

Abstract
In the decade following their initial discovery, the suppressor of cytokine signaling (SOCS) proteins have been studied for their potential use as immunomodulators in disease. SOCS proteins, especially SOCS1 and SOCS3, are expressed by immune cells and cells of the central nervous system (CNS) and have the potential to impact immune processes within the CNS, including inflammatory cytokine and chemokine production, activation of microglia, macrophages and astrocytes, immune cell infiltration and autoimmunity. We describe CNS-relevant in vitro and in vivo studies that have examined the function of SOCS1 or SOCS3 under various neuroinflammatory or neuropathological conditions, including exposure of CNS cells to inflammatory cytokines or bacterial infection, demyelinating insults, stroke, spinal cord injury, multiple sclerosis and glioblastoma multiforme.
The SOCS family
Suppressor of cytokine signaling (SOCS) proteins are intracellular, cytokine-inducible proteins that inhibit cytokine signaling in numerous cell types, including cells of the immune and central nervous systems (CNS). The SOCS family is composed of eight members: cytokine inducible SRC homology 2 (SH2)-domain-containing protein (CIS) and SOCS1 to SOCS7 [1,2]. To exert their function, SOCS proteins associate with phosphorylated tyrosine residues on Janus kinases (JAKs) and/or cytokine receptor subunits through a central SH2 domain. A C-terminal SOCS box then interacts with components of the ubiquitin ligase machinery and mediates proteosomal degradation of associated proteins [3]. In addition, the N-terminus of SOCS1 and SOCS3, specifically, contains a kinase-inhibitory region (KIR) (Figure 1), which acts as a pseudosubstrate for JAKs, conferring inhibition of JAK kinase activity [1]. Through these interactions, SOCS proteins attenuate responses to cytokines and growth factors. Because studies of SOCS family members have established SOCS1 and SOCS3 as the most important in regulating innate and adaptive immune responses, they are the focus of this review. There is limited information regarding the role of other SOCS family members in CNS immunity. Therefore, they are not discussed here.
Figure 1.

Domain structure of SOCS1 and SOCS3. SOCS1 and SOCS3 proteins are comprised of an N-terminal variable region, a 12-amino acid (aa) kinase-inhibitory region (KIR), a 12-aa extended SH2 subdomain (ESS), a classical SH2 domain and a C-terminal SOCS box. Four residues of the KIR (F56, F59, D64 and Y65) are crucial for inhibiting the kinase activity of JAK2. The I68 and L75 residues of the ESS region are crucial for interaction with pY1007 of JAK2. The SOCS box associates with elongins C and B, which interact with components of the ubiquitin-ligase machinery, to target SOCS1, SOCS3 and associated proteins for degradation by the proteosome.
SOCS regulation of JAK/STAT signaling
Signal transduction through the JAK/signal transducer and activator of transcription (STAT) signaling pathway is a crucial mediator of inflammatory and immune responses in the CNS [4]. Activation of the JAK/STAT pathway is achieved by cytokines binding to their associated cell-surface receptors, leading to a series of phosphorylation events, culminating in phosphorylation of the STAT transcription factors (Figure 2). Activated STATs promote expression of immune molecules by binding to cis-elements in the promoters of target genes and activating transcription. Along with many proinflammatory targets, STATs also induce SOCS1 and SOCS3, which feed back to negatively regulate JAK/STAT signaling (Figure 2). Expression of SOCS1 and SOCS3 is regulated primarily by activation of STAT1 and STAT3, respectively, although their expression can be mediated through other signaling cascades, including the mitogen activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB) pathways. SOCS1 and SOCS3 have short half-lives (1–2 h) and their stability can be regulated by phosphorylation or association with other proteins, including the serine and threonine kinase PIM1 and ubiquitin [5–9]. Other regulatory mechanisms probably exist but have not been characterized. Due to their rapid induction and quick turnover, SOCS proteins present a robust and self-regulated mechanism to modulate cytokine signaling in various disease states of the CNS.
Figure 2.
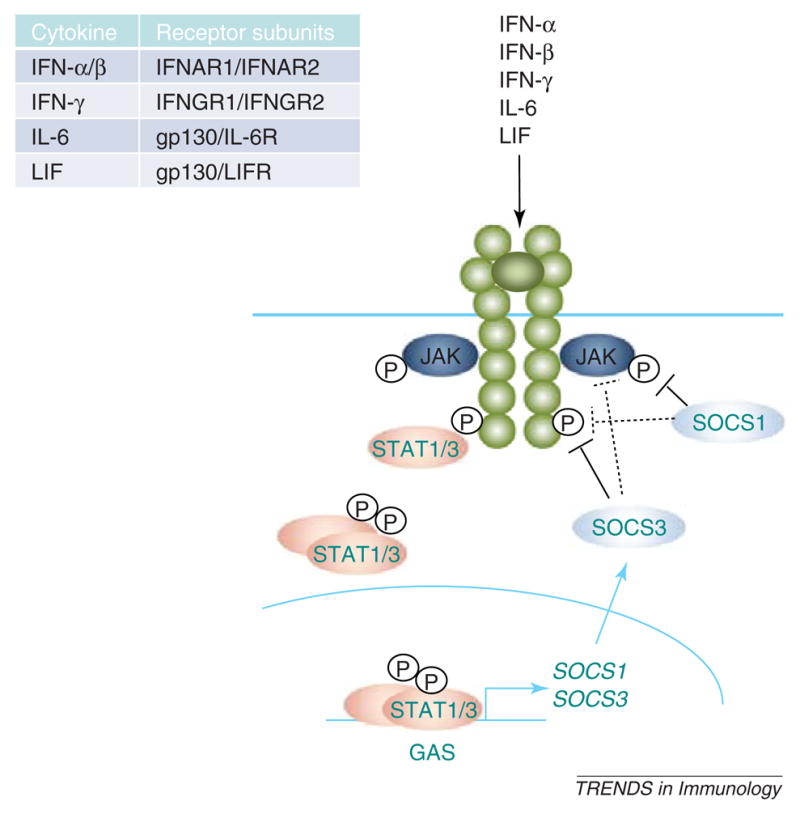
Cytokine signaling through the JAK/STAT pathway. Cytokines, including IFNs -α, -β and -γ, IL-6 and LIF, signal through receptor complexes (see inset) that activate the JAK/STAT pathway. Activation of STAT transcription factors, particularly STAT1 and STAT3, induces SOCS1 and SOCS3 gene expression. Both SOCS1 and SOCS3 inhibit JAK activity through their KIR. However, SOCS1 binds primarily to phosphorylated, receptor-associated JAK proteins through its SH2 domain, whereas the SH2 domain of SOCS3 binds to phosphorylated tyrosine residues in the cytoplasmic domain of receptors. In some cases, however, SOCS1 can bind to phosphorylated receptors and SOCS3 can interact directly with JAKs. All of these interactions terminate STAT activation and suppress downstream gene expression.
