

Abstract
Astrocytes play a number of important physiological roles in Central Nervous System (CNS) homeostasis. Inflammation stimulates astrocytes to secrete cytokines and chemokines that guide macrophages/microglia and T-cells to sites of injury/inflammation, and herein we describe how these processes are controlled by the Suppressor Of Cytokine Signaling (SOCS) proteins, a family of proteins that negatively regulate adaptive and innate immune responses. In this study, we describe that the immunomodulatory cytokine IFN-β induces SOCS-1 and SOCS-3 expression in primary astrocytes at the transcriptional level. SOCS-1 and SOCS-3 transcriptional activity is induced by IFN-β through GAS elements within their promoters. Studies in STAT-1α deficient astrocytes indicate that STAT-1α is required for IFN-β-induced SOCS-1 expression, while STAT-3 siRNA studies demonstrate that IFN-β-induced SOCS-3 expression relies on STAT-3 activation. Specific siRNA inhibition of IFN-β-inducible SOCS-1 and SOCS-3 in astrocytes enhances their pro-inflammatory responses to IFN-β stimulation, such as heightened expression of the chemokines CCL2 (MCP-1), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL5 (RANTES) and CXCL10 (IP-10), and promoting chemotaxis of macrophages and CD4+ T-cells. These results indicate that IFN-β induces SOCS-1 and SOCS-3 in primary astrocytes in order to attenuate its own chemokine-related inflammation in the CNS.
INTRODUCTION
Astrocytes are the major glial cell type within the CNS and are critical for CNS homeostasis (1–3). Astrocytes regulate neuronal function by releasing neurotrophic factors, guiding neuronal development, and contributing to neurotransmitter metabolism (4, 5). Astrocytes also participate in synaptic plasticity and influence the formation of the blood-brain-barrier (BBB) (1, 4). Chemokines produced by astrocytes, such as MCP-1, MIP-1α and IP-10, attract macrophages/microglia and T-cells to CNS inflammatory sites (3, 4, 6, 7). Aberrant expression of chemokines accompanies CNS disorders such as Multiple Sclerosis (MS), Alzheimer’s Disease, HIV-1-Associated Dementia and brain injury/trauma (3). Astrocytes are involved in these disorders, but to date, mechanisms by which astrocytes modulate inflammation within the CNS remain sketchy.
Type I Interferons (IFNs) elicit anti-viral, anti-proliferative and immunomodulatory responses upon interaction with their receptors (8, 9). IFN-β primarily signals through the tyrosine kinases JAK1 and TYK2 to activate Signal Transducers and Activators of Transcription-1α (STAT-1α) and STAT-2, which associate with IRF-9 to form the Interferon-Stimulated Gene Factor-3 (ISGF-3) complex. ISGF-3 upregulates IFN-responsive genes by interacting with interferon-responsive sequence elements (ISRE) in their promoters (8). Less frequently, IFN-β induces STAT-1α to form homodimers that bind to IFN-γ Activation Site (GAS) elements (8). IFN-β also activates STAT-3, and the resulting STAT-1/STAT-3 heterodimers or STAT-3 homodimers bind to GAS elements to induce gene expression (10, 11). IFN-β-deficient mice develop a more severe form of experimental autoimmune encephalomyelitis (EAE) than wild-type littermates, suggesting IFN-β may inhibit CNS inflammation (12, 13).
Inflammatory infiltrates in demyelinating lesions consist primarily of infiltrating macrophages and resident microglia, as well as infiltrating T-cells (7). Chemokines, particularly IP-10, MCP-1, RANTES and MIP-1α, play important roles in the initial recruitment of inflammatory cells to the CNS (7, 14–16). Studies in MCP-1-deficient mice indicate that MCP-1 has a nonredundant role in modulating monocyte and T-cell infiltration during inflammation (17). MCP-1 has also been shown to attract neural progenitors to sites of neuroinflammation (18). IP-10 also participates in leukocyte recruitment to CNS inflammatory sites, and blocking IP-10 with neutralizing antibodies markedly diminished the severity of EAE and reduced the levels of inflammatory T-cells in the CNS (19). Astrocytes are also a source of RANTES, which functions as a chemoattractant and activator of T cells and monocytes, and induces cytokine and chemokine expression, suggesting that it can promote inflammatory cascades in the brain (20, 21).
Suppressors Of Cytokine Signaling (SOCS) proteins (SOCS-1–7 and CIS) negatively regulate cytokine signaling pathways (22–24). The inducible SOCS-1 and SOCS-3 proteins inhibit the JAK-STAT pathway in a negative feedback loop, utilizing a variety of mechanisms (25). Targeted deletion of SOCS-3 in macrophages results in markedly enhanced IL-6-induced STAT-3 activation (26). Mice deficient in SOCS-3 in hematopoietic and endothelial cells demonstrate exacerbated IL-1 dependent arthritis (27), while forced expression of SOCS-3 in mouse arthritis models suppressed the induction and/or development of disease (22, 28). Administration of a cell-penetrating SOCS-3 suppressed cytokine-mediated signal transduction associated with acute inflammation (29). Studies from the EAE animal model and MS patients suggest that lower levels of SOCS-3 are associated with relapsing EAE and MS (30, 31). Treatment with a mimetic of SOCS-1 (TKip) inhibited STAT-1 activation, and had a protective effect on EAE disease, suggesting SOCS-1 exerts a beneficial effect in the EAE model (32). We previously demonstrated that IFN-β induces SOCS-1 expression in macrophages/microglia, which attenuates IFN-β-induced expression of the costimulatory protein CD40 (33). Similarly, LPS induces SOCS-3 expression in macrophages/microglia (34), and overexpression of SOCS-3 attenuates LPS-induced gene expression in these cells (35). These finding indicate that SOCS-1 and SOCS-3 inhibit inflammatory responses in macrophages/microglia.
