

Abstract
Adaptive phenotypic plasticity in life history traits, behaviours, and strategies is ubiquitous in biological systems. It is driven by variation in selection pressures across environmental gradients and operates under constraints imposed by trade-offs. Phenotypic plasticity has been thoroughly documented for multicellular taxa, such as insects, birds and mammals, and in many cases the underlying selective pressures are well understood. Whilst unicellular parasites face many of the same selective pressures and trade-offs, plasticity in their phenotypic traits has been largely overlooked and remains poorly understood. Here, we demonstrate that evolutionary theory, developed to explain variation observed in the life-history traits of multicellular organisms, can be applied to parasites. Though our message is general – we can expect the life-histories of all parasites to have evolved phenotypic plasticity – we focus our discussion on malaria parasites. We use an evolutionary framework to explain the trade-offs that parasites face and how plasticity in their life history traits will be expressed according to changes in their in-host environment. Testing whether variation in parasites traits is adaptive will provide new and fundamental insights into the basic biology of parasites, their epidemiology and the processes of disease during individual infections.
Keywords: gametocyte conversion, genotype by environment interaction, life history trade-off, malaria, phenotypic plasticity, Plasmodium, red blood cell preference, sex allocation, virulence
Understanding how natural selection shapes the traits of infectious disease causing organisms is central to evaluating the likely success of intervention strategies and virulence management (Stearns and Koella 2007). Evolutionary theory has been successfully developed as a tool for investigating the ultimate causation of biological variation in multicellular organisms, and is increasingly being applied to explain why genetic variation is maintained for traits, such as virulence, in unicellular parasites. However, other parasite life-history traits and behaviours of considerable clinical and epidemiological importance also vary extensively. Both the ultimate and proximate causes of this variation are poorly understood but available data indicate that parasites employ remarkably sophisticated strategies for maximizing their transmission success (e.g. Paul et al. 2000; Koella et al. 2002; Reece et al. 2008). In this article, we consider how evolutionary theory can be applied to explain why traits associated with growth and reproduction in malaria (Plasmodium) parasites vary throughout infections and in response to changes in the in-host environment. We explain the relevant evolutionary concepts, review recent success in explaining a key parasite trait (sex ratio), and outline how an evolutionary framework will provide novel and important insights into parasite biology. Our discussion focuses on malaria parasites because within-infection processes have been most thoroughly examined in this group but our message is general: the traits exhibited by parasites and pathogens are a result of natural selection optimizing resource allocation strategies.
Evolution of life history traits: trade-offs and phenotypic plasticity
Trade-offs and adaptive phenotypic plasticity are central to our evolutionary understanding of biological variation in metazoan organisms. Fundamental to life history theory is the idea that, whilst natural selection acts to maximize some measure of fitness, the possible combinations of phenotypic traits that can be expressed are limited by trade-offs and constraints (Roff 1992). Trade-offs can take many and diverse forms, but theoreticians have most commonly considered the role of resource allocation trade-offs: organisms have limited resources to invest in the processes (e.g. growth, maintenance, and reproduction) required to successfully transmit gene copies to future generations (Roff 1992; Stearns 1992). Natural selection is expected to optimize the allocation of resources between different processes so as to maximize fitness, assuming that sufficient genetic variation exists to allow the optimum to be reached.
Understanding life history decisions and the reasons for phenotypic variability becomes more complicated when one considers that phenotypes do not evolve under constant environment conditions. The concept of adaptive phenotypic plasticity has been central to our understanding of the evolutionary consequences of environmental variation (Schlichting and Pigliucci 1998). Phenotypic plasticity is broadly defined as a change in the phenotype of a given genotype in response to a change in environmental conditions (Schlichting and Pigliucci 1998). If natural selection favours different phenotypes under different environmental conditions, we expect the evolution of plastic phenotypic responses to information or cues that predict environmental change (Gotthard and Nylin 1995; Via et al. 1995; Pigliucci 1996). Phenotypic plasticity can evolve providing that the environment is predictable and suitable genetic variation exists. Plastic responses may involve facultative changes in a single trait, whole suites of life history, behaviours, or resource allocation strategies (Stearns 1992; Pigliucci 2001).
Phenotypic plasticity is expected to be costly, in that its expression will require the diversion of resources away from other functions such as reproduction or maintenance. Costs are diverse and can include the expenditure of resources on maintaining sensory systems, gathering and processing information, or expressing alternative phenotypes. The ability to respond appropriately to changes in the environment will also be limited by organisms’ ability to detect reliable cues and process the resulting information (de Witt et al. 1998). Selection will act to maximize the fitness advantages of plasticity whilst minimizing the costs involved. This trade-off could result in the evolution of a single, fixed plastic response across a population, variation in plastic responses across genotypes within a population (genotype-by-environment interactions; ‘GxE’), or nonplastic, environmental specialist phenotypes (Pigliucci 2001). In the event that environmental variation is completely unpredictable, selection may instead favour the production of randomly or alternately varying phenotypes (bet hedging strategies).
Whilst individual organisms are readily identifiable as targets of natural selection in metazoans, the genotype within an infection represents the comparable target in parasites (West et al. 2006). When infections consist of a single genotype, all parasites are genetically identical and trade-off resolution will maximize the fitness of all co-infecting parasites (Hamilton 1963; Foster 2005). When infections are composed of multiple genotypes, parasites from the same genotype are more closely related to each other than to con-specifics. Each genotype could have different optimal strategies because factors such as variation in competitive ability or immune-evasion may influence the relative importance of investing in different processes.
Trait variation in the life cycle of malaria parasites
Malaria (Plasmodium) and related Apicomplexan parasites comprise a diverse group of pathogens that are responsible for some of the most serious infectious diseases of humans, wildlife, livestock and companion animals (Garnham 1966). Despite more than a century of research, these parasites have resisted efforts to eradicate and control them and remain responsible for between 1 and 3 million deaths per year. There are over 170 described Plasmodium species, which infect mammals, birds and a wide variety of reptiles (Garnham 1966). Despite their diversity, their life-cycle (Fig. 1) always includes several rounds of asexual replication in a vertebrate host and sexual reproduction in a dipteran vector. When an infected vector bites a host, infective stages (sporozoites) are released and start the pre-erythrocytic phase by invading a specific tissue. After several rounds of replication, erythrocyte-invading stages are released into the blood. These blood-stage parasites replicate asexually and disease symptoms develop (anaemia, weight loss and cerebral malaria in some cases). Every cell cycle a small proportion of asexually produced parasites develop into nonreplicating, male or female sexual stages, termed gametocytes (Smith et al. 2002). Asexual parasites do not survive when taken up by a vector in a blood meal, but the gametocytes rapidly differentiate into gametes and fertilization occurs within an hour (Micks et al. 1948; Sinden 1983a,b; Janse et al. 1986). Each male gametocyte can differentiate into up to eight gametes whereas each female gametocyte differentiates into a single gamete. To make their gametes, males must leave their red blood cells, undergo three rounds of genome replication/mitotic division, and construct eight flagella, each with a genome (Janse et al. 1986). Conversely, female gametocytes become gametes once they have condensed and left their red blood cells. Zygotes undergo a series of morphological transformations to establish infections in their vector, leading to the formation of thousands of host-infective sporozoites that migrate to the salivary glands ready for transmission (Baton and Ranford-Cartwright 2005).
Figure 1.