SOCS1 and SOCS3 expression is cell-type- and stimulus-specific. In CNS cells, such as astrocytes, microglia, oligodendrocytes and neurons, SOCS1 and SOCS3 expression is induced by stimuli such as IL-4, -6, -10, interferon (IFN)-β, -γ and lipopolysaccharide (LPS). SOCS proteins are also inducibly expressed in immune cells, including dendritic cells (DCs), T cells and macrophages, which are recruited to the CNS under inflammatory conditions (Tables 1 and 2). As we discuss later, the effects of SOCS1 and SOCS3 on immune responses and neurological outcomes vary greatly depending on the neuroinflammatory context. However, these outcomes are linked in that they are a result of SOCS1 or SOCS3 inhibition of the JAK/STAT pathway.
Table 1.
Cells of the CNS and their functions
| Cell type | Function |
|---|---|
| Microglia | Resident macrophages within the brain parenchyma that actively survey the extracellular environment for damage or infection. Under neuroinflammatory conditions, they have phagocytic capacity, secrete numerous cytokines and chemokines and present CNS antigens to T cells. |
| Astrocytes | Control local blood flow (oxygen and glucose) to active brain regions, regulate formation and activity of synapses, induce formation of the blood-brain barrier and secrete many pro- and anti-inflammatory cytokines under neuroinflammatory conditions. |
| Neurons | Communicate information through the generation of action potentials and release of neurotransmitters. Axons in white matter are wrapped in myelin by oligodendrocytes which is essential for proper conduction of action potentials. Neurodegenerative disease, such as MS, cause demyelination of axons, impairing conduction of action potentials. |
| Oligodendrocytes | Cells of the CNS that insulate axons by wrapping them in layers of myelin. These cells are targets in MS, wherein myelin is destroyed and oligodendrocytes are lost. |
| Macrophages | Phagocytic immune cell of myeloid lineage that can infiltrate the CNS under inflammatory conditions. They engulf pathogens and cellular debris, process and present foreign antigens to T cells and produce cytokines and chemokines to regulate immune responses. |
| Dendritic cells | Professional antigen-presenting cells (APCs) that regulate differentiation and maturation of T cells through secretion of cytokines. DCs can traffic into the CNS under neuroinflammatory conditions. |
| T cells | T-cell subsets include Th1, Th2, Th17, T regulatory cells and memory T cells. Engagement of the T-cell receptor with antigen-bound MHC on APCs activates T cells, leading to cytokine production by T cells and APCs, inducing T-cell maturation and expansion. T cells also infiltrate the CNS under inflammatory conditions, where they are implicated in CNS autoimmune diseases, such as MS. |
Table 2.
Mechanism of SOCS1 and SOCS3 expression in CNS cells
| Cell type | SOCS1 Inducer | Pathway | SOCS3 Inducer | Pathway |
|---|---|---|---|---|
| Microglia | IFN-γ | JAK/STAT | IFN-γ | JAK/STAT |
| IFN-β | JAK/STAT1 | IFN-β | JAK/STAT3 | |
| LPS | ? | LPS | IL-10-JAK/STAT3, MAPK | |
| IL-4 | JAK/STAT | IL-10 | JAK/STAT3 | |
| HIV-1 Tat | NF-κB | |||
| Thrombin | PKC-™ | |||
| Astrocytes | 15d-PGJ2 | Non-JAK/STAT | 15d-PGJ2 | Non-JAK/STAT |
| Rosiglitazone | Non-JAK/STAT | Rosiglitazone | Non-JAK/STAT | |
| IFN-γ | JAK/STAT | IFN-γ | JAK/STAT | |
| IFN-β | JAK/STAT | IFN-β | JAK/STAT | |
| OSM | JAK/STAT1 | OSM | JAK/STAT3, MAPK | |
| CNTF | ? | |||
| Neurons | IFN-γ | JAK/STAT | IGF-1 | JAK/STAT3 |
| IL-6 | JAK/STAT | |||
| OSM | JAK/STAT | |||
| LPS | ? | |||
| Oligodendrocytes | IFN-γ | JAK/STAT | LIF | JAK/STAT |
| Macrophages | IFN-γ | JAK/STAT | IFN-γ | JAK/STAT |
| IFN-β | JAK/STAT | IFN-β | JAK/STAT | |
| LPS | ? | LPS | IL-10-JAK/STAT, MAPK | |
| IL-10 | JAK/STAT3 | IL-10 | JAK/STAT3 | |
| CpG | MAPK | CpG | MAPK | |
| IL-4 | JAK/STAT6 | HIV-1 Tat | NF-κB | |
| FMLP | Non-JAK/STAT | IL-6 | JAK/STAT3 | |
| IL-8 | Non-JAK/STAT | TNF-α | MAPK (RNA stabilization) | |
| IL-21 | ? | |||
| Dendritic cells | CpG | MAPK | CpG | MAPK |
| IL-21 | ? | IL-21 | ? | |
| FMLP | Non-JAK/STAT | |||
| IL-8 | Non-JAK/STAT | |||
| T cells | IL-6 | JAK/STAT | IL-6 | JAK/STAT |
| IL-4 | JAK/STAT1 | IL-4 | JAK/STAT1 | |
| IL-12 | JAK/STAT | IL-12 | JAK/STAT1 | |
| IFN-γ | JAK/STAT | IFN-γ | JAK/STAT1 | |
| IFN-α | ? | IFN-α | ? | |
| Flagellin | ? |
SOCS1
SOCS1 functions most classically to inhibit IFN signaling by interacting with the IFN-α receptor 1 (IFNAR1) and IFN-γ receptor (IFNGR) subunits, limiting IFN-activation of STATs (namely STAT1, STAT2 and STAT3) [10,11]. However, SOCS1 has more recently been shown to disrupt a broader range of signaling pathways by facilitating degradation of p65 [12], a crucial component of the NF-κB pathway, and ASK1, a kinase upstream of the JNK and p38 pathways [13]. SOCS1-deficient mice (SOCS1−/−) develop multi-organ failure and death within 3 weeks of birth, owing to a hyper-responsiveness to IFN-γ [14–16]. During this time, however, heightened IFN responses also render SOCS1−/− mice resistant to viral and parasitic infections. Accordingly, overexpression of SOCS1 results in reduced responsiveness to IFN-α, -β and -γ in many cell types [10,17–21]. Additionally, macrophages, DCs and fibroblasts from SOCS1−/− mice are hypersensitive to LPS and other Toll-like receptor ligands, as measured by increased production of proinflammatory cytokines, including TNF-α, IL-6, IL-12 and IFN-γ (Table 3) [22,23]. Clearly, SOCS1 is a powerful attenuator of IFN- and Toll-like receptor-mediated responses in immune cells.
Table 3.