Elucidating the function of SOCS-1 and SOCS-3 in astrocytes, and how they are expressed, is critical for understanding how astrocytes regulate inflammation in the CNS. Results from this study indicate that in primary murine astrocytes, IFN-β-mediated activation of STAT-1 and STAT-3 are responsible for SOCS-1 and SOCS-3 induction, respectively. Studies using astrocytes transfected with SOCS-1 or SOCS-3 siRNA revealed that SOCS-1 and SOCS-3 inhibit IFN-β-induced expression of chemokines such as MCP-1, MIP-1α, MIP-1β, RANTES and IP-10, and inhibit chemotaxis of macrophages and T-cells. These findings connect SOCS-1 and SOCS-3 induction in astrocytes with negative regulation of CNS inflammation.
MATERIALS AND METHODS
Recombinant Proteins and Reagents
Recombinant murine IFN-β was from R&D Systems (Minneapolis, MN). Antibodies against phospho-STAT-1Tyr701, phospho-STAT-2Tyr690 and phospho-STAT-3Tyr705 were from Cell Signaling Technology (Beverly, MA). Antibodies against STAT-1, STAT-2, STAT-3 and actin were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against SOCS-1 and SOCS-3 were from Invitrogen Corporation (Carlsbad, CA).
Primary Astrocyte Cultures, Generation of Bone Marrow Derived Macrophages (BMDM) and CD4+ T-Cell Isolation
All animal studies were approved by the Animal Studies Committee at the University of Alabama at Birmingham (Birmingham, AL). Primary astrocyte cultures from C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) and WT and STAT-1α deficient mice on the 129S6/SvEv background (Taconic, Germantown, NY) were established from neonatal mice cerebra as described previously (36). Cells were monitored for purity by immunofluorescence and were routinely >97% positive for glial fibrillary acidic protein (36). Bone marrow cells were isolated by flushing the marrow cavities of mouse femurs from C57BL/6J mice as previously described (34). Bone marrow cells were cultured for 7 days in the presence of 20 ng/ml of murine M-CSF. Adherent cells were detached with 1 mM EDTA and purity of the macrophage cultures was > 95%, as determined by staining for CD11b. Spleens were collected from C57BL/6J mice and single-cell suspensions prepared by mechanical disruption. CD4+ T-cells were isolated by magnetic sorting with anti-mouse CD4-magnet beads (BD Pharmingen, San Diego, CA) according to the manufacturer’s directions (37).
RNA Isolation, Riboprobes, and Ribonuclease Protection Assay (RPA)
Total cellular RNA was isolated from unstimulated or IFN-β-treated astrocytes. Riboprobes for murine SOCS-1, SOCS-3, MCP-1, MIP-1α, MIP-1β, RANTES, IP-10, and GAPDH were prepared as described previously (35), and the riboprobes were hybridized with 20 μg of total RNA at 42°C overnight. The hybridized mixture was treated with RNase A/T1 (1:200) and analyzed by 5% denaturing (8 M urea) polyacrylamide gel electrophoresis. Values for SOCS-1, SOCS-3, MCP-1, MIP-1α, MIP-1β, RANTES, and IP-10 mRNA expression levels were normalized to GAPDH mRNA levels for each experimental condition.
Immunoblotting
Proteins (30 μg) in cell lysates were separated on 8% SDS-PAGE and probed with phospho-STAT-1, phospho-STAT-2, or phospho-STAT-3 antibodies as described previously (35). Membranes were stripped and reprobed for total STAT-1, STAT-2, STAT-3, or actin. SOCS-1 and SOCS-3 protein expression was analyzed using 15% SDS-PAGE, followed by transfer of proteins to PDVF membranes and immunoblotting (35).
SOCS-1 and SOCS-3 Promoter Constructs, Transient Transfection, and Luciferase Assays
Primary astrocytes were seeded in 6-well plates (5 × 105 cells/well) and transiently transfected using Lipofectamine Plus as described previously (36). Cells received the 1478-bp (−1380 to +98 bp) murine SOCS-1 promoter, the 1619-bp (−1492 to +127 bp) murine SOCS-3 promoter, or serial deletion mutants (see Figure 2). Transfected cells were treated with medium or IFN-β for 12 h, and luciferase activity of each sample normalized to the total protein concentration in each well. Luciferase activity from the untreated sample was arbitrarily set at 1 for calculation of fold induction.
Figure 2. IFN-β-induced activation of the murine SOCS-1 and SOCS-3 promoters requires GAS elements.

Serial deletion and mutagenesis constructs of the SOCS-1 (A) and SOCS-3 (B) promoters are represented by line diagrams. Primary astrocytes were transiently transfected with 0.2 μg of the indicated SOCS-1 or SOCS-3 promoter constructs, allowed to recover for 4 h, treated with medium or IFN-β (100 U/ml) for 12 h, and analyzed for luciferase activity. Values were normalized to total protein levels, and the fold inductions shown are the IFN-β treatment values divided by untreated values. Data are the mean ± S.D. of three experiments.
RNA Interference
RNA interference followed a protocol provided by Dharmacon (Lafayette, CO), with previously described modifications (33). DharmaFECT™ 1 siRNA transfection reagent, SMARTpool small interfering RNAs (siRNAs) specific for murine STAT-3, STAT-2, SOCS-1, or SOCS-3, and CONTROL nontargeting siRNA were purchased from Dharmacon. Primary astrocytes (5 × 105 cells/well in six-well plates) were transfected with 100 nM of CONTROL or one of the specific siRNAs using the DharmaFECT™ 1 reagent. Cells were analyzed after 48 h of transfection as follows: 30 μg of cell lysate was immunoblotted to determine STAT-3 and STAT-2 expression and phosphorylation levels, or 5 μg of RNA was subjected to RPA to determine SOCS-1 or SOCS-3 mRNA expression.