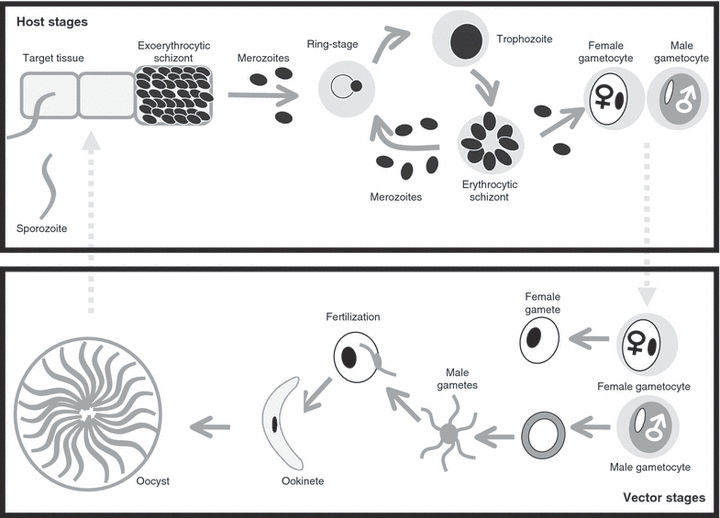
The life-cycle of malaria (Plasmodium) parasites.
Whilst the basic features of their life-cycle are conserved, malaria parasites exhibit considerable variation in traits associated with growth and reproduction. For example, marked variation in sex ratio patterns (Fig. 2; proportion of gametocytes that are male) occurs across species, within species, and during infections (Paul et al. 1999, 2000; West et al. 2001; Paul et al. 2002; Read et al. 2002; Paul et al. 2003; Robert et al. 2003; Reece et al. 2005, 2008). Whilst striking species differences in some traits are well documented, most have been largely overlooked. In Table 1, we highlight examples of traits for which variation has been observed between and within species and during infections. Empirical data are scarce but do show that there is interesting and important biological variation to be explained.
Figure 2.
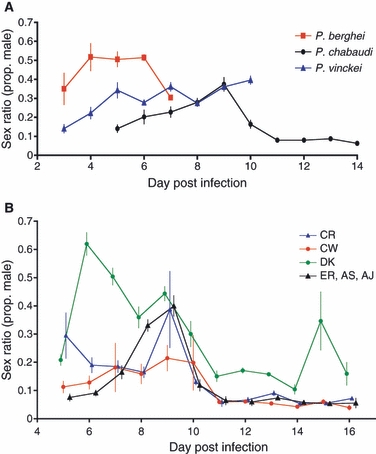
Sex ratios of malaria parasites vary considerably across species (A), within species (B), and during infections (A) and (B). Data shown are mean ± SE for infections of: (A) three different rodent malaria species, followed until either the infections were cleared or the experiment was terminated; and (B) six different Plasmodium chabaudi genotypes that follow four significantly different patterns throughout infections (Reece et al. 2008). Means are calculated from between 6 and 30 independent infections and all were initiated by 106 parasites in the same host genotype (MF1). Thus, this variation is not due to infective dose or host genotype.
Table 1.
Variation in life-history traits associated with growth and reproduction exhibited by malaria (Plasmodium) parasites. We highlight traits for which variation is observed across species (species), within species (genotype), and during infections (infection). This list is by no means exhaustive, but is indicative of the empirical literature to date
| Function | Trait | Examples of variation | Refs |
|---|---|---|---|
| Growth | Cell cycle duration and synchronicity | Species. The species of parasite infecting humans can be diagnosed from the regularity of fevers resulting from each synchronous replication cycle (P. vivax and P. falciparum for fevers every 48 h and P. malariae every 72 h). In the rodent malarias, P. berghei is asynchronous with cell cycle duration of 22–23 h, whereas mature P. chabaudi parasites rupture synchronously every 24 h | Mons et al. (1985), Carter and Walliker (1975), Mackinnon et al. (2002a,b), Reilly et al. (2007) and Garnham (1966) |
| Genotype. In-vitro evidence that two clones of P. falciparum have an average difference of ∼5 h in their cell cycle duration | |||
| Infection. Observations suggest that P. chabaudi synchronicity decreases as infections progress | |||
| Number of merozoites per schizont | Species. Species-specific data are normally presented as ranges (e.g. P.falciparum 8–32 merozoites per schizont), but it is not known if this represents genetic or within-infection variation. Species-specific estimates for the number of merozoites produced per schizont range from 4 to 5 in P. juxtanucleare (an avian malaria) to over 90 for P. giganteum (a lizard malaria) | Reilly et al. (2007), Garnham (1966), Kissinger et al. (2002) and Schall (1990) | |
| Genotype. Different clones of P. falciparum produce different modal numbers of merozoites per schizont, and this may also vary with the type of tissue in which parasites sequester | |||
| Infection. P. berghei produces more merozoites per schizont in reticulocytes than in mature RBC | |||
| Red blood cell preference | Species. In human malarias, P. ovale and P. vivax prefer reticulocytes, P. malariae prefers mature RBCs and P. falciparum is able to infect both age classes. Similar patterns occur in the rodent malarias; preference spans the continuum from strict preference for reticulocytes (e.g. P. berghei) to specialists for mature RBCs (e.g. P. vinckei) with generalists (e.g. P. chabaudi) in between. Similar examples can be found in bird malarias | Antia et al. (2008), Simpson et al. (1999), Paul and Brey (2003),Killick-Kendrick and Peters (1978) and Taylor-Robinson and Phillips (1994) | |
| Genotype. Some evidence that P. chabaudi genotypes may vary in the age range of cells they can infect | |||
| Infection. P. falciparum may become less selective as infections progress. A clone of P. chabaudi changes from strict preference for mature RBCs to reticulocytes around peak parasitaemia, changing again to strict preference for mature RBCs during the chronic stage of the infection | |||
| Cytoadherence | Species. Sequestration* occurs in most (e.g. P. falciparum and P. berghei) but not all (e.g. P. malariae and P. knowlesii) species investigated. Rosetting† rates vary across species, but has been observed in all species analysed | Mackinnon et al. (2002a,b), Garnham (1966), Sherman et al. (2003) | |
| Genotype. Genetic variation for rosetting rates observed in P. chabaudi (from 10% to 40%) | |||
| Infection. Highest rosetting rates observed before peak parasitaemia in P. chabaudi. Some lines of P. falciparum lose the ability to sequester with serial culture | |||
| Reproduction | Conversion | Species. Rarely calculated, potentially because data on cell cycle duration, gametocyte development time and longevity are required | Graves et al. (1984), Reece et al. (2005), Buckling et al. (1997, 1999) |
| Genotype. In vitro data suggest genetic variation for conversion in P. falciparum | |||
| Infection. P. chabaudi increases conversion in response to EPO and antimalarial drugs. P. falciparum also increases conversion in response to antimalarial drugs | |||
| Sex ratio | Species, genotype and infection. See Fig. 2 and text (Explaining sex ratio variation in malaria parasites) | West et al. (2001), Reece et al. (2008) and Paul et al. (2000) |
Sequestration: withdrawal of infected RBCs from the peripheral circulation to the microvasculature where they adhere to endothelial cells.
Rosetting: adherence of uninfected RBCs to infected cells leading to the formation of small clusters of cells.