Expression and function of molecules involved in regulation of the immune response
| Molecule | Secreted by | Function |
|---|---|---|
| IFN-α/β | DCs, MΦs, NK cells, fibroblasts, T cells, B cells, MG, astrocytes, neurons | Produced in response to viral infection or microbial pathogens, type I IFNs initiate antiviral responses, including inhibition of cell growth, regulation of proinflammatory cytokine production, upregulation of MHC class I expression and activation of NK cells. |
| IFN-γ | NK cells, cytotoxic T cells, Th1 cells | Produced in response to mitogenic or antigenic stimuli, IFN-γ promotes Th1-mediated immune responses, upregulates MHC class II expression, increases phagocytic activity of macrophages and induces cytokine and chemokine expression. |
| IL-1 | MG, MΦs, DCs | Produced in response to inflammatory stimuli, such as proinflammatory cytokines, β-amyloid, HIV-1, hypoxia and bacterial infection. IL-1 acts on astrocytes and microglia to induce expression of itself, IL-6, TNF-α, NO and COX-2 and is thought to promote astrogliosis, microglial activation, brain inflammation and neuronal injury. |
| IL-2 | T cells | Produced and secreted by T cells on antigen recognition by a specific T-cell receptor, leading to clonal expansion of T-cells. IL-2 also promotes growth and differentiation of B cells, NK cells and MΦs, partially through induction of IFN-γ and IL-4. |
| IL-4 | Th2 cells, NK cells | Promotes Th2 differentiation and is largely anti-inflammatory. |
| IL-6 | MΦs, astrocytes, MG, neurons | Induced in response to inflammatory cytokines and tissue injury. Activates NK and cytotoxic T cells and, in conjunction with TGF-β, promotes development of Th17 cells. IL-6 promotes astrogliosis and can be neurotoxic or neuroprotective, depending on the disease context. |
| IL-8 | MΦs, T cells, astrocytes | Chemokine upregulated by IL-1, TNF-α and IL-17. IL-8 is a potent chemoattractant for neutrophils and, through distinct mechanisms, promotes angiogenesis in brain tumors. |
| IL-10 | Th2 cells, CD8+, MΦs, B cells, DCs, astrocytes, MG | Classical Th2 cytokine that inhibits IFN-γ production by Th1 cells and is considered primarily anti- inflammatory. IL-10 inhibits expression of proinflammatory cytokines and chemokines, MHC class II and B7. |
| IL-12 | DCs, T cells, monocytes, MΦs, neutrophils | Promotes proliferation and cytotoxicity of NK cells and T cells (especially γ™ T cells) and has antitumor effects in glioma. Promotes development of IFN-γ-producing Th1 cells. |
| IL-17 | Th17 cells, astrocytes | Upregulates cytokines and chemokines, such as IL-6, IL-8 and MCP-1, activates T cells and promotes trafficking of macrophages and neutrophils. Crucial in autoimmunity. |
| IL-23 | DCs, MΦs, MG | Stimulates development of Th17 cells, which secrete inflammatory cytokines, such as IL-17, IL-6 and TNF-α, and is believed to be crucial in autoimmunity. IL-23 also recruits neutrophils and macrophages and protects the host against pathogen infection. |
| IL-27 | MΦs, DCs, astrocytes | Synergizes with IL-12 to induce IFN-γ production and promote Th1 differentiation. Suppresses Th17 effector function. |
| OSM | MΦs, T cells, DCs, monocytes | Proinflammatory cytokine that induces expression of IL-6, TNF-α and NOS2. |
| LIF | Astrocytes, MΦs oligodendrocytes, neurons | Promotes astrogliogenesis, oligodendrocyte survival and regulates neurite outgrowth. |
| CNTF | Astrocytes, neurons | Promotes oligodendrocyte and neuron survival. |
| IGF-1 | Neurons, astrocytes | Increases proliferation of neural precursor cells during development, promotes neurite outgrowth and stimulates myelination of axons by oligodendrocytes. |
| TGF-β | Astrocytes, regulatory T cells | Inhibits IFN-γ and TNF-α production and decreases expression of MHC class II and costimulatory molecules. TGFβ, in combination with IL-6, induces Th17 differentiation, whereas co-expression with IL-2 stimulates development of regulatory T cells. |
| TNF-α | MG, MΦs | Increases cytotoxic T-cell development, NK-cell toxicity and MHC class II expression. TNF-α also induces expression of proinflammatory cytokines, such as IL-1, IL-6, IL-8 and IFN-γ, and induces oligodendrocyte cell death. |
Abrreviations: MG, microglia; MΦs, macrophages; DC, dendritic cells
In vitro effects of SOCS1 in cells of the CNS
Several in vitro studies have highlighted important functions of SOCS1 in cells of the CNS. For example, negative feedback by SOCS1 in astrocytes might limit chemokine-induced migration of immune cells within the brain. IFN-β treatment of astrocyte cultures induces robust expression of chemokines, such as CCL2 (monocyte chemotactic protein-1; MCP-1), CCL3 (macrophage inflammatory protein 1 α; MIP-1α), CCL4 (macrophage inflammatory protein 1 β; MIP-1β), CCL5 (regulated upon activation, normal T cell expressed and secreted; RANTES) and CXCL10 (interferon-inducible protein 10; IP-10) [24]. However, this expression is self-regulated by concurrent IFN-β-induced expression of SOCS1, as shown by increased production of these chemokines when SOCS1 is experimentally depleted using small interfering RNA (siRNA). Functionally, this increase in chemokine expression correlates with enhanced migration of macrophages and CD4+ T cells in an in vitro assay, indicating that SOCS1 might limit inflammatory cell migration within the CNS.
SOCS1 also inhibits expression of crucial immune molecules on the cell surface, including MHC class I, class II and CD40. MHC class I mediates the presentation of intracellular pathogens to the immune system and its expression is enhanced following IFN-γ stimulation. Interestingly, dorsal root ganglia (DRG) neurons of the adult mouse, which unlike most cell types do not express MHC class I, express high levels of SOCS1 [25]. However, in SOCS1−/− DRG neurons, stimulation with IFN-γ induces strong MHC class I expression [26]. By contrast, myelinating cells of the nervous system do express MHC class I in response to IFN-γ. However, SOCS1 overexpression in both Schwann cells and oligodendrocytes inhibits this expression [26,27]. Therefore, SOCS1 inhibits IFN-γ-induced MHC class I expression in CNS cells and its expression represents a mechanism whereby intracellular pathogens could evade detection by the immune system. SOCS1 also inhibits IFN-γ-induced expression of MHC class II in macrophages and microglia by inhibiting STAT1-mediated expression of the transcription factor CIITA [18]. In addition, by blocking STAT1 activation, SOCS1 inhibits IFN-γ-induced CD40 expression in macrophages [19] and IFN-β-induced CD40 expression in macrophages and microglia [28]. Thus, SOCS1 inhibits surface expression of crucial immune molecules on CNS cells, promoting a general dampening of T-cell activation and immune responses in the brain.