ELISA
Supernatants were collected from unstimulated or IFN-β-stimulated primary astrocytes and assayed for IP-10 secretion using ELISA (R&D Systems, Inc., Minneapolis, MN). IP-10 levels were normalized to total protein levels.
Chemotaxis Assay
Primary astrocytes were transfected with control, SOCS-1, or SOCS-3 siRNA for 48 h, and then cultured with medium or IFN-β for 16 and 24 h. Chemotaxis assays were performed in a Transwell system (5-μm pore size; Costar, Corning, NY) as described previously (38). Briefly, single-cell suspensions of primary macrophages or CD4+ T-cells (1 × 106) were loaded into the upper well inserts of a Transwell system, and supernatants from the transfected primary astrocyte cultures loaded into the bottom chambers. After incubation for 4 h at 37°C, cells that had migrated to the lower chamber were collected and counted by flow cytometry (38). The chemotaxis index was calculated as the number of cells that migrated in response to factors in the IFN-β-treated astrocyte supernatant, divided by the number of cells that migrated in response to untreated astrocyte supernatant.
Statistical Analysis
All experiments were repeated a minimum of three times. For comparisons between samples, levels of significance were determined by Student’s t test distribution. P values of ≤0.05 were considered to be statistically significant.
RESULTS
IFN-β Induces SOCS-1 and SOCS-3 Expression in Primary Astrocytes
It is clear from previous work that IFN-β can function as a neuroprotective agent by modulating the inflammatory responses of astrocytes (3, 15), but specific elements of the underlying mechanism remain unclear. To determine whether IFN-β affected the inflammatory potential of astrocytes through the SOCS proteins, we began by analyzing whether IFN-β regulated SOCS-1 and SOCS-3 gene expression in primary astrocytes. IFN-β induced SOCS-1 and SOCS-3 expression after 30 minutes of treatment. SOCS-1 mRNA levels peaked at 4 h of IFN-β treatment (13.5-fold induction), and diminished afterward (Figure 1A). SOCS-3 mRNA expression peaked at 2 h (10.3-fold induction), and diminished at 4 h (Figure 1A). In accordance with the increases in mRNA levels, SOCS-1 and SOCS-3 proteins were detected after IFN-β stimulation. SOCS-1 protein levels peaked at 4 – 8 h, and were still elevated at 16 h (Figure 1B), while SOCS-3 protein expression was detectable 30 min after IFN-β stimulation, peaked at 4 h, and returned to basal levels at 24 h (Figure 1B).
Figure 1. IFN-β induces SOCS-1 and SOCS-3 gene expression in primary astrocytes.

The images shown are representative of at least three experiments. (A) To measure IFN-β-induced increases in SOCS mRNA levels, primary astrocytes were treated with medium (UN) or IFN-β (100 U/ml) for up to 12 h, and total RNA analyzed by RPA to measure SOCS-1, SOCS-3, and GAPDH mRNA levels. mRNA levels in the untreated sample were set at 1.0, and results are expressed as fold induction over these control levels. (B) To determine the IFN-β-induced increases in SOCS protein levels, primary astrocytes were treated with IFN-β for up to 24 h and cell lysates immunoblotted with anti-SOCS-1 or anti-SOCS-3. The blots were stripped and reprobed with anti-actin as a loading control.
IFN-β Induction of SOCS-1 and SOCS-3 Promoter Activity Requires Functional GAS Elements
Since IFN-β induced SOCS-1 and SOCS-3 expression, we sought to define which cis-acting regulatory elements in their promoters were necessary for this induction. The SOCS-1 promoter (−1380 to +98 bp) contains multiple regulatory elements: three GAS elements (designated as GAS#1, GAS#2, and GAS#3), one GATA-1 site, one AP-1 site, two Sp1 sites, one AP-2 site, and one C/EBP element (Figure 2A). We generated luciferase reporter-based constructs that sequentially deleted regulatory elements from the 5′ end of the SOCS-1 promoter. The reporter constructs were transiently transfected into primary astrocytes and treated with medium or IFN-β for 12 h. A luciferase construct containing the full-length SOCS-1 promotor served as a positive control. Our results indicate that IFN-β stimulated a 6.1-fold increase in WT SOCS-1 promoter activity. Deletion of the GATA-1, AP-1, Sp1, AP-2, GAS#1, and C/EBP elements (Δ1) did not significantly affect IFN-β-induced SOCS-1 promoter activity (Figure 2A). Deletion of the GAS#2 site (Δ2) reduced IFN-β-induced SOCS-1 promoter activity by ~48% compared to Δ1 (Figure 2A). In construct Δ3, deletion of both GAS#2 and GAS#3 abrogated IFN-β-induced SOCS-1 promoter activity (Figure 2A). To further define the relative contributions of the GAS#2 and GAS#3 elements, assays were performed using constructs of the murine SOCS-1 promoter that specifically mutated these elements, but left the rest of the promoter intact (mGAS#2 and mGAS#3; Figure 2A). Mutation of the GAS#2 element led to a ~53% inhibition of SOCS-1 promoter activity compared to the WT promoter, while mutation of the GAS#2 element inhibited IFN-β activation of the SOCS-1 promoter by ~67% (Figure 2A). These results indicated that of all the potential cis-acting regulatory elements within the SOCS-1 promoter, these two GAS elements (GAS#2 and GAS#3) made the most important contributions to IFN-β-induced promoter activity in astrocytes.