A fundamental question in evolutionary biology is whether or not genetic variation exists in a focal trait. Natural selection cannot produce an evolutionary response without appropriate genetic variation underlying phenotypic variation. In malaria parasites, within-species genetic variation can be tested for when genotypes (clonal isolates) can be detected (e.g. Schall and Vardo-Zalik 2007). One source of such isolates is the bank of P. chabaudi genotypes in the WHO Registry of Standard Malaria Parasites, held at the University of Edinburgh, UK. Experimental work has revealed that whilst these con-specific genotypes follow broadly similar infection dynamics, they vary in the maximum parasite and gametocyte densities achieved, and the degree of harm caused to their hosts in terms of weight loss, anaemia and mortality risk (Mackinnon and Read 1999; Timms et al. 2001; Mackinnon et al. 2002a,b;de Roode et al. 2003; Grech et al. 2006). Analogous field experiments using wild caught parasites of the lizard malaria (P. mexicanum) have also revealed evidence for genetic variation in these traits (Eisen and Schall 2000). The details of infection patterns exhibited by a given parasite genotype can also be influenced by host genotype, as observed in P. chabaudi (de Roode et al. 2004; Grech et al. 2006; Raberg et al. 2007). These studies clearly show there is significant genetic variation underlying both important parasite life history traits and their expression in response to changes in the in-host environment.
Another key evolutionary question is how variation in the environment will influence selection on life history trade-offs. The ‘environment’ that malaria parasites experience throughout their life-cycle is remarkably complex as it encompasses the internal biotic environment of at least two other living organisms (the host and vector), and the external environment experienced by those organisms. Parasites will encounter different types of host, for which there could be different optimal solutions to resource allocation problems. During infections, the in-host environment shifts radically due to complex interactions between factors driven by both parasites and hosts (e.g. immune factors, erythropoietic state, presence of competing parasites). To thrive in such variable conditions, parasites should alter their life history decisions and behaviours to best suit each situation they encounter. The ability of natural selection to shape how parasites respond to environmental variation will depend on: the availability of reliable information on environmental conditions and the ability of parasites process it, the presence of suitable genetic variation in parasite populations (GxE), and the costs of that particular form of plasticity.
In the next section, we describe how evolutionary theory has been successfully used to understand variation in sex ratio, an important fitness-determining trait. In the subsequent sections we outline how this approach can be applied to explain other parasite life history traits that are central to survival and transmission. Whilst the concepts we present are applicable to all host and vector stages of the life-cycle, we focus on parasite traits expressed during the erythrocytic phase of infections because the necessary tools and techniques to experimentally test our predictions have recently become available.
Explaining sex ratio variation in malaria parasites
Sex allocation is one of the most well understood topics in evolutionary biology (Charnov 1982; Hardy 2002; West 2009). In many cases, simple theory can successfully predict when, why and by how much organisms should adjust their offspring sex ratio in response to a change in their circumstances. As shown in Fig. 2, sex allocation of malaria parasites varies considerably, but the application of evolutionary theory to explain this variation has been controversial (Shutler et al. 1995; Ferguson 2002, 2003; West et al. 2003).
Sex ratios in malaria parasites are generally female-biased and evolutionary theory predicts that sex ratios reflect the inbreeding rate (Read et al. 1992; Dye and Godfray 1993; West et al. 2000; Nee et al. 2002). Gametocytes taken up in a blood meal represent the genetic composition of their host's infection. The inbreeding rate is high when mating occurs between gametocytes from one or a small number of genotypes, but the inbreeding rate is low when multiple genotypes are present in a mating group. Female-biased sex ratios are expected when inbreeding occurs because this represents the most efficient allocation of resources to maximize the fertilization success of a mating group. Because each male can fertilize more than one female, a female-biased sex ratio reduces competition between related males and maximizes the number of females available to mate with (Taylor 1981). In contrast, when outbreeding, the greatest fitness returns come from increasing investment in males. In this situation, because unrelated females belonging to other genotypes are present, a genotype that produces more males will have the greatest genetic representation in the zygote population. This is ‘Hamilton's Local Mate Competition’ theory (LMC; Hamilton 1967) and it explains why female-biased sex ratios are favoured in spatially structured populations for a variety of taxa, from plants to insects to snakes (West et al. 2005).
The application of LMC theory to malaria parasites has proved controversial because it does not explain why sex ratios vary throughout infections or why population sex ratios in some related Apicomplexans do not correspond to their inbreeding rate (Shutler and Read 1998; West et al. 2001; Paul et al. 2003). However, cases where the data do not correspond to LMC can be explained by a simple extension of the theory, known as ‘fertility insurance’ (West et al. 2002; Gardner et al. 2003). Fertility insurance is important when there is a risk that not all females in a blood meal will be fertilized. This problem can arise when gametocyte density is low, due to red cell limitation in anaemic hosts or low conversion, or when transmission-blocking immune factors impair the ability of male gametocytes to make viable gametes. When male gamete viability is low or few gametocytes are able to interact in a blood meal, parasites are expected to alter their sex ratio by increasing their investment in male gametocytes, resulting in a less female-biased sex ratio than their inbreeding rate suggests. By increasing investment in males, parasites can ensure that they have produced enough males to fertilize their females.
The recent development of reverse transcription quantitative PCR assays for malaria parasites that are both genotype- and sex-specific (Drew and Reece 2007) have enabled the predictions of LMC and fertility insurance theory to be tested. Recently conducted, conceptually simple experiments in which the sex ratios produced by focal P. chabaudi genotypes were followed in single- and double-genotype infections, showed that parasites can evaluate their likely inbreeding rate and faculatively adjust their sex ratio according to the predictions of LMC theory (Reece et al. 2008). Furthermore, the double-infection data revealed a negative correlation between sex ratio and the proportion of gametocytes contributed by focal genotypes that quantitatively fits LMC theory. Analysis of sex ratio patterns followed throughout infections by six different P. chabaudi genotypes support the predictions of fertility insurance theory (Reece et al. 2008). First, female-biased sex allocation in response to LMC decreases as infections progress and parasite densities decline due to resource competition, anaemia and development of the host's immune response. Second, within-infection sex ratio patterns, for which there is significant genetic variation, can be explained by variation in anaemia and parasite densities. Third, sex ratios are adjusted in response to erythropoetin (EPO; a hormone produced by anaemic hosts) and subsequent changes in the availability of RBC resources (Paul et al. 2000; Reece et al. 2005).
Facultative sex-ratio adjustment in response to changes in the genetic diversity of infections and within-host environment confirm that: (i) sex allocation strategies in malaria parasites are as sophisticated as those observed in multi-cellular taxa; (ii) malaria parasites can discriminate kin from non-kin and infer their own relative frequency in infections; (iii) malaria parasites are able to detect and respond to changes in host anaemia and the availability of preferred RBCs; and (iv) crucially, sex ratio variation can be explained by evolutionary theory (Shutler and Read 1998; Knowles and Sheldon 2008; Reece et al. 2008).
Adaptive plasticity in other parasite traits: keeping up with the environment?
In this section, we develop this successful application of evolutionary theory to predict how parasites should solve other trade-off problems. Specifically, we consider how plasticity in the allocation of resources to reproduction and growth should be expressed, discuss evidence for plasticity in the underlying traits, and describe how our predictions can be tested.