SOCS1 in pathogenic diseases of the CNS
IFNs serve as the first line of defense against pathogen invasion [29]. Recently, it has been shown that several CNS pathogens can induce SOCS proteins as a method of evading IFN-mediated innate immune responses. Toxoplasma gondii, an intracellular parasite that causes encephalitis, induces SOCS1 directly in macrophages [30]. This expression correlates with a reduction in IFN-γ-stimulated target genes, which is rescued by SOCS1 deletion. However, CNS pathogen-induced SOCS proteins are also beneficial for the host. Borrelia burgdorferi, the pathogen that causes Lyme disease, inflicts widespread damage to the joints, heart and CNS following massive induction of inflammatory cytokines in these tissues [31]. Concurrently, host macrophages are induced to express both SOCS1 and SOCS3 proteins directly in response to this pathogen, resulting in diminished production of inflammatory cytokines, including IL-1β, IL-6 and TNF-α 31. Therefore, pathogen-induced SOCS1 can strongly influence inflammatory responses within the CNS during infection.
SOCS1 in demyelinating diseases
Multiple sclerosis (MS) is an autoimmune disease of the CNS that is characterized by regions of inflammation and demyelination of axons, in addition to neuron and oligodendrocyte cell loss [32]. Several experimental observations suggest that IFN-γ might have an important role in the pathogenesis of this disease. Increased levels of IFN-γ are observed in MS lesions and cerebrospinal fluid (CSF) levels of IFN-γ correlate with disease severity [33]. In vitro, exposure of oligodendrocytes to IFN-γ results in cell death [33,34]. During mouse development, overexpression of IFN-γ in the CNS results in motor tremors, hypomyelination and oligodendrocyte loss, effects similar to those seen in MS [27,35,36]. Not surprisingly, SOCS1 dampens the effects of IFN-γ in demyelinating disease in mice. For instance, CNS-IFN-γ overexpressors are protected against demyelination by concurrent overexpression of SOCS1 in oligodendrocytes [27]. In the context of experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, the early CNS presence of IFN-γ provides protection to oligodendrocytes but IFN-γ expression later in disease is harmful [37]. SOCS1 overexpression in oligodendrocytes suppresses the early, protective effects of IFN-γ on oligodendrocytes, but also blocks its later, deleterious actions in EAE [38]. Finally, in several studies, administration of the SOCS1 mimetic, tyrosine kinase-inhibitor peptide (Tkip), prevents EAE. Tkip binds to the JAK2 autophosphorylation site, preventing JAK2-mediated phosphorylation of STAT1 and STAT3, and functionally blocking STAT1-mediated IFN-γ and TNF-α signaling, in addition to STAT3-mediated IL-6 signaling [39]. Administration of this peptide before EAE induction in NZW mice prevents acute EAE [40]. In SJL/J mice, administration of Tkip blocks the acute and relapse phases of EAE, even when given after the establishment of disease [40]. This protection correlates with decreased expression of IL-2, IL-5, TNF-α and IFN-γ in the CNS of Tkip-treated mice. These findings collectively indicate that SOCS1 can attenuate neuroinflammatory responses and might therefore have therapeutic value in MS.
SOCS1 in CNS malignancy
A therapeutic role for SOCS1 might also be found in CNS malignancy. In glioblastoma multiforme (GBM), a highly aggressive and fatal brain tumor, SOCS1 might function as a tumor suppressor. The SOCS1 promoter is hypermethylated in ~25% of brain tumors and in many glioma cell lines [41,42], which corresponds to a fivefold reduction in SOCS1 mRNA expression. However, reintroduction of SOCS1 into glioma cells sensitizes them to radiation-induced destruction [41].
SOCS1 might also be protective in the context of CNS metastasis of melanoma. Injection of melanoma cells into the carotid artery results in metastasis to the brain in 100% of cases, whereas injection of melanoma cells overexpressing SOCS1 leads to metastasis in only 39% of cases [43]. SOCS1 overexpressing melanoma cells have decreased activation of STAT3 and lower expression of matrix metalloproteinase 2 (MMP-2) and vascular endothelial growth factor (VEGF), molecules important for invasion and angiogenesis, respectively [43]. Therefore, in the context of CNS malignancy, SOCS1 expression might have therapeutic value in CNS malignancy by sensitizing glioma cells to radiation and inhibiting expression of tumor-promoting genes.
SOCS1 summary
SOCS1 is a powerful attenuator of JAK/STAT signaling, most notably when initiated by IFNs. As has been reviewed, however, this discrete action of SOCS1 has diverse effects throughout the CNS depending on the inflammatory microenvironment or disease context. SOCS1 can limit inflammation by dampening expression of cytokines and chemokines, suppress surface expression of molecules that mediate immune responses, accommodate pathogen infiltration or protect against demyelination and CNS malignancy, depending on the context of its expression. SOCS1 has a crucial role in modulating cytokine responses and might hold promise as a therapeutic modulator in CNS disease states.
SOCS3
The predominant function of SOCS3 is inhibition of signaling by the IL-6 family of cytokines (Figure 2) [2]. SOCS3 accomplishes this by interacting with the common receptor subunit of this family, gp130, through its SH2 domain, inhibiting receptor-associated JAK activity through its KIR and targeting JAKs for degradation via the SOCS box [44–46]. Together, these actions prevent JAK-mediated activation of STAT3. However, SOCS3 exerts a much broader effect on immune responses by inhibiting signaling of additional immune molecules, such as LPS, the type I and type II IFNs, IL-2 and IL-12 [24,47–49]. Furthermore, SOCS3 inhibits the NF-κB pathway [50], antagonizes cAMP-mediated signaling [51] and enhances signaling through the MAPK pathway [52]. Given this broad range of effects, it is not surprising that SOCS3-deficient mice are embryonic lethal. Embryos exhibit dysregulated signaling by leukemia inhibitory factor (LIF), an IL-6 family member, and demonstrate extensive erythrocytosis [53].
In vitro effects of SOCS3 in cells of the CNS
As would be expected from the broad impact of SOCS3 on cytokine signaling, SOCS3 can exert several effects on CNS and immune cells. IFN-β induces the expression of SOCS3 in astrocytes in a STAT3 activation-dependent manner [24]. Disruption of SOCS3 expression enhances the production of chemokines by IFN-β, promoting the migration of microglia and T cells. Thus, astrocyte production of chemokines is inhibited by SOCS3, decreasing immune-cell migration within the CNS. Studies involving the manipulation of astrocyte-derived SOCS3 expression must be performed to determine if SOCS3 has a functional role in immune-cell trafficking into the brain in vivo.
In neurons, however, the effects of SOCS3 expression are very different. For example, in human neuroblastoma cells, overexpression of SOCS3 reverses the STAT3-mediated protective effects of insulin-like growth factor (IGF-1) against TNF-α-induced cell death [54]. In cultures of DRG neurons, lentiviral expression of SOCS3 significantly decreases STAT3-mediated neurite outgrowth, whereas lentiviral expression of a dominant-negative SOCS3 construct increases the median neurite length [55]. Similarly, neuronal SOCS3 expression following sciatic nerve transection in rats is thought to hamper the beneficial effects of STAT3 activation in neurons. Thus, activation of STAT3 by growth factors, such as IGF-1, or following nerve injury might support neuron viability, which is opposed by the expression of SOCS3.