The 1619 bp (−1492 – +127 bp) murine SOCS-3 promoter also contains many cis-regulatory targets: three AP-1 sites, one C/EBP site, one GATA-1 site, two Sp1 sites, one NF-κB site, and two GAS elements (GAS#1 and GAS#2). To ascertain the functional roles of these elements, we generated luciferase reporter-based deletion constructs from the 5′ end of the SOCS-3 promoter (35) (Figure 2B). The WT SOCS-3 promoter or one of the deletion constructs were transiently transfected into astrocytes and treated with IFN-β for 12 h prior to assaying for luciferase activity. IFN-β stimulation mediated a 7.9-fold induction of WT SOCS-3 promoter activity. Deletion of the distal and medial AP-1 elements, the C/EBP element, the GATA-1 element, the NF-κB binding site, and the two Sp1 elements (Δ1) did not affect IFN-β-induced SOCS-3 promoter activity (Figure 2B). Similarly, deletion of the proximal AP-1 element (Δ2) did not affect IFN-β-induced SOCS-3 promoter activity (Figure 2B). In contrast, deletion of the distal GAS element (GAS#1; Δ3) reduced IFN-β-induced SOCS-3 promoter activity by 47% compared to Δ2. Deletion of both the GAS elements (GAS#1 and GAS#2; Δ4) abolished IFN-β-induced SOCS-3 promoter activity (Figure 2B). Site-directed mutagenesis was used to individually mutate each GAS element in the murine SOCS-3 promoter (Figure 2B). Mutation of the distal GAS#1 or the proximal GAS#2 element reduced IFN-β-induced SOCS-3 promoter activity by ~80% or 82%, respectively, compared to the Δ1 construct (Figure 2B). These data indicated that among the proximal regulatory elements in the SOCS-3 promoter, the two GAS elements were most important for IFN-β-induced SOCS-3 promoter activity in astrocytes.
STAT-1α is Important for IFN-β-induced SOCS-1, but not SOCS-3, Expression in Primary Astrocytes
In T-cells and macrophages, numerous stimuli, including IFN-γ, IFN-β, IL-6, IL-10 and Growth Hormone, are capable of inducing SOCS-1 and SOCS-3 expression by activating various STAT proteins (22, 28). To investigate the activation of STATs by IFN-β in primary astrocytes, the cells were treated in the absence or presence of IFN-β for up to 4 h, then protein lysates analyzed to detect the phosphorylation status of STAT-1α and STAT-3 (Figure 3A). STAT-1αTyr701 was strongly phosphorylated after 0.25–2 h of IFN-β treatment, which diminished at 4 h. Similarly, phosphorylation of STAT-3Tyr705 was detected at 0.25 h of IFN-β treatment, peaked between 0.5 and 1 h, then diminished at 2–4 h. These results indicate that IFN-β activates the STAT-1/STAT-3 signaling pathways in primary astrocytes.
Figure 3. STAT-1α is important for IFN-β-induced SOCS-1, but not SOCS-3, expression in primary astrocytes.

Images shown are representative of at least three experiments. (A) To evaluate STAT activation in response to IFN-β treatment, astrocytes were incubated with IFN-β for up to 4 h, then cell lysates immunoblotted with anti-phospho-Tyr-STAT-1α or anti-phospho-Tyr-STAT-3. The membranes were stripped and reprobed with anti-STAT-1α, anti-STAT-3 or anti-actin as a loading control. (B) Primary astrocytes from WT- and STAT-1α-deficient mice were treated with IFN-β (100 U/ml) for up to 2 h. Total cellular mRNA was analyzed for SOCS-1, SOCS-3, and GAPDH mRNA expression by RPA. Basal levels in the untreated sample were set at 1.0, and results are expressed as fold induction over these control levels. (C) To evaluate STAT activation levels in response to IFN-β, primary astrocytes from WT and STAT-1α deficient mice were treated with IFN-β for up to 2 h, and cell lysates immunoblotted with anti-phospho-Tyr-STAT-1 or anti-phospho-Tyr-STAT-3. The membranes were stripped and reprobed with anti-STAT-1, anti-STAT-3 or anti-actin as a loading control.
To determine whether STAT-1α was involved in IFN-β-induced SOCS-1 and SOCS-3 expression in astrocytes, we examined primary astrocytes from STAT-1α-deficient mice. After stimulation with IFN-β, these astrocytes did not express SOCS-1 mRNA, but SOCS-3 mRNA expression was much stronger than in WT astrocytes (Figure 3B). These experiments demonstrated that IFN-β-mediated induction of SOCS-1 expression occurred in a STAT-1α-dependent manner, but IFN-β induction of SOCS-3 expression did not involve STAT-1α. In fact, the stronger SOCS-3 expression in STAT-1α-deficient astrocytes suggest that STAT-1α may negatively regulate SOCS-3 expression.
We next examined IFN-β activation of STAT-1α and STAT-3 in WT and STAT-1α-deficient primary astrocytes. In WT astrocytes, IFN-β induced strong and sustained STAT-1α tyrosine phosphorylation, while STAT-3 phosphorylation was much weaker (Figure 3C). It should be noted that the STAT-3 phosphorylation status in WT astrocytes from 129s6/SVEV mice is reduced (Figure 3C) compared to that from astrocytes from C57BL/6J mice (Figure 3A). IFN-β-induced STAT-3 phosphorylation was much stronger and prolonged in STAT-1α-deficient astrocytes than in WT cells (Figure 3C). This further supports the idea that STAT-1α negatively regulates STAT-3, and suggests that enhanced STAT-3 signaling in the absence of STAT-1α may have contributed to SOCS-3 expression.