Investment into reproduction: conversion rate
From a life history perspective, the asexual replication of a single-genotype malaria infection can be viewed as ‘growth and maintenance’ and gametocyte density as ‘reproduction’. As in metazoans, a fundamental trade-off exists – resources are invested into growth at the expense of reproduction and vice-versa (Stearns 1992). In metazoans, quantifying how an individual has resolved this trade-off in terms of reproductive effort is rarely possible. In contrast, the reproductive effort of malaria parasites can easily be calculated as the proportion of a cohort of parasites that develop into gametocytes (conversion rate). Though rarely measured, conversion rate frequently varies throughout infections and is increased in response to EPO and the presence of antimalarial drugs (Buckling et al. 1997, 1999; Reece et al. 2005). Here, we outline explanations for these observations and predict how parasites are expected to alter conversion in response to changes in their in-host and social circumstances. These scenarios are not mutually exclusive and in reality we expect that parasites will simultaneously be resolving many of these trade-offs.
Why should parasites increase their reproductive effort when under attack from drugs? This is a ‘stressful’ situation in which parasites face elimination, so the currently held belief is that parasites have nothing to lose, and should increase conversion as a last attempt at transmission before being cleared. However, theory could also predict the opposite: parasites in stressful circumstances should reduce their conversion to maximize their chances of survival (Schneider et al. 2008). In this scenario, by investing in asexual replication, parasites benefit from the future reproductive opportunities that can be attained from surviving (Mideo and Day 2008). If this is the case, why do the data show the opposite pattern? To adopt the most appropriate strategy, parasites need the ability to determine when continued survival is impossible (e.g. due to terminally decreasing parasite density or inevitable host death). For novel situations like exposure to drugs, parasites may not have been under selection for long enough to be able to evaluate their survival chances. In contrast, parasites will frequently experience severely anaemic hosts, so the ability to infer when host death is inevitable should have evolved. Available data are encouraging: P. gallinaceum parasites can determine whether their infections are lethal or will be resolved by hosts (Paul et al. 1999). In metazoans the analogous hypothesis (known as ‘terminal investment’; Williams 1966) is very difficult to test because measurements of relative reproductive effort are required and confounding variables (e.g. decline due to senescence or infection) make it hard to measure the intrinsic state of individuals. These issues can be overcome with malaria parasites because the performance of replicate infections of genetically identical parasites can be followed in progressively deteriorating in-host conditions or drug treatment regimes.
The terminal investment hypothesis was developed from the prediction that the reproductive strategy an organism adopts will depend on the value of the fitness gains made from current reproductive effort, relative to future reproductive effort. In humans, relatively few malaria infections result in host death during the acute phase and parasites enter a longer-term (chronic) phase in which they can persist for years (Snounou et al. 2000). Because these infections have a long ‘life-span’, resource allocation strategies will reflect the trade-off between current and future reproductive success. More data on transmission success from acute and chronic stage infections are needed to test for this trade-off, but parasites at extremely low densities in chronic P. falciparum infections can infect mosquitoes, (Schneider et al. 2007).
The idea that current reproductive effort should be traded-off against future reproductive effort applies to two other situations: in-host competition and resource availability. Parasites frequently experience in-host competition with con-specific and con-generic parasites (Paul et al. 2003; de Roode et al. 2003, 2005; Mayxay et al. 2004; Bell et al. 2006). Because the best competitors are predicted to be genotypes that acquire the greatest share of RBC resources, a decrease in conversion will result in faster replication and increased survival (Mideo and Day 2008). This prediction has been tested but the data are inconclusive: focal P. chabaudi genotypes monitored in single- and double-genotype infections revealed only a small reduction in conversion in the competitively inferior genotype in one of the two host strains used (Wargo et al. 2007). In addition to competition, parasites should alter their conversion when hosts become anaemic. If preferred RBCs are scarce, parasites should invest in survival, but only if their resource limitation situation is temporary. Depending on their preference for young or old RBCs, host anaemia can mean either the loss or production of preferred RBCs. In line with this prediction, P. chabaudi, a species that can infect young and old RBCs, increases investment in gametocytes when hosts receive EPO, but P. vinckei, which can only infect mature RBCs, does not alter conversion and maintains transmission by adjusting sex ratio instead (Reece et al. 2005).
Virulence and replication
Parasites must maximize survival and transmission in the face of competition and the inevitable attack of the immune response. Theory predicts that success in these endeavors is correlated with parasite virulence (damage caused to hosts) because fast growing parasites benefit from a larger pool of mature parasites to produce gametocytes. A trade-off between the rate and duration of transmission occurs because virulent parasites risk killing their host and losing future transmission opportunities (Frank 1996). However, as slow growing parasites are poor competitors and at risk from rapid clearance by the host immune response, intermediate levels of virulence are favoured by natural selection (Read 1994; Frank 1996; Day 2003; Day and Proulx 2004; Frank and Schmid-Hempel 2008). Of all parasite traits, virulence is unique in having a large body of theory but, like most traits, the data lag far behind. The idea that parasites should adjust their virulence by facultatively altering traits that underlie replication rate is both theoretically and empirically unexplored. In addition to conversion rate, plasticity in components of replication rate would also regulate virulence. Traits likely to exhibit adaptive plasticity include the number of daughter cells (merozoites) produced by mature parasites (schizonts), cell cycle duration and synchronicity, and the ability of merozoites to invade different types of RBC. In this section, we explore how parasites should adjust these traits in response to the type of host they infect and changes in their in-host environment.
The type of host that malaria parasites encounter will vary substantially in health and immune status. This variation is likely to shape how selection acts on parasite life histories. If parasites face a trade-off between growth/maintenance and reproduction, it follows that changes in conversion rate will influence replication rate and virulence. Could parasite use their conversion rate to regulate virulence? For example, reducing conversion to prioritize replication will be the best strategy when attempting to establish an infection in an adult host with an already up-regulated immune response, and with competing parasites. But, if the next host is an immunologically naïve infant, parasites will be provided with plentiful resources and no competition or immediate danger of immune attack. Uncontrolled replication in this host type will result in premature host death, and instead, we expect parasites to adopt high conversion rates to prevent host death and prolong the lifespan of their infection, with the added bonus that plenty of gametocytes result (Alizon and van Baalen 2008). Could this be the evolutionary explanation for why more gametocytes are observed in children than adults? The use of conversion rate to regulate virulence may seem intuitively unlikely because gametocytes usually represent a small (<1%) proportion of the parasites present in an infection. Indeed, a frequently posed question is ‘why are there so few gametocytes?’ (Taylor and Read 1997; Mideo and Day 2008). The answer may lie in the trade-off between investing resources into growth/maintenance and reproduction: the fitness benefits of increasing gametocyte number may rarely outweigh the survival costs incurred in immune hosts and in competition with con-specifics.
Alternatively, parasites could also regulate virulence by altering a key component of their cell cycle: the number of merozoites released by each mature schizont. The pre-requisites for plasticity to evolve in this trait are met: there is genetic variation for the number of merozoites produced by P. falciparum schizonts (Reilly et al. 2007) and this trait is under fine-scale control because each nucleus within a developing schizont can divide a different number of times (Leete and Rubin 1996). Despite the vast species differences in the number of merozoites produced by schizonts, remarkably little is known about the ecological factors and evolutionary pressures shaping variation in this trait. However, increasing merozoite number would clearly be advantageous under deteriorating environmental conditions (e.g. immune attack), when survival is at risk. Data on variation in the number of merozoites in mature schizonts are extremely scarce, which is partly due to the challenges of collecting them – in many species, they sequester in tissues. New cell and molecular methods can overcome these difficulties because parasites can be collected prior to sequestration and incubated in vitro to maturity (Reece and Thompson 2008), or located during sequestration by whole-organism imaging methods (Franke-Fayard et al. 2006). Also, a variety of methods can be used to identify mature schizonts (e.g. fluorescent microscopy or cell sorting), which can be prevented from rupturing by incubation with cysteine protease inhibitors (Rosenthal et al. 1987; Sijwali et al. 2004). These tools can now be used to investigate when maturing parasites commit to the number of nuclear divisions they will undertake and which factors influence this. For example, does merozoite number vary according to whether schizonts contain parasites committed to asexual or sexual differentiation? There may also be trade-offs involved between the quantity of merozoites produced and their invasion capabilities, or time required to complete a cell cycle.