In macrophages and microglia, SOCS3 meditates the anti-inflammatory effects of IL-10. LPS induces CD40 expression, which is inhibited by co-incubation with IL-10 [47]. LPS and IL-10 synergistically induce SOCS3 and overexpression of SOCS3 inhibits LPS-induced CD40 expression. These data suggest that one mechanism by which IL-10 exerts its anti-inflammatory properties in macrophages and microglia is through the induction of SOCS3.
SOCS3 in pathogenic diseases of the CNS
In addition to SOCS1, CNS pathogens also induce SOCS3 in an attempt to evade immune defenses. Persistent infection of macrophages with Listeria monocytogenes, an intracellular bacterium that causes meningitis, results in robust expression of SOCS3 [56]. L. monocytogenes induction of SOCS3 is both direct and indirect, partially relying on an unidentified pathogen-secreted protein for maximal induction. This expression correlates with decreased STAT-1 tyrosine phosphorylation, STAT-1 dimerization and STAT-1-mediated transcriptional activity in response to IFN-γ stimulation, impairing the antiviral host response [56].
SOCS3 in traumatic brain and spinal cord insults
Acute spinal cord injury (SCI) causes death of neurons, oligodendrocytes and astrocytes and the interruption of ascending and descending axonal tracts. An inflammatory component develops within the spinal cord secondary to the initial trauma, consisting of neutrophil, microglia, macrophage and T-cell infiltration, in addition to the production of proinflammatory molecules, which contribute to further tissue damage [57]. STAT3 is activated in astrocytes, neurons and other cell types during the first several days after SCI [58,59]. STAT3 activation is crucial for astrocyte migration and glial-scar formation, which limits infiltration of inflammatory cells and subsequent neuron and oligodendrocyte death. Mice with conditionally deleted SOCS3 expression in nestin-expressing cells have increased STAT3 activation, thereby limiting infiltration of inflammatory cells and subsequent neuron and oligodendrocyte death [58]. This leads to improved functional recovery after SCI. By contrast, CNS deletion of STAT3 in this SCI model delays glial-scar formation, enhances infiltration of inflammatory cells, increases demyelination and impairs functional recovery. Similarly, compared with wild-type mice, those lacking STAT3 in astrocytes exhibit decreased SCI-induced expression of glial fibrillary acidic protein (GFAP) and vimentin, two important markers of astrogliosis [59]. Glial-scar formation and organization is also severely disrupted in mice with STAT3-deficient astrocytes. Phenotypically, these mice have larger lesions, increased numbers of inflammatory cells in the lesion and surrounding brain parenchyma and impaired motor function compared with WT mice. Therefore, SOCS3 expression following SCI might be detrimental because STAT3 activation is crucial for limiting the death of oligodendrocytes and neurons and improving functional recovery.
Alterations in SOCS3 expression have also been noted in other models of neuronal injury. Expression of SOCS3 is increased in rat cerebral cortex following a cortical impact injury and in the hippocampus following lithium-pilocarpine-induced seizure [60,61]. Unfortunately, the functional importance of SOCS3 expression in these models was not studied. In addition, SOCS3 expression is upregulated after middle cerebral-artery occlusion (MCAO) in the rat [62,63]. Infusion of antisense SOCS3 into the ventricle before MCAO increases lesion volumes and worsens neurological outcomes compared with control-infused mice [62], suggesting that upregulation of SOCS3 in this model of stroke is neuroprotective. Clearly, SOCS3 regulation of neuronal recovery is complex and differs depending on the type of neurological insult.
SOCS3 in demyelinating diseases
The IL-6 family member, LIF, is an endogenous survival factor for oligodendrocytes [64]. Therefore, its function has been evaluated in multiple models of demyelination [65,66]. In an EAE model of demyelination, LIF administration reduces oligodendrocyte apoptosis and disease severity. Similarly, in a cuprizone model of demyelination, LIF expression is induced in the corpus callossum region of the brain and cuprizone induces greater oligodendrocyte loss in LIF−/− mice compared with WT mice. [67]. However, LIF, in turn, induces SOCS3 expression in oligodendrocytes, which dampens its protective effects. Mice lacking SOCS3 in oligodendrocytes display less oligodendrocyte loss after cuprizone exposure compared with WT mice [67]. Therefore, SOCS3 expression in oligodendrocytes in the cuprizone model limits the protective effect of LIF on oligodendrocytes and inhibition of SOCS3 in the context of demyelination might enable greater LIF-mediated protection. Ciliary neurotrophic factor (CNTF), IGF-1 and neurotrophin (NT)-3 are also trophic factors for oligodendrocytes, and SOCS3, as an inhibitor of signaling by these cytokines, might also limit their protective effects in the context of demyelination.
However, the effects of SOCS3 in demyelinating disease are not straightforward because SOCS3 is also protective. Injection of SOCS3-expressing DCs at EAE induction or at disease onset reduces the clinical severity of EAE compared with injection with control DCs [68]. The splenocytes from mice injected with SOCS3-overexpressing DCs express reduced amounts of IFN-γ and IL-17 but more IL-4 compared with control DC-injected mice. Thus, SOCS3 expression in DCs has the capacity to limit T-cell differentiation into Th1 and Th17 cells, promoting Th2 differentiation and providing protection in EAE. Also in the context of EAE, loss of STAT3 in CD4+ T cells promotes resistance to CNS inflammation [69]. STAT3 is required for IL-17 production by Th17 cells and for T-cell trafficking into the CNS. Because SOCS3 functions as an attenuator of STAT3 activation, SOCS3 expression in T cells will probably result in the same phenotypic outcome as deletion of STAT3 in T cells.
SOCS3 in human disease
Multiple sclerosis
Monocytes, CD4+ and CD8+ T cells from patients with a relapsing-remitting (RR) form of MS express less SOCS3 during relapse than do cells from MS patients in remission [70]. This correlates, during relapse, with increased levels of activated STAT3, suggesting an association among decreased SOCS3 expression, increased STAT3 activation and MS relapse [70,71]. In addition to activated STAT3, the hormone leptin has been implicated in MS relapses [72]. Leptin is increased in MS lesions and CSF of MS patients [73,74] and autoreactive T cells isolated from MS patients upregulate leptin and the leptin receptor on activation [74]. Leptin also induces TNF-α, IL-6 and IL-10 expression in monocytes from relapsing patients but not in cells from patients with stable disease [72]. Because SOCS3 inhibits leptin signaling robustly [75], lower expression of SOCS3 during relapses might account for enhanced leptin-mediated cytokine expression. Indeed, basal and leptin-induced STAT3 activation is much higher in monocytes from relapsing MS patients than those in remission [70]. As such, SOCS3 might be an important regulator of STAT3- and leptin-mediated inflammatory responses in MS.
