Scholar]
- 18.Lemieux M E, Chappel S M, Miller C L, Eaves C J. Exp Hematol. 1997;25:951–957. [PubMed] [Google Scholar]
- 19.Holyoake T L, Freshney M G, McNair L, Parker A N, McKay P J, Steward W P, Fitzsimons E, Graham G J, Pragnell I B. Blood. 1996;87:4589–4595. [PubMed] [Google Scholar]
- 20.Hawley R G, Hawley T S, Fong A Z C, Quinto C, Collins M, Leonard J P, Goldman S J. Proc Natl Acad Sci USA. 1996;93:10297–10302. doi: 10.1073/pnas.93.19.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unkeless J C. J Exp Med. 1979;150:580–596. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rebel V I, Miller C L, Thornbury G R, Dragowska W H, Eaves C J, Lansdorp P M. Exp Hematol. 1996;24:638–648. [PubMed] [Google Scholar]
- 23.Miller C L, Rebel V I, Lemieux M E, Helgason C D, Lansdorp P M, Eaves C J. Exp Hematol. 1996;24:185–194. [PubMed] [Google Scholar]
- 24.Lemieux M E, Rebel V I, Lansdorp P M, Eaves C J. Blood. 1995;86:1339–1347. [PubMed] [Google Scholar]
- 25.Landreth K S, Dorshkind K. J Immunol. 1988;140:845–852. [PubMed] [Google Scholar]
- 26.Fazekas De St. Groth S. J Immunol Methods. 1982;49:R11–R23. doi: 10.1016/0022-1759(82)90269-1. [DOI] [PubMed] [Google Scholar]
- 27.Szilvassy S J, Humphries R K, Lansdorp P M, Eaves A C, Eaves C J. Proc Natl Acad Sci USA. 1990;87:8736–8740. doi: 10.1073/pnas.87.22.8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Osawa M, Hanada K I, Hamada H, Nakauchi H. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 29.Pawliuk R, Eaves C, Humphries R K. Blood. 1996;88:2852–2858. [PubMed] [Google Scholar]
- 30.Capel B, Hawley R G, Mintz B. Blood. 1990;75:2267–2270. [PubMed] [Google Scholar]
- 31.Morrison S J, Weissman I L. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 32.Humphries R K, Eaves A C, Eaves C J. Proc Natl Acad Sci USA. 1981;78:3629–3633. doi: 10.1073/pnas.78.6.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sutherland H J, Lansdorp P M, Henkelman D H, Eaves A C, Eaves C J. Proc Natl Acad Sci USA. 1990;87:3584–3588. doi: 10.1073/pnas.87.9.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zandstra P W, Conneally E, Petzer A L, Piret J M, Eaves C J. Proc Natl Acad Sci USA. 1997;94:4698–4703. doi: 10.1073/pnas.94.9.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siminovitch L, McCulloch E A, Till J E. J Cell Physiol. 1963;62:327–336. doi: 10.1002/jcp.1030620313. [DOI] [PubMed] [Google Scholar]
- 36.Miller C L, Rebel V I, Helgason C D, Lansdorp P M, Eaves C J. Blood. 1997;89:1214–1223. [PubMed] [Google Scholar]
- 37.Lemieux M E, Eaves C J. Blood. 1996;88:1639–1648. [PubMed] [Google Scholar]
- 38.Micklem H S, Ford C E, Evans E P, Ogden D A, Papworth D S. J Cell Physiol. 1972;79:293–298. doi: 10.1002/jcp.1040790214. [DOI] [PubMed] [Google Scholar]
- 39.Jordan C T, Astle C M, Zawadzki J, Mackarehtshian K, Lemischka I R, Harrison D E. Exp Hematol. 1995;23:1011–1015. [PubMed] [Google Scholar]
- 40.Rebel V I, Miller C L, Eaves C J, Lansdorp P M. Blood. 1996;87:3500–3507. [PubMed] [Google Scholar]