In addition to variation in the duration of cell cycles, the synchronicity and development time of parasite cohorts varies substantially (Gautret et al. 1994). Conventional wisdom assumes that cell cycles are timed to match gametocyte maturation with the diurnal patterns of vectors, and synchronous development enables parasites to withstand immune attack through ‘safety in numbers’ (Hawking et al. 1968). Yet, these explanations are unsatisfactory because it is precisely the synchronous bursting of schizonts that elicits a rapid and short-lived immune response that sterilizes gametocytes for several hours and attacks merozoites (Naotunne et al. 1991). Such high costs to transmission suggest that synchronicity yields substantial fitness benefits or is the outcome of a constraint imposed either by host factors or parasite physiology. The latter is unlikely because parasites increase their synchronicity and developmental rate in response to host melatonin (P. berghei;Hotta et al. 2000) and co-culture with other parasites (P. falciparum;Dyer and Day 1993). Our own observations suggest that synchronicity decreases in P. chabaudi as infections progress. This could allow parasites to avoid attack from immune factors or be directly caused by host immunity. Experiments to distinguish between parasite and host factors are possible using mice without immune systems or by blocking the actions of specific factors. The fitness costs and benefits of synchronicity can be quantified by assaying the expression of stage specific genes in synchronous (using purified merozoites), or asynchronous (a mix of stages) infections (Mons et al. 1985). Comparing cohort development in wild type parasites with transformed parasites in which genes underlying cell cycle regulation have been deleted would also be extremely useful.
Replication rate and virulence are also determined by parasites’ ability to acquire RBCs for replication. Cell and molecular data indicate there is substantial scope for adaptive plasticity in RBC preference, but variation in this trait is yet to be integrated into an evolutionary framework. The RBC preference of different species ranges from reticulocytes for P. vivax and P. ovale to mature RBCs for P. malariae, P. falciparum has the ability to switch its invasion strategy, and similar variation is found in the rodent malarias (Stubbs et al. 2005). Given the variation in the age and density of RBCs available in healthy and anaemic hosts during infections, the ability to facultatively alter RBC preference would be advantageous (Paul et al. 2003). The ability to invade all available RBC types may seem preferable, but different merozoite surface machinery is required for invading RBC of different ages (Cowman and Crabb 2006), and because the genes involved in invasion are also a source of antigenic variation, is it important for parasites to avoid expressing them simultaneously (Meunier 2001). This could explain why each P. yoelii merozoite only expresses one of the 25 genes that determine its RBC preference at a time (Preiser et al. 1999) and P. falciparum parasites can switch their invasion strategy (Stubbs et al. 2005). If so, invasion strategies will reflect the trade-off between the benefits of being a generalist that is able to invade a range of RBCs, versus the costs of exposing antigens to the immune system. An additional trade-off may be operating between the range of RBCs parasites can invade and their ability to grow within them. For example, P. berghei, parasites have a stronger preference for reticulocytes, and produce more merozoites per schizont in reticulocytes (Killick-Kendrick and Peters 1978). The molecular tools (e.g. reverse transcription quantitative PCR) required to measure gene expression and invasion abilities are available and can be applied to experiments in which host haemopoetic state is manipulated. However, to understand how parasites cope with changes in resource availability, quantitative data on RBC preference throughout infections are required. Here, mathematical models would prove particularly useful because complex erythropeotic processes determine what RBCs are available to parasites and this cannot accurately be inferred from snapshot observations of the type of RBCs that parasites have developed within (Cromer et al. 2006).
Conclusions and future directions
In addition to the specific investigations we have suggested, a substantially broader understanding of how within-infection dynamics are influenced by host factors, parasite traits, and interactions between them is required. This can be readily achieved for malaria parasites because: (i) focal genotypes can be followed during replicate infections in a range of scenarios; (ii) multiple focal genotypes can be investigated to establish whether GxE underlies patterns of trait variation and plasticity; (iii) multiple parasite and host variables can be measured and their potentially confounding influence on traits of interest can be statistically controlled for; and (iv) the use of mathematical models to examine within-host dynamics offers a new tool for hypothesis testing: iterations of theoretical developments and empirical testing promise to be a powerful approach (Mideo et al. 2008).
Evolutionary theory has been developed as a tool for investigating the ultimate causes of variation in life history traits and can provide new directions in the search for the proximate (mechanistic) processes. For example, evolutionary approaches have revealed adaptive phenotypic plasticity in sex ratio strategies, informing the search for underlying mechanistic processes (Paul et al. 2002). For other traits, such as red blood cell preference, proximate approaches have identified mechanisms likely to be involved in trade-offs and adaptive responses to environmental change. Experimental manipulations can be used to investigate what environmental information parasites use, whether these are direct or indirect cues, and what costs are paid for phenotypic plasticity. By understanding how, when and why parasites respond to different cues could enable interventions to induce parasites to make suboptimal choices that are clinically or epidemiologically beneficial. Identifying the physiological and genetic constraints on plasticity is of central importance in predicting how parasites will respond in the short- and long-term to drug and vaccine interventions. A synergy between research directed at evolutionary and mechanistic explanations for trait variation is surely the most efficient way to progress both fields.
Acknowledgments
We thank the Wellcome Trust (SR; WT082234MA), NERC (DN) and Fundação para a Ciência e Tecnologia Portugal (RR; SFRH/BD/39960/2007) for funding, DE Allen and two reviewers for improving the paper.