Statins, best known as reducers of cholesterol synthesis, have been studied, more recently, for their immunomodulatory and anti-inflammatory effects [76]. Interestingly, simvastatin induces SOCS3 expression in monocytes from RR MS patients, which is associated with decreased activation of STAT1 and STAT3, and diminished production of IL-6 and IL-23, cytokines which drive Th17-cell development [77]. This decrease in IL-6 and IL-23 production leads to suppression of the IL-17 transcription factor RAR-related orphan receptor C (RORC) and decreased IL-17 production by CD4+ T cells in culture. SOCS3 might partially mediate this effect by suppressing STAT3 activation and blocking IL-6 and IL-23 induction [78]. Thus, simvastatin-induced SOCS3 expression in monocytes blocks the development of Th17 cells and might be protective in autoimmune diseases, such as MS. Currently, statins are being tested in clinical trials in MS patients [79].
Glioblastoma
A hallmark of GBMs and other tumors is the presence of activated STAT3, as indicated by tyrosine and serine phosphorylation of STAT3 [80]. Because SOCS3 is a negative regulator of STAT3 activation, it was assumed initially that SOCS3 might function as a tumor suppressor and its expression might be repressed in GBM tissues. The tumor-suppressing function of SOCS3 was suggested by reports describing the hypermethylation of the SOCS3 promoter, subsequent loss of expression and worse patient outcomes in GBM [42]. This defective expression of SOCS3, with the loss of feedback inhibition of STAT3 activation, might therefore contribute to tumor progression. However, overexpression of SOCS3 has also been observed in GBM tissues [41,80]. This aberrant expression of SOCS3 correlates with enhanced GBM-cell growth and survival, in addition to radioresistance [41]. Because SOCS3 is a transcriptional target of activated STAT3, the parallel, elevated expression of both activated STAT3 and SOCS3 in GBM is plausible and might contribute to the anti-apoptotic and radio-resistant phenotype of GBMs. More comprehensive studies are needed to definitively determine the status and function of SOCS3 in GBM.
Use of a small-molecule inhibitor of STAT3 (WP1066) reverses immunosuppression of macrophages and microglia isolated from peripheral blood or glioma tissue from GBM patients [81]. Specifically, expression of the co-stimulatory molecules CD80 and CD86 is increased, in addition to phosphorylation of Syk and ZAP-70, which are important molecules for monocyte and T-cell activation, respectively. WP1066 treatment also enhances expression of immunostimulatory molecules, such as IL-2, IL-4 and IL-12, and WP1066-treated peripheral blood mononuclear cells (PBMCs) isolated from GBM patients exhibit enhanced T-cell proliferation responses. Although the function of SOCS3 was not examined directly, these results implicate SOCS3 as a potential immunomodulatory molecule that can promote immune responses in GBM patients whose CNS immune responses are suppressed.
SOCS3 summary
SOCS3 therefore regulates numerous disease states through inhibition of the JAK/STAT pathway. However, because this pathway is a key mediator of signaling in response to a broad range of CNS ligands, generalized SOCS3 expression or repression can lead to competing effects within a single disease state. For instance, in the context of SCI, SOCS3 might limit inflammation by suppressing cytokine and chemokine production, but also might inhibit recovery by decreasing neurite outgrowth and blocking glial-scar formation. In the context of MS, SOCS3 inhibits protective LIF signaling in oligodendrocytes, but also inhibits detrimental leptin signaling. Likewise, in CNS malignancy, SOCS3 expression has been reported to have both protective and deleterious effects. Therefore, although strong evidence exists to support SOCS3 as a crucial regulator of many disease processes, further studies are needed to elucidate its overall function within each disease state.
Conclusions and perspectives
To date, there are a limited number of studies that have examined the role of SOCS1 or SOCS3 in the regulation of human neuroinflammatory diseases (Table 4). However, these studies, in conjunction with in vitro data, highlight the potential clinical importance of SOCS1 and SOCS3 in CNS diseases. SOCS1, owing to its potent regulation of IFN responses, is likely to be crucial in governing CNS responses to infection. To our knowledge, studies involving SOCS1 regulation of CNS responses to viral or bacterial infection in vivo have not been conducted. This is an exciting area of future research because the ability to suppress viral replication and bacterial infection in the CNS is of great importance. As an inhibitor of IFN-γ signaling, SOCS1 might also confer some protection in autoimmune diseases, such as MS, by blocking expression of MHC class II and CD40 on immune cells and by also inhibiting production of proinflammatory cytokines and chemokines. However, blocking responses to IFN-γ, a Th1 cytokine, might promote the development of Th2 and/or Th17 T cells, which might, conversely, have negative implications in the context of MS. In addition to better defining the role of SOCS1 in MS, the functions of SOCS1 in SCI and stroke need to be explored.
Table 4.
Summary of SOCS1 and SOCS3 function in CNS insults
| CNS disease | SOCS1 | SOCS3 |
|---|---|---|
| EAE | Oligodendrocytes – detrimental early and protective late SOCS1 mimetic is protective | Oligodendrocytes – detrimental DCs – beneficial by limiting Th17 development |
| GBM | Beneficial by sensitizing glioma cells to radiation | Controversial – lack of SOCS3 expression might contribute to tumor progression. Aberrant SOCS3 expression promotes radioresistance |
| SCI | Unknown | Astrocytes – detrimental by limiting STAT3 activation |
| MCAO | Unknown | Neuroprotective |
| MS | Unknown | Protective - decreased expression associated with relapses |
Studies examining SOCS3 suggest perhaps an even broader potential for clinical application in the CNS. However, in light of conflicting functional effects of SOCS3, much work is needed to clarify SOCS3 function in different neurological insults. In the context of SCI, SOCS3 inhibits growth factor signaling, promotes inflammatory cell infiltration and appears refractory to repair. By contrast, SOCS3 is protective in a model of stroke. Thus, the function of SOCS3 varies depending on the neurological insult. SOCS3, as a negative regulator of IL-6 cytokines, might be detrimental in the context of MS because signaling by LIF and CNTF is protective for oligodendrocytes. However, SOCS3 might exert protective effects by blocking development of Th17 cells. Therefore, more studies are needed to determine the function of SOCS3 in MS. Finally, the function of SOCS proteins in GBM remains to be clarified. Conflicting reports have described SOCS3 loss or overexpression. Large-scale, systematic studies are necessary to categorize the status of SOCS3 in GBM subtypes and elucidate the function of SOCS3 in GBM progression.
Precise regulation of STAT activation is crucial for mediating proper growth signals and immune responses within the CNS. As regulators of many cytokines that signal through the JAK/STAT pathway, SOCS proteins have powerful roles in both health and disease and have great potential as therapeutic targets in many CNS diseases. It is important to note, however, that although the prevailing and most well-studied function of SOCS1 and SOCS3 proteins is to inhibit JAK/STAT signaling, additional signaling pathways, such as the MAPK and NF-κB pathways, are also modulated by these proteins. Therefore, future studies will need to examine the function of SOCS1 and SOCS3 in regulating these pathways to develop a better understanding of these proteins in CNS diseases.