STAT-3 is Important for IFN-β-induced SOCS-3, but not SOCS-1, Expression in Primary Astrocytes
IFN-β induced the activation of both STAT-1 and STAT-3 in primary astrocytes, but the absence of STAT-1α enhanced STAT-3 activation and IFN-β-induced SOCS-3 mRNA (Figures 3B and 3C). Therefore, we investigated whether activation of STAT-3 was required for IFN-β to induce SOCS-3 gene expression. Using primary astrocytes from WT and STAT-1α deficient mice, STAT-3 expression was inhibited by transfecting cells with a STAT-3-specific siRNA construct (or siRNA control) for 48 h. This inhibited constitutive STAT-3 protein levels in WT astrocytes by ~90% (Figure 4A, compare lanes 1–3 to lanes 4–6) and IFN-β-induced STAT-3 phosphorylation by ~85% (Figure 4A, compare lanes 2–3 to lanes 5–6). Similar inhibition was achieved for STAT-3 protein expression and STAT-3 phosphorylation in STAT-1α deficient astrocytes (Figure 4A, compare lanes 7–9 to lanes 10–12). Furthermore, STAT-3 siRNA did not affect constitutive STAT-1 protein levels or IFN-β-induced STAT-1α activation in WT astrocytes (Figure 4A, lanes 1–6).
Figure 4. STAT-3 is important for IFN-β-induced SOCS-3, but not SOCS-1, expression in primary astrocytes.

Images shown are representative of at least three experiments. (A) Primary astrocytes from WT and STAT-1-deficient mice were transfected with STAT-3 siRNA (SMARTpool) or an siRNA control (100 nM) for 48 h. Transfected cells were then treated with medium (UN) or IFN-β for 1 or 2 h and cell lysates immunoblotted with anti-STAT-3, anti-phospho-Tyr-STAT-3, anti-STAT-1, anti-phospho-Tyr-STAT-1 or anti-actin. (B) Using astrocytes treated as in (A), total RNA was analyzed for SOCS-1, SOCS-3, and GAPDH mRNA expression using RPA. Basal levels in the untreated sample were set at 1.0, and results are expressed as fold induction over these control levels. (C) To determine the effects of STAT-2 inhibition, primary astrocytes were transfected with STAT-2 siRNA (SMARTpool) or an siRNA control (100 nM). Cell lysates were immunoblotted with anti-phospho-Tyr-STAT-1, anti-phospho-Tyr-STAT-2, anti-phospho-Tyr-STAT-3, anti-STAT-1, anti-STAT-2, anti-STAT-3, and anti-actin, where indicated. (D) Total RNA from cells treated as in (C) was analyzed for SOCS-1, SOCS-3, and GAPDH mRNA using RPA. Basal levels in the untreated sample were set at 1.0, and results are expressed as fold induction over these control levels.
We next examined whether inhibition of STAT-3 affected IFN-β-induced SOCS-3 expression. Primary astrocytes from WT or STAT-1α-deficient mice were incubated with STAT-3-specific or control siRNA for 48 h, then IFN-β added for 1 or 2 h to induce SOCS-1 and SOCS-3 mRNA expression. IFN-β-induced SOCS-3 expression was higher in STAT-1α-deficient astrocytes than in WT cells (Figure 4B, compare lanes 2–3 to lanes 8–9). In WT astrocytes, STAT-3 knockdown inhibited IFN-β-induced SOCS-3 expression by ~65% (Figure 4B, compare lanes 2–3 to lanes 5–6), and the STAT-3 siRNA effect was more pronounced in STAT-1α −/− astrocytes (~82% inhibition of SOCS-3 expression; Figure 4B, compare lanes 8–9 to lanes 11–12). IFN-β-induced SOCS-1 mRNA induction was abolished in STAT-1α-deficient cells (Figure 4B), and STAT-3 siRNA did not significantly affect IFN-β-induced SOCS-1 mRNA expression in WT astrocytes (Figure 4B, compare lanes 2–3 to lanes 5–6). These results collectively indicate that STAT-3 activation plays a critical role in IFN-β-induced SOCS-3 gene expression, but that it is dispensable for SOCS-1 expression.
STAT-2 is not Critical for IFN-β Induction of SOCS-1 and SOCS-3 Expression
IFN-β primarily signals through the STAT-1/STAT-2/IRF-9 heterotrimer (8). Our data strongly implicated STAT-1 in IFN-β-mediated SOCS-1 expression, and since STAT-2 is also a component of the ISGF-3 complex, we sought to determine whether STAT-2 was involved in IFN-β-induced SOCS-1 gene expression. By transfecting primary astrocytes with siRNA against STAT-2, constitutive STAT-2 protein expression was suppressed by ~95% (Figure 4C, compare lanes 1–3 and 4–6), without affecting STAT-1 or STAT-3 expression. STAT-2 siRNA also inhibited IFN-β-induced STAT-2 phosphorylation by ~90% (Figure 4C) without affecting IFN-β-induced STAT-1α and STAT-3 activation (Figure 4C, compare lanes 2–3 vs. lanes 5–6). Upon evaluating SOCS-1 and SOCS-3 mRNA expression levels following IFN-β stimulation of STAT-2-depleted primary astrocytes, we found that SOCS-1 mRNA was slightly decreased (~30%) compared to siRNA control treated cells, and expression of SOCS-3 mRNA was actually enhanced 1.9 fold by STAT-2 knockdown (Figure 4D). These results indicate that IFN-β-induced SOCS-1 and SOCS-3 gene expression were moderately but not critically influenced by the STAT-2 transcription factor.