Literature cited
- Alizon S, Van Baalen M. Transmission-virulence trade-offs in vector-borne diseases. Theoretical Population Biology. 2008;74:6–15. doi: 10.1016/j.tpb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Antia R, Yates A, De Roode JC. The dynamics of acute malaria infections. I. Effect of the parasite's red blood cell preference. Proceedings of the Royal Society B: Biological Sciences. 2008;275:1449–1458. doi: 10.1098/rspb.2008.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baton LA, Ranford-Cartwright LC. Spreading the seeds of million murdering death: metamorphoses of malaria in the mosquito. Trends in Parasitology. 2005;21:573–580. doi: 10.1016/j.pt.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Bell AS, De Roode JC, Sim D, Read AF. Within-host competition in genetically diverse malaria infections: parasite virulence and competitive success. Evolution. 2006;60:1358–1371. [PubMed] [Google Scholar]
- Buckling AGJ, Taylor LH, Carlton JMR, Read AF. Adaptive changes in Plasmodium transmission strategies following chloroquine chemotherapy. Proceedings of the Royal Society of London Series B-Biological Sciences. 1997;264:553–559. doi: 10.1098/rspb.1997.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckling A, Ranford-Cartwright LC, Miles A, Read AF. Chloroquine increases Plasmodium falciparum gametocytogenesis in vitro. Parasitology. 1999;118:339–346. doi: 10.1017/s0031182099003960. [DOI] [PubMed] [Google Scholar]
- Carter R, Walliker D. New observations on the malaria parasites of rodents of the Central African Republic – Plasmodium vinckei petteri subsp. nov. and Plasmodiumc habaudi Landau, 1965. Annals of Tropical Medicine and Parasitology. 1975;69:187–196. doi: 10.1080/00034983.1975.11687000. [DOI] [PubMed] [Google Scholar]
- Charnov EL. The Theory of Sex Allocation. Princeton: Princeton University Press; 1982. [PubMed] [Google Scholar]
- Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124:755–766. doi: 10.1016/j.cell.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Cromer D, Evans KJ, Schofield L, Davenport MP. Preferential invasion of reticulocytes during late-stage Plasmodium berghei infection accounts for reduced circulating reticulocyte levels. International Journal for Parasitology. 2006;36:1389–1397. doi: 10.1016/j.ijpara.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Day T. Virulence evolution and the timing of disease life-history events. Trends in Ecology & Evolution. 2003;18:113–118. [Google Scholar]
- Day T, Proulx SR. A general theory for the evolutionary dynamics of virulence. American Naturalist. 2004;163:E40–E63. doi: 10.1086/382548. [DOI] [PubMed] [Google Scholar]
- Drew DR, Reece SE. Development of reverse-transcription PCR techniques to analyse the density and sex ratio of gametocytes in genetically diverse Plasmodium chabaudi infections. Molecular and Biochemical Parasitology. 2007;156:199–209. doi: 10.1016/j.molbiopara.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye C, Godfray HCF. On sex ratio and inbreeding in malaria parasite populations. Journal of Theoretical Biology. 1993;161:131–134. doi: 10.1006/jtbi.1993.1045. [DOI] [PubMed] [Google Scholar]
- Dyer M, Day KP. Regulation of the rate of asexual growth and commitment to sexual development by diffusible factors from in vitro cultures of Plasmodium falciparum. American Journal of Tropical Medicine and Hygiene. 1993;68:403–409. [PubMed] [Google Scholar]
- Eisen RJ, Schall JJ. Life history of a malaria parasite (Plasmodium mexicanum): independent traits and basis for variation. Proceedings of the Royal Society of London Series B-Biological Sciences. 2000;267:793–799. doi: 10.1098/rspb.2000.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DJP. Toxoplasma gondii and sex: essential or optional extra. Trends in Parasitolology. 2002;18:355–359. [PubMed] [Google Scholar]
- Ferguson DJP. More on Toxoplasma gondii, sex and premature rejection. Trends in Parasitolology. 2003;19:157–158. doi: 10.1016/s1471-4922(03)00033-3. [DOI] [PubMed] [Google Scholar]
- Foster KR. Hamiltonian medicine: why the social lives of pathogens matter. Science. 2005;308:1269–1270. doi: 10.1126/science.1108158. [DOI] [PubMed] [Google Scholar]
- Frank SA. Models of parasite virulence. Quarterly Review of Biology. 1996;71:37–78. doi: 10.1086/419267. [DOI] [PubMed] [Google Scholar]
- Frank S, Schmid-Hempel P. Mechanisms of pathogenesis and evolution of parasite virulence. Journal of Evolutionary Biology. 2008;21:396–404. doi: 10.1111/j.1420-9101.2007.01480.x. [DOI] [PubMed] [Google Scholar]
- Franke-Fayard B, Waters AP, Janse CJ. Real-time in vivo imaging of transgenic bioluminescent blood stages of rodent malaria parasites in mice. Nature Protocols. 2006;1:476–485. doi: 10.1038/nprot.2006.69. [DOI] [PubMed] [Google Scholar]
- Gardner A, Reece SE, West SA. Even more extreme fertility insurance and the sex ratios of protozoan blood parasites. Journal of Theoretical Biology. 2003;223:515–521. doi: 10.1016/s0022-5193(03)00142-5. [DOI] [PubMed] [Google Scholar]
- Garnham PCC. Malaria Parasites and Other Haemosproridia. Oxford: Blackwell Science; 1966. [Google Scholar]
- Gautret P, Deharo E, Chabaud AG, Ginsburg H, Landau I. Plasmodium vinckei vinckei, Pv-lentum and P-yoelii-yoelii - chronobiology of the asexual cycle in the blood. Parasite-Journal de la Societe Francaise de Parasitologie. 1994;1:235–239. doi: 10.1051/parasite/1994013235. [DOI] [PubMed] [Google Scholar]
- Gotthard K, Nylin S. Adaptive plasticity and plasticity as an adaptation: a selective review of plasticity in animal morphology and life history. Oikos. 1995;74:3–17. [Google Scholar]
- Graves PM, Carter R, McNeill KM. Gametocyte production in cloned lines of Plasmodium falciparum. American Journal of Tropical Medicine and Hygeine. 1984;33:1045. doi: 10.4269/ajtmh.1984.33.1045. [DOI] [PubMed] [Google Scholar]
- Grech K, Watt K, Read AF. Host-parasite interactions for virulence and resistance in a malaria model system. Journal of Evolutionary Biology. 2006;19:1620–1630. doi: 10.1111/j.1420-9101.2006.01116.x. [DOI] [PubMed] [Google Scholar]
- Hamilton WD. The evolution of altruistic behaviour. American Naturalist. 1963;97:354–356. [Google Scholar]
- Hamilton WD. Extraordinary sex ratios. Science. 1967;156:477–488. doi: 10.1126/science.156.3774.477. [DOI] [PubMed] [Google Scholar]
- Hardy ICW. Sex Ratios: Concepts and Research Methods. Cambridge: Cambridge University Press; 2002. [Google Scholar]
- Hawking F, Worms ME, Gammage K. 24 and 48 hour cycles of malaria parasites in the blood: their purpose and control. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1968;62:731–760. doi: 10.1016/0035-9203(68)90001-1. [DOI] [PubMed] [Google Scholar]
- Hotta CT, Gazarini ML, Beraldo FH, Varotti FP, Lopes C, Markus RP, Pozzan T, et al. Calcium dependent modulation by melatonin of the circadian rhythm in malaria parasites. Nature Cell Biology. 2000;2:466–468. doi: 10.1038/35017112. [DOI] [PubMed] [Google Scholar]
- Janse CJ, Vanderklooster PFJ, Vanderkaay HJ, Vanderploeg M, Overdulve JP. Rapid repeated DNA-replication during microgametogenesis and DNA-synthesis in young zygotes of Plasmodium-berghei. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1986;80:154–157. doi: 10.1016/0035-9203(86)90219-1. [DOI] [PubMed] [Google Scholar]