Acknowledgments
We thank members of the Benveniste laboratory for helpful discussions on SOCS proteins. This work was funded in part by grants from the National Institutes of Health (NS-57563, NS-50665) and the National Multiple Sclerosis Society (RG 3892-A-12). B.J.B. is supported by NIH T32-NS-48039 and L.N.A. is supported by the UAB Medical Scientist Training Program, by NIH T32-AI-07493 and by NIH F30-NS-65600.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Croker BA, et al. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 3.Babon JJ, et al. The SOCS box domain of SOCS3: structure and interaction with the elonginBC-cullin5 ubiquitin ligase. J Mol Biol. 2008;381:928–940. doi: 10.1016/j.jmb.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell IL. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res Brain Res Rev. 2005;48:166–177. doi: 10.1016/j.brainresrev.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Haan S, et al. Tyrosine phosphorylation disrupts elongin interaction and accelerates SOCS3 degradation. J Biol Chem. 2003;278:31972–31979. doi: 10.1074/jbc.M303170200. [DOI] [PubMed] [Google Scholar]
- 6.Siewert E, et al. Different protein turnover of interleukin-6-type cytokine signalling components. Eur J Biochem. 1999;265:251–257. doi: 10.1046/j.1432-1327.1999.00719.x. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki A, et al. The N-terminal truncated isoform of SOCS3 translated from an alternative initiation AUG codon under stress conditions is stable due to the lack of a major ubiquitination site, Lys-6. J Biol Chem. 2003;278:2432–2436. doi: 10.1074/jbc.C200608200. [DOI] [PubMed] [Google Scholar]
- 8.Chen XP, et al. Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc Natl Acad Sci U S A. 2002;99:2175–2180. doi: 10.1073/pnas.042035699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peltola KJ, et al. Pim-1 kinase inhibits STAT5-dependent transcription via its interactions with SOCS1 and SOCS3. Blood. 2004;103:3744–3750. doi: 10.1182/blood-2003-09-3126. [DOI] [PubMed] [Google Scholar]
- 10.Fenner JE, et al. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol. 2006;7:33–39. doi: 10.1038/ni1287. [DOI] [PubMed] [Google Scholar]
- 11.Qing Y, et al. Role of tyrosine 441 of interferon-gamma receptor subunit 1 in SOCS-1-mediated attenuation of STAT1 activation. J Biol Chem. 2005;280:1849–1853. doi: 10.1074/jbc.M409863200. [DOI] [PubMed] [Google Scholar]
- 12.Ryo A, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 13.He Y, et al. SOCS1 inhibits tumor necrosis factor-induced activation of ASK1-JNK inflammatory signaling by mediating ASK1 degradation. J Biol Chem. 2006;281:5559–5566. doi: 10.1074/jbc.M512338200. [DOI] [PubMed] [Google Scholar]
- 14.Alexander WS, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 15.Starr R, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A. 1998;95:14395–14399. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marine JC, et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 17.Federici M, et al. Impaired IFN-gamma-dependent inflammatory responses in human keratinocytes overexpressing the suppressor of cytokine signaling 1. J Immunol. 2002;169:434–442. doi: 10.4049/jimmunol.169.1.434. [DOI] [PubMed] [Google Scholar]
- 18.O’Keefe GM, et al. IFN-gamma regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J Immunol. 2001;166:2260–2269. doi: 10.4049/jimmunol.166.4.2260. [DOI] [PubMed] [Google Scholar]
- 19.Wesemann DR, et al. Suppressor of cytokine signaling 1 inhibits cytokine induction of CD40 expression in macrophages. J Immunol. 2002;169:2354–2360. doi: 10.4049/jimmunol.169.5.2354. [DOI] [PubMed] [Google Scholar]
- 20.Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem. 1998;273:35056–35062. doi: 10.1074/jbc.273.52.35056. [DOI] [PubMed] [Google Scholar]
- 21.Zimmerer JM, et al. IFN-alpha-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. J Immunol. 2007;178:4832–4845. doi: 10.4049/jimmunol.178.8.4832. [DOI] [PubMed] [Google Scholar]
- 22.Hanada T, et al. Induction of hyper Th1 cell-type immune responses by dendritic cells lacking the suppressor of cytokine signaling-1 gene. J Immunol. 2005;174:4325–4332. doi: 10.4049/jimmunol.174.7.4325. [DOI] [PubMed] [Google Scholar]
- 23.Gingras S, et al. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J Biol Chem. 2004;279:54702–54707. doi: 10.1074/jbc.M411043200. [DOI] [PubMed] [Google Scholar]
- 24.Qin H, et al. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176. doi: 10.4049/jimmunol.181.5.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polizzotto MN, et al. Expression of “suppressor of cytokine signalling” (SOCS) genes in the developing and adult mouse nervous system. J Comp Neurol. 2000;423:348–358. [PubMed] [Google Scholar]
- 26.Turnley AM, et al. Failure of sensory neurons to express class I MHC is due to differential SOCS1 expression. J Neuroimmunol. 2002;123:35–40. doi: 10.1016/s0165-5728(01)00480-5. [DOI] [PubMed] [Google Scholar]
- 27.Balabanov R, et al. Suppressor of cytokine signaling 1 expression protects oligodendrocytes from the deleterious effects of interferon-gamma. J Neurosci. 2006;26:5143–5152. doi: 10.1523/JNEUROSCI.0737-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin H, et al. IFN-beta-induced SOCS-1 negatively regulates CD40 gene expression in macrophages and microglia. FASEB J. 2006;20:985–987. doi: 10.1096/fj.05-5493fje. [DOI] [PubMed] [Google Scholar]
- 29.Borden EC, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmermann S, et al. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-gamma signaling. J Immunol. 2006;176:1840–1847. doi: 10.4049/jimmunol.176.3.1840. [DOI] [PubMed] [Google Scholar]
- 31.Dennis VA, et al. Interleukin-10 anti-inflammatory response to Borrelia burgdorferi, the agent of Lyme disease: a possible role for suppressors of cytokine signaling 1 and 3. Infect Immun. 2006;74:5780–5789. doi: 10.1128/IAI.00678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 33.Vartanian T, et al. Interferon-gamma-induced oligodendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Mol Med. 1995;1:732–743. [PMC free article] [PubMed] [Google Scholar]
- 34.Baerwald KD, Popko B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J Neurosci Res. 1998;52:230–239. doi: 10.1002/(SICI)1097-4547(19980415)52:2<230::AID-JNR11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 35.Corbin JG, et al. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci. 1996;7:354–370. doi: 10.1006/mcne.1996.0026. [DOI] [PubMed] [Google Scholar]
- 36.Lin W, et al. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol. 2005;169:603–612. doi: 10.1083/jcb.200502086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin W, et al. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117:448–456. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balabanov R, et al. Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci. 2007;27:2013–2024. doi: 10.1523/JNEUROSCI.4689-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flowers LO, et al. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J Immunol. 2004;172:7510–7518. doi: 10.4049/jimmunol.172.12.7510. [DOI] [PubMed] [Google Scholar]