IFN-β Induces Expression of CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES, and CXCL10/IP-10 in Primary Astrocytes
Chemokines are 8–14 kDa proteins responsible for the maturation and trafficking of leukocytes, particularly during inflammation (6, 7). We examined the effect of IFN-β on expression of several chemokines with essential roles in neuroinflammation. IFN-β induced expression of MCP-1, MIP-1α, MIP-1β, RANTES, and IP-10, although the kinetics differed among the chemokines (Figure 5A). We observed MCP-1, MIP-1α, MIP-1β, and IP-10 mRNA after 1 h of IFN-β treatment; expression peaked between 2 and 4 h and decreased after 8 h (Figure 5A). Although the astrocytes constitutively expressed RANTES, IFN-β enhanced RANTES mRNA expression after 2 h of treatment, and RANTES expression continued to increase over time (Figure 5A). These data indicate that in primary astrocytes, IFN-β rapidly induces the expression of several chemokines that are relevant to the inflammatory process.
Figure 5. IFN-β induces CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES, and CXCL10/IP-10 expression in primary astrocytes.

Images shown are representative of at least three experiments. (A) Primary astrocytes were treated with medium (UN) or IFN-β for up to 8 h, then total RNA was assayed by RPA to determine mRNA expression levels of MCP-1, MIP-1α, MIP-1β, RANTES, IP-10, and GAPDH. Basal levels in the untreated sample were set at 1.0, and results are expressed as fold induction over these control levels. (B) Primary astrocytes from WT and STAT-1α deficient mice were treated with medium (UN) or IFN-β for up to 8 h, and then mRNA was analyzed by RPA for MCP-1, MIP-1α, MIP-1β, RANTES, IP-10, and GAPDH expression. The basal level of the untreated sample was set at 1.0, and fold induction upon IFN-β treatment compared to that.
To determine whether STAT-1 is involved in IFN-β-induced chemokine expression, primary astrocytes from STAT-1α deficient mice were examined. The absence of STAT-1α abrogated IFN-β induction of MCP-1, MIP-1α, MIP-1β and RANTES mRNA expression (Figure 5B). IFN-β induction of IP-10 was partially inhibited in STAT-1 deficient astrocytes compared to WT mice (Figure 5B). These experiments demonstrate that IFN-β induction of chemokine expression occurred in a STAT-1α dependent manner.
SOCS-1 and SOCS-3 Influence IFN-β-induced Chemokine Expression in Primary Astrocytes
To determine whether SOCS-1 and/or SOCS-3 contribute to the regulation of these chemokines during IFN-β signaling, chemokine mRNA expression was evaluated in astrocytes transfected with SOCS-1 or SOCS-3 siRNA. Following a 48 h transfection, cells were treated in the absence or presence of IFN-β for up to 8 h and analyzed for SOCS-1, SOCS-3 and chemokine mRNA expression. Both targeting siRNA constructs effectively attenuated induction of the SOCS mRNA by IFN-β. SOCS-1 siRNA inhibited SOCS-1 induction by 57%, 86%, and 70% at 1, 2, and 4 h of IFN-β treatment, respectively (Figure 6A, compare lanes 3–5 to lanes 9–11). SOCS-3 siRNA inhibited SOCS-3 induction by ~60% (1 h) and ~66% (2 h) (Figure 6A, compare lanes 3–4 to lanes 15–16). Compared to astrocytes expressing siRNA-Control, we observed enhanced IFN-β-induced SOCS-3 mRNA induction in the astrocytes transfected with SOCS-1 siRNA (Figure 6A, compare lanes 3–4 to lanes 9–10) and stronger SOCS-1 mRNA expression in cells harboring SOCS-3 siRNA (Figure 6A, compare lanes 3–5 to lanes 15–17). These results indicate that SOCS-1 negatively regulates the expression of SOCS-3, and vice versa.
Figure 6. SOCS-1 and SOCS-3 negatively influence IFN-β-induced chemokine expression in primary astrocytes.


Images shown are representative of at least three experiments. (A) After transfecting primary astrocytes with siRNA Control, SOCS-1 siRNA or SOCS-3 siRNA (100 nM) for 48 h, cells were treated with medium (UN) or IFN-β for up to 8 h. RPA was used to analyze SOCS-1, SOCS-3 and GAPDH mRNA levels. Basal levels in the untreated sample were set at 1.0, and results are expressed as fold induction over these control levels. (B) RNA from astrocytes transfected as in (A) was analyzed for MCP-1, MIP-1α, MIP-1β, RANTES, IP-10, and GAPDH mRNA levels. Basal levels in the untreated sample were set at 1.0, and results are expressed as fold induction over these control levels. (C) represents a tabulation of the data in (B). Each arrow indicates an increase of at least 100% in cells harboring SOCS-1 or SOCS-3 siRNA compared to cells with control siRNA. (D) Astrocytes transfected as in (A) were treated with medium (UN) or IFN-β for up to 36 h, and supernatants were analyzed for IP-10 protein using ELISA. Data are the mean ± S.D. of three experiments. *P ≤ 0.05.