- Killick-Kendrick R, Peters W. Rodent Malaria. London: Academic Press; 1978. [Google Scholar]
- Kissinger JC, Souza PCA, Soares CO, Paul REL, Wahl AM, Rathores D, McCutchan TF, et al. Molecular phylogenetic analysis of the avian malaiarial parasite Plasmodium (Novyella) juxtanucleare. Journal of Parasitology. 2002;88:769. doi: 10.1645/0022-3395(2002)088[0769:MPAOTA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Knowles SCL, Sheldon BC. Evolutionary biology: parasite, know thyself. Current Biology. 2008;18:R655–R657. doi: 10.1016/j.cub.2008.06.032. [DOI] [PubMed] [Google Scholar]
- Koella JC, Rieu L, Paul REL. Stage-specific manipulation of a mosquito's host-seeking behavior by the malaria parasite Plasmodium gallinaceum. Behavioral Ecology. 2002;13:816–820. [Google Scholar]
- Leete TH, Rubin H. Malaria and the cell cycle. Trends in Parasitology. 1996;12:442–444. doi: 10.1016/0169-4758(96)10068-5. [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Read AF. Genetic relationships between parasite virulence and transmission in the rodent malaria Plasmodium chabaudi. Evolution. 1999;53:689–703. doi: 10.1111/j.1558-5646.1999.tb05364.x. [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Walker PR, Rowe JA. Plasmodium chabaudi: rosetting in a rodent malaria model. Experimental Parasitology. 2002a;101:121–128. doi: 10.1016/s0014-4894(02)00103-0. (Jun-Jul, 2002) [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Gaffney DJ, Read AF. Virulence in rodent malaria: host genotype by parasite genotype interactions. Infection, Genetics and Evolution. 2002b;1:287–296. doi: 10.1016/s1567-1348(02)00039-4. [DOI] [PubMed] [Google Scholar]
- Mayxay M, Pukrittayakamee S, Newton PN, White NJ. Mixed-species malaria infections in humans. Trends in Parasitology. 2004;20:233–240. doi: 10.1016/j.pt.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Meunier L. Clonal variation of gene expression as a source of phenotypic diversity in parasitic protozoa. Trends in Parasitology. 2001;17:475–479. doi: 10.1016/s1471-4922(01)02017-7. [DOI] [PubMed] [Google Scholar]
- Micks DW, De Caires PF, Franco LB. The relationship of exflagellation in avian Plasmodia to pH and immunity in the mosquito. American Journal of Hygeine. 1948;48:182–190. doi: 10.1093/oxfordjournals.aje.a119234. [DOI] [PubMed] [Google Scholar]
- Mideo N, Day T. On the evolution of reproductive restraint in malaria. Proceedings of the Royal Society B-Biological Sciences. 2008;275:1217–1224. doi: 10.1098/rspb.2007.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mideo N, Day T, Read AF. Modelling malaria pathogenesis. Cellular Microbiology. 2008;10:1947–1955. doi: 10.1111/j.1462-5822.2008.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons B, Janse CJ, Boorsma EG, Vanderkaay HJ. Synchronized erythrocytic schizogony and gametocytogenesis of Plasmodium-berghei in vivo and in vitro. Parasitology. 1985;91:423–430. doi: 10.1017/s0031182000062673. [DOI] [PubMed] [Google Scholar]
- Naotunne TS, Karunaweera ND, Del Giudice G, Kularatne MU, Grau GE, Carter R, Mendis KN. Cytokines kill malaria parasites during infection crisis: extracellular complementary factors are essential. Journal of Experimental Medicine. 1991;173:523–529. doi: 10.1084/jem.173.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nee S, West SA, Read AF. Inbreeding and parasite sex ratios. Proceedings of the Royal Society of London Series B-Biological Sciences. 2002;269:755–760. doi: 10.1098/rspb.2001.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul REL, Brey PT. Malaria parasites and red blood cells: from anaemia to transmission. Molecules and Cells. 2003;15:139–149. (Apr 30, 2003) [PubMed] [Google Scholar]
- Paul REL, Raibaud A, Brey PT. Sex ratio adjustment in Plasmodium gallinaceum. Parassitologia. 1999;41:153–158. [PubMed] [Google Scholar]
- Paul REL, Coulson TN, Raibaud A, Brey PT. Sex determination in malaria parasites. Science. 2000;287:128–131. doi: 10.1126/science.287.5450.128. [DOI] [PubMed] [Google Scholar]
- Paul REL, Brey PT, Robert V. Plasmodium sex determination and transmission to mosquitoes. Trends in Parasitology. 2002;18:32–38. doi: 10.1016/s1471-4922(01)02122-5. [DOI] [PubMed] [Google Scholar]
- Paul REL, Ariey F, Robert V. The evolutionary ecology of Plasmodium. Ecology Letters. 2003;6:866–880. [Google Scholar]
- Pigliucci M. How organisms respond to environmental changes: From phenotypes to molecules (and vice versa) Trends in Ecology & Evolution. 1996;11:168–173. doi: 10.1016/0169-5347(96)10008-2. [DOI] [PubMed] [Google Scholar]
- Pigliucci M. Phenotypic Plasticity: Beyond Nature and Nurture. Baltimore: Johns Hopkins University Press; 2001. [Google Scholar]
- Preiser PR, Jarra W, Capiod T, Snounou G. A rhoptry-protein-associated mechanism of clonal phenotypic variation in rodent malaria. Nature. 1999;398:618–622. doi: 10.1038/19309. [DOI] [PubMed] [Google Scholar]
- Raberg L, Sim D, Read AF. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science. 2007;318:812–814. doi: 10.1126/science.1148526. [DOI] [PubMed] [Google Scholar]
- Read AF. The evolution of virulence. Trends in Microbiology. 1994;2:73–76. doi: 10.1016/0966-842x(94)90537-1. [DOI] [PubMed] [Google Scholar]
- Read AF, Narara A, Nee S, Keymer AE, Day KP. Gametocyte sex-ratios as indirect measures of outcrossing rates in malaria. Parasitology. 1992;104:387–395. doi: 10.1017/s0031182000063630. [DOI] [PubMed] [Google Scholar]
- Reece SE, Thompson J. Transformation of the rodent malaria parasite, Plasmodium chabaudi and generation of stable fluorescent lines. Malaria Journal. 2008;7:183. doi: 10.1186/1475-2875-7-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece SE, Duncan AB, West SA, Read AF. Host cell preference and variable transmission strategies in malaria parasites. Proceedings of the Royal Society B-Biological Sciences. 2005;272:511–517. doi: 10.1098/rspb.2004.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece SE, Drew DR, Gardner A. Sex ratio adjustment and kin discrimination in malaria parasites. Nature. 2008;453:609–614. doi: 10.1038/nature06954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly HB, Wang H, Steuter JA, Marx AM, Ferdig MT. Quantitative dissection of clone-specific growth rates in cultured malaria parasites. International Journal for Parasitology. 2007;37:1599. doi: 10.1016/j.ijpara.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert V, Sokhna CS, Rogier C, Ariey F, Trape JF. Sex ratio of Plasmodium falciparum gametocytes in inhabitants of Dielmo, Senegal. Parasitology. 2003;127:1–8. doi: 10.1017/s0031182003003299. [DOI] [PubMed] [Google Scholar]
- Roff DA. The Evolution of Life Histories: Theory and Analysis. New York: Chapman and Hall; 1992. [Google Scholar]
- De Roode J, Read AF, Chan BH, Mackinnon MJ. Rodent malaria parasites suffer from the presence of conspecific clones in three-clone Plasmodium chabaudi infections. Parasitology. 2003;127:411–418. doi: 10.1017/s0031182003004001. [DOI] [PubMed] [Google Scholar]