- 40.Mujtaba MG, et al. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J Immunol. 2005;175:5077–5086. doi: 10.4049/jimmunol.175.8.5077. [DOI] [PubMed] [Google Scholar]
- 41.Zhou H, et al. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res. 2007;13:2344–2353. doi: 10.1158/1078-0432.CCR-06-2303. [DOI] [PubMed] [Google Scholar]
- 42.Martini M, et al. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int J Cancer. 2008;123:2955–2960. doi: 10.1002/ijc.23805. [DOI] [PubMed] [Google Scholar]
- 43.Huang FJ, et al. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res. 2008;68:9634–9642. doi: 10.1158/0008-5472.CAN-08-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lang R, et al. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 45.Lehmann U, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem. 2003;278:661–671. doi: 10.1074/jbc.M210552200. [DOI] [PubMed] [Google Scholar]
- 46.Boyle K, et al. Deletion of the SOCS box of suppressor of cytokine signaling 3 (SOCS3) in embryonic stem cells reveals SOCS box-dependent regulation of JAK but not STAT phosphorylation. Cell Signal. 2009;21:394–404. doi: 10.1016/j.cellsig.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin H, et al. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006;177:7761–7771. doi: 10.4049/jimmunol.177.11.7761. [DOI] [PubMed] [Google Scholar]
- 48.Pauli EK, et al. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008;4:e1000196. doi: 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohney SJ, et al. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 1999;19:4980–4988. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baetz A, et al. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279:54708–54715. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- 51.Bellezza I, et al. Suppressor of cytokine signaling-3 antagonizes cAMP effects on proliferation and apoptosis and is expressed in human prostate cancer. Am J Pathol. 2006;169:2199–2208. doi: 10.2353/ajpath.2006.060171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cacalano NA, et al. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 2001;3:460–465. doi: 10.1038/35074525. [DOI] [PubMed] [Google Scholar]
- 53.Marine JC, et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98:617–627. doi: 10.1016/s0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- 54.Yadav A, et al. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem. 2005;280:31830–31840. doi: 10.1074/jbc.M501316200. [DOI] [PubMed] [Google Scholar]
- 55.Miao T, et al. Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J Neurosci. 2006;26:9512–9519. doi: 10.1523/JNEUROSCI.2160-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoiber D, et al. Listeria monocytogenes modulates macrophage cytokine responses through STAT serine phosphorylation and the induction of suppressor of cytokine signaling 3. J Immunol. 2001;166:466–472. doi: 10.4049/jimmunol.166.1.466. [DOI] [PubMed] [Google Scholar]
- 57.Thuret S, et al. Therapeutic interventions after spinal cord injury. Nat Rev Neurosci. 2006;7:628–643. doi: 10.1038/nrn1955. [DOI] [PubMed] [Google Scholar]
- 58.Okada S, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 59.Herrmann JE, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raghavendra Rao VL, et al. Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J Neurosci Res. 2003;71:208–219. doi: 10.1002/jnr.10486. [DOI] [PubMed] [Google Scholar]
- 61.Rosell DR, et al. Differential expression of suppressors of cytokine signaling-1, -2, and -3 in the rat hippocampus after seizure: implications for neuromodulation by gp130 cytokines. Neuroscience. 2003;122:349–358. doi: 10.1016/s0306-4522(03)00594-3. [DOI] [PubMed] [Google Scholar]
- 62.Raghavendra Rao VL, et al. Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J Neurochem. 2002;83:1072–1086. doi: 10.1046/j.1471-4159.2002.01208.x. [DOI] [PubMed] [Google Scholar]
- 63.Bates S, et al. Characterisation of gene expression changes following permanent MCAO in the rat using subtractive hybridisation. Brain Res Mol Brain Res. 2001;93:70–80. doi: 10.1016/s0169-328x(01)00186-3. [DOI] [PubMed] [Google Scholar]
- 64.Mayer M, et al. Ciliary neurotrophic factor and leukemia inhibitory factor promote the generation, maturation and survival of oligodendrocytes in vitro. Development. 1994;120:143–153. doi: 10.1242/dev.120.1.143. [DOI] [PubMed] [Google Scholar]
- 65.Butzkueven H, et al. LIF receptor signaling limits immune-mediated demyelination by enhancing oligodendrocyte survival. Nat Med. 2002;8:613–619. doi: 10.1038/nm0602-613. [DOI] [PubMed] [Google Scholar]
- 66.Linker RA, et al. CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat Med. 2002;8:620–624. doi: 10.1038/nm0602-620. [DOI] [PubMed] [Google Scholar]
- 67.Emery B, et al. Suppressor of cytokine signaling 3 limits protection of leukemia inhibitory factor receptor signaling against central demyelination. Proc Natl Acad Sci U S A. 2006;103:7859–7864. doi: 10.1073/pnas.0602574103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y, et al. Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol. 2006;177:1679–1688. doi: 10.4049/jimmunol.177.3.1679. [DOI] [PubMed] [Google Scholar]
- 69.Liu X, et al. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol. 2008;180:6070–6076. doi: 10.4049/jimmunol.180.9.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frisullo G, et al. The effect of disease activity on leptin, leptin receptor and suppressor of cytokine signalling-3 expression in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2007 doi: 10.1016/j.jneuroim.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 71.Frisullo G, et al. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. J Neurosci Res. 2006;84:1027–1036. doi: 10.1002/jnr.20995. [DOI] [PubMed] [Google Scholar]
- 72.Frisullo G, et al. Leptin enhances the release of cytokines by peripheral blood mononuclear cells from relapsing multiple sclerosis patients. J Clin Immunol. 2004;24:287–293. doi: 10.1023/B:JOCI.0000025450.48267.a5. [DOI] [PubMed] [Google Scholar]
- 73.Lock C, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 74.Matarese G, et al. Leptin increase in multiple sclerosis associates with reduced number of CD4(+)CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2005;102:5150–5155. doi: 10.1073/pnas.0408995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bjorbaek C, et al. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 76.Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci. 2002;23:482–486. doi: 10.1016/s0165-6147(02)02077-1. [DOI] [PubMed] [Google Scholar]
- 77.Zhang X, et al. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol. 2008;180:6988–6996. doi: 10.4049/jimmunol.180.10.6988. [DOI] [PubMed] [Google Scholar]
- 78.Chen Z, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weber MS, Zamvil SS. Statins and demyelination. Curr Top Microbiol Immunol. 2008;318:313–324. doi: 10.1007/978-3-540-73677-6_12. [DOI] [PubMed] [Google Scholar]
- 80.Brantley EC, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. 2008;14:4694–4704. doi: 10.1158/1078-0432.CCR-08-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hussain SF, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]