We next examined the effect of SOCS-1 and SOCS-3 knockdown on induction of chemokine expression by IFN-β. Primary astrocytes transfected with SOCS-1 siRNA (Figure 6B, middle panel) exhibited more robust IFN-β-mediated chemokine induction (accompanied by slightly altered kinetics in some cases) than cells transfected with non-targeting siRNA (left panel). Throughout the IFN-β stimulation period, SOCS-1 inhibition enhanced CCL2/MCP-1, CCL4/MIP-1β, CCL5/RANTES, and CXCL10/IP-10 expression compared to siRNA control astrocytes. In addition, SOCS-1 inhibition substantially enhanced the marginal CCL3/MIP-1α induction after a 1 to 2 h IFN-β treatment. For all chemokines, we observed similar but less pronounced results in astrocytes expressing SOCS-3 siRNA (Figure 6B, right panel). Figure 6C summarizes the effect of SOCS-1 or SOCS-3 knockdown on chemokine mRNA expression. We chose one chemokine (IP-10) to analyze at the protein level. A significant enhancement of IFN-β-induced IP-10 expression was observed in SOCS-1 siRNA or SOCS-3 siRNA astrocytes compared to astrocytes with control siRNA (Figure 6D). These results indicate that both SOCS-1 and SOCS-3 negatively regulate IFN-β-mediated induction of chemokines that are relevant to neuroinflammation.
Knockdown of SOCS-1 and SOCS-3 Enhances Chemotaxis of Macrophages and T-cells to Supernatants from IFN-β-treated Astrocytes
Astrocytic chemokine expression contributes to proinflammatory responses within the CNS by attracting resident microglia and infiltrating macrophages, and by promoting the infiltration of T-cells into the CNS (7, 39–41). Our data demonstrate that SOCS-1 and SOCS-3 attenuate the induction of several chemokines in response to IFN-β. Therefore, we investigated whether this negative regulation was functionally significant; specifically, whether SOCS-1 or SOCS-3 levels negatively affected chemoattraction of immune cells following IFN-β stimulation in primary astrocytes. In these experiments, we examined the ability of bone marrow derived primary macrophages or spleen CD4+ T-cells to migrate toward supernatants from IFN-β-stimulated astrocytes that were depleted in either SOCS-1 or SOCS-3. After transfecting primary astrocytes with SOCS-1 siRNA, SOCS-3 siRNA, or control siRNA for 48 h, cells were treated with medium or IFN-β for 16 and 24 h. Supernatants from the astrocytes were utilized in Transwell chemotaxis assays. Consistent with enhanced chemokine induction in the absence of SOCS proteins (Figure 6), we observed significant enhancement of macrophage chemotaxis toward supernatants from SOCS-1 siRNA or SOCS-3 siRNA transfected astrocytes compared to control siRNA-transfected cells (Figure 7A). The same effect was observed for CD4+ T-cell chemotaxis (Figure 7B). These results indicate that enhanced chemokine expression in SOCS-1- and SOCS-3-depleted astrocytes enhances their chemoattractant properties for macrophages and CD4+ T-cells.
Figure 7. SOCS-1 and SOCS-3 negatively regulate IFN-β-induced macrophage and CD4+ T-cell chemotaxis.

(A)Astrocytes transfected as in Figure 6A were treated with medium (UN) or IFN-β for 16 or 24 h, and their supernatants used in a chemotactic assay with bone marrow derived primary macrophages in a Transwell migration chamber. Data are the mean ± S.D. of three experiments. *P ≤ 0.05. (B) The same supernatants were used in a chemotactic assay with spleen CD4+ T-cells in a Transwell migration chamber. Data are the mean ± S.D. of three experiments. *P ≤ 0.05 and **P ≤ 0.001.
DISCUSSION
IFN-β is one of the most successful MS therapies because it decreases exacerbation rates and delays disease progression (42–44). In addition, IFN-β treatment diminishes the clinical signs of EAE due to reduced cellular infiltration in the CNS (14, 45). IFN-β functions as an immunosuppressive agent by inhibiting class II MHC, MMP-9 and IL-12 expression, blocking Th1 development, inducing expression of the anti-inflammatory cytokines IL-10 and IL-4, inhibiting the inflammatory responses of macrophages/microglia, and improving BBB integrity (9, 46, 47). However, IFN-β also induces expression of chemokines that modulate inflammatory and immune responses in the CNS (15, 21). These chemokines recruit leukocytes to inflammatory sites, and may participate in regulating cytokine production by naive T helper cells (48). Thus, IFN-β has both anti- and pro-inflammatory activities (9, 49).
In the present study, IFN-β induced the expression of SOCS-1 and SOCS-3 in astrocytes, and this induction was critical for controlling inflammatory responses in these cells. IFN-β utilizes distinct STAT proteins for induction of SOCS-1 and SOCS-3. The induction of SOCS-1 is dependent on STAT-1, while SOCS-3 induction relies on STAT-3 (Figure 8). In support of STAT involvement, the results from analysis of the SOCS-1 and SOCS-3 promoters reveal that GAS elements are critical for IFN-β induced SOCS-1 and SOCS-3 expression in astrocytes. We have recently shown that Oncostatin M, an IL-6 family member, induces SOCS-3 expression in a manner dependent on STAT-3, ERK1/2 and JNK in astrocytes (50). In primary astrocytes, SOCS-1 and SOCS-3 expression is induced by IFN-γ (51), and also by PPAR-γ agonists such as 15d-PGJ2 and rosiglitazone (52). PPAR-γ agonist induction of SOCS-1 and SOCS-3 occurs in a STAT-1 independent manner, with the possible involvement of PKA and PKC (52). Thus, STAT-dependent and independent pathways lead to SOCS-1 and SOCS-3 expression in astrocytes in a stimulus-specific manner. Furthermore, there are CNS cell-type specific responses to IFN-β with respect to SOCS expression. Microglia are induced to express both SOCS-1 and SOCS-3 in response to IFN-β, similar to astrocytes, while in primary neurons, IFN-β induces SOCS-3, but not SOCS-1 expression (unpublished observation, H.Q. and E.N.B.).
Figure 8. Proposed model of inhibitory function of SOCS-1 and SOCS-3 on chemokine expression in astrocytes.