- De Roode JC, Culleton R, Cheesman SJ, Carter R, Read AF. Host heterogeneity is a determinant of competitive exclusion or coexistence in genetically diverse malaria infections. Proceedings of the Royal Society of London Series B-Biological Sciences. 2004;271:1073–1080. doi: 10.1098/rspb.2004.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roode JC, Pansini R, Cheesman SJ, Helinski MEH, Huijben S, Wargo AR, Bell AS, et al. Virulence and competitive ability in genetically diverse malaria infections. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:7624–7628. doi: 10.1073/pnas.0500078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PJ, Kim K, McKerrow JH, Leech JH. Identification of three stage specific proteinases of Plasmodium falciparum. Journal of Experimental Medicine. 1987;166:816–821. doi: 10.1084/jem.166.3.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schall JJ. Virulence of lizard malaria – the evolutionary ecology of an ancient parasite host association. Parasitology. 1990;100:S35–S52. doi: 10.1017/s0031182000073005. [DOI] [PubMed] [Google Scholar]
- Schall JJ, Vardo-Zalik AM. Identification of microsatellite markers in Plasmodium mexicanum, a lizard malaria parasite that infects nucleated erythrocytes. Molecular Ecology Notes. 2007;7:227–229. [Google Scholar]
- Schlichting CD, Pigliucci M. Phenotypic Evolution: A Reaction Norm Perspective. Sunderland, MA: Sinauer; 1998. [Google Scholar]
- Schneider P, Bousema JT, Gouagna LC, Otieno S, Van De Vegte-Bolmer M, Omar SA, Sauerwein RW. Submicroscopic Plasmodium falciparum gametocyte densities frequently result in mosquito infection. American Journal of Tropical Medicine and Hygiene. 2007;76:470–474. [PubMed] [Google Scholar]
- Schneider P, Chan HK, Reece SE, Read AF. Does the drug sensitivity of malaria parasites depend on their virulence? Malaria Journal. 2008;7:257. doi: 10.1186/1475-2875-7-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman IW, Eda S, Winograd E. Cytoadherence and sequestration in Plasmodium falciparum: defining the ties that bind. Microbes and Infection. 2003;5:897–909. doi: 10.1016/s1286-4579(03)00162-x. [DOI] [PubMed] [Google Scholar]
- Shutler D, Read AF. Local mate competition, and extraordinary and ordinary blood parasite sex ratios. Oikos. 1998;82:417–424. [Google Scholar]
- Shutler D, Bennett GF, Mullie A. Sex proportions of Haemoproteus blood parasites and local mate competition. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:6748–6752. doi: 10.1073/pnas.92.15.6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijwali PS, Kato K, Seydel KB, Gut J, Lehman J, Klemba M, Goldberg DE, et al. Plasmodium falciparum cysteine protease falcipain-1 is not essential in erythrocytic stage malaria parasites. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8721–8726. doi: 10.1073/pnas.0402738101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JA, Silamut K, Chotivanich K, Pukrittayakamee S, White NJ. Red cell selectivity in malaria: a study of multiple-infected erythrocytes. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:165–168. doi: 10.1016/s0035-9203(99)90295-x. [DOI] [PubMed] [Google Scholar]
- Sinden RE. The cell biology of sexual development in Plasmodium. Parasitology. 1983a;86:7–28. doi: 10.1017/s0031182000050824. [DOI] [PubMed] [Google Scholar]
- Sinden RE. Sexual development of malarial parasites. Advances in Parasitology. 1983b;22:153–216. doi: 10.1016/s0065-308x(08)60462-5. [DOI] [PubMed] [Google Scholar]
- Smith TG, Ranford-Cartwright L, Walliker D. Sexual differentiation and sex determination in the apicomplexa. Parasitology Today. 2002;18:315–323. doi: 10.1016/s1471-4922(02)02292-4. [DOI] [PubMed] [Google Scholar]
- Snounou G, Jarra W, Preiser P. Malaria multigene families: the price of chronicity. Trends in Parasitology. 2000;16:28–30. doi: 10.1016/s0169-4758(99)01546-x. [DOI] [PubMed] [Google Scholar]
- Stearns SC. The Evolution of Life Histories. Oxford: Oxford University Press; 1992. [Google Scholar]
- Stearns SC, Koella JC. Evolution in Health and Disease. Oxford: Oxford University Press; 2007. [Google Scholar]
- Stubbs J, Simpson KM, Triglia T, Plouffe D, Tonkin CJ, Duraisingh MT, Maier AG, et al. Molecular mechanism for switching of P. falciparum invasion pathways into human erythrocytes. Science. 2005;309:1384–1387. doi: 10.1126/science.1115257. [DOI] [PubMed] [Google Scholar]
- Taylor PD. Intra-sex and inter-sex sibling interactions as sex-ratio determinants. Nature. 1981;291:64–66. [Google Scholar]
- Taylor LH, Read AF. Why so few transmission stages? Reproductive restraint by malaria parasites. Parasitolology Today. 1997;13:135–140. doi: 10.1016/s0169-4758(97)89810-9. [DOI] [PubMed] [Google Scholar]
- Taylor-Robinson AW, Phillips RS. Predominance of infected reticulocytes in the peripheral-blood of CD4(+) T-cell-depleted mice chronically infected with Plasmodium-chabaudi chabaudi. Parasitology Research. 1994;80:614–619. doi: 10.1007/BF00933011. (Oct) [DOI] [PubMed] [Google Scholar]
- Timms R, Colegrave N, Chan BHK, Read AF. The effect of parasite dose on disease severity in the rodent malaria Plasmodium chabaudi. Parasitology. 2001;123:1–11. doi: 10.1017/s0031182001008083. [DOI] [PubMed] [Google Scholar]
- Via S, Gomulkiewicz R, Dejong G, Scheiner SM, Schlichting CD, Vantienderen PH. Adaptive phenotypic plasticity - consensus and controversy. Trends in Ecology & Evolution. 1995;10:212–217. doi: 10.1016/s0169-5347(00)89061-8. [DOI] [PubMed] [Google Scholar]
- Wargo AR, De Roode JC, Huijben S, Drew DR, Read AF. Transmission stage investment of malaria parasites in response to in-host competition. Proceedings of the Royal Society B-Biological Sciences. 2007;274:2629–2638. doi: 10.1098/rspb.2007.0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SA. Sex Allocation. Princeton: Princeton University Press; 2009. [Google Scholar]
- West SA, Smith TG, Read AF. Sex allocation and population structure in apicomplexan (protozoa) parasites. Proceedings of the Royal Society of London Series B-Biological Sciences. 2000;267:257–263. doi: 10.1098/rspb.2000.0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SA, Reece SE, Read AF. Evolution of gametocyte sex ratios in malaria and related apicomplexan (protozoan) parasites. Trends in Parasitology. 2001;17:525–531. doi: 10.1016/s1471-4922(01)02058-x. [DOI] [PubMed] [Google Scholar]
- West SA, Smith TG, Nee S, Read AF. Fertility insurance and the sex ratios of malaria and related hemospororin blood parasites. Journal of Parasitology. 2002;88:258–263. doi: 10.1645/0022-3395(2002)088[0258:FIATSR]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- West SA, Reece SE, Read AF. Toxoplasma gondii, sex and premature rejection. Trends in Parasitolology. 2003;19:155–157. doi: 10.1016/s1471-4922(03)00033-3. [DOI] [PubMed] [Google Scholar]
- West SA, Shuker DM, Sheldon BC. Sex-ratio adjustment when relatives interact: a test of constraints on adaptation. Evolution. 2005;59:1211–1228. [PubMed] [Google Scholar]
- West SA, Griffin AS, Gardner A, Diggle SP. Social evolution theory for microorganisms. Nature Reviews Microbiology. 2006;4:597–607. doi: 10.1038/nrmicro1461. [DOI] [PubMed] [Google Scholar]
- Williams GC. Adaptation and Natural Selection. Princeton: Princeton University Press; 1966. [Google Scholar]
- De Witt TJ, Sih A, Wilson DS. Costs and limits of phenotypic plasticity. Trends in Ecology & Evolution. 1998;13:77–81. doi: 10.1016/s0169-5347(97)01274-3. [DOI] [PubMed] [Google Scholar]