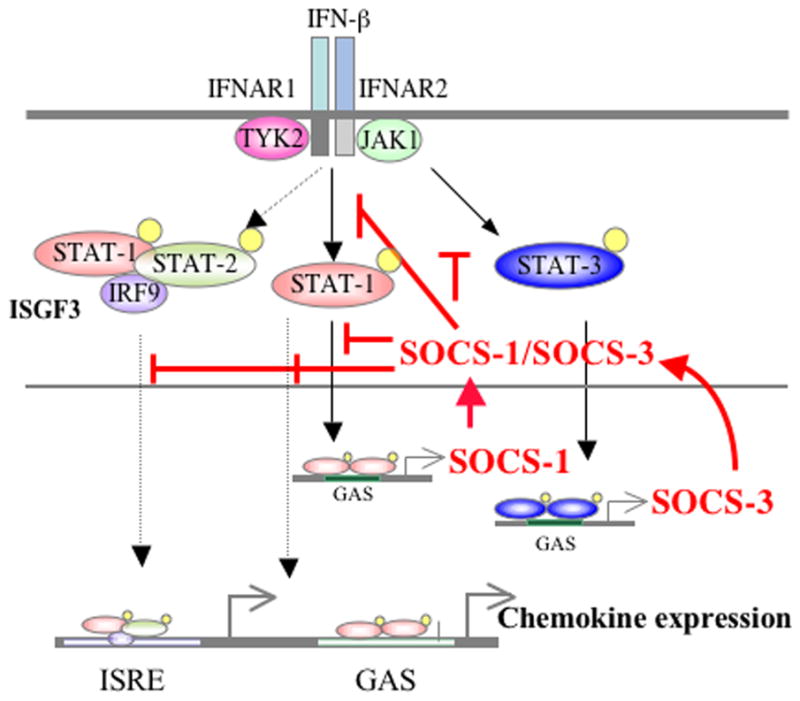
IFN-β activates STAT-1 and STAT-3, which then induce SOCS-1 and SOCS-3 expression, respectively. IFN-β induces SOCS-1 gene expression in a STAT-1-dependent manner and SOCS-3 gene expression in a STAT-3-dependent manner. Induction of the chemokines CCL2, CCL3, CCL4 and CCL5 upon IFN-β treatment occurs in a STAT-1-dependent manner. IFN-β-induced CXCL10 expression is partially dependent on STAT-1. SOCS-1 and SOCS-3 inhibit IFN-β-induced chemokine expression in a negative feedback regulatory manner. See text for details.
Studies in which the abundance of SOCS-1 and SOCS-3 have been enhanced or abrogated have established that these proteins are negative regulators of adaptive and innate immune responses (23, 24). SOCS-1 is a key regulator of IFN-γ signaling by its inhibitory effects on STAT-1 activation (53–55). As well, SOCS-1 inhibits other signaling pathways including those activated by IFN-α/β, LPS, IL-2 and insulin (33, 53, 56–59). Conditional gene-targeting has been used to elucidate the function of SOCS-3, given that SOCS-3 deficiency is embryonic lethal (60). The major function of SOCS-3 is to regulate signaling by the IL-6 family of cytokines by inhibiting STAT-3 activation (61, 62). Targeted deletion of SOCS-3 in macrophages results in an enhancement of IL-6-induced STAT-3 activation (26). In addition, SOCS-3 inhibits signal transduction induced by LPS, IL-12, IL-4 and IFN-α/β (63–71). Furthermore, SOCS-3 has a broad range of actions as it inhibits the NF-κB pathway (72), antagonizes cAMP-mediated signaling (73), and enhances signaling through the ERK/MAPK pathways (74). The results from our study indicate that in astrocytes, SOCS-1 and SOCS-3 function as negative regulators of the IFN-β signaling pathway. Reducing IFN-β-induced SOCS-1 and SOCS-3 expression levels in astrocytes with siRNA significantly enhanced IFN-β-induced expression of chemokines, including MCP-1, MIP-1α, MIP-1β, RANTES and IP-10. This translated to functional effects as demonstrated by enhanced chemotaxis of primary T-cells and macrophages to supernatants from SOCS-1 and SOCS-3 knockdown astrocytes, compared to those with intact SOCS-1 or SOCS-3. Thus, IFN-β induction of SOCS-1 and SOCS-3 negatively regulates IFN-β-induced expression of chemokines such as MCP-1, MIP-1α, MIP-1β, RANTES and IP-10, which likely is the result of attenuation of STAT-1, STAT-2 and STAT-3 activation (Figure 8). Through these functions, SOCS proteins may modulate disease-related inflammation in the CNS, particularly leukocyte migration.
In addition to inhibiting chemokine expression, SOCS proteins, particularly SOCS-3, inhibit chemokine-mediated chemotaxis in a variety of cell types, including T-cells (75). This is due to the ability of SOCS-3 to bind to chemokine receptors, which then attenuates the chemotactic response. Thus, it will be of interest to determine whether the induction of SOCS-3 in glial cells (astrocytes, microglia) attenuates chemokine-mediated chemotaxis of these cells. Furthermore, this is an illustration of signaling cross-talk between cytokine and chemokine responses that impact a multiple of neuroinflammatory responses (7).
Polizzotto et al., (76) examined the temporal expression of SOCS-1 and SOCS-3 mRNA in the developing and adult nervous system, and observed maximal expression from embryonic day 14 to postnatal day 8, which declined thereafter. During a variety of CNS disease states, both SOCS-1 and SOCS-3 are expressed. In the EAE model, SOCS-1 and SOCS-3 mRNA expression increased significantly in the cerebellum and spinal cord at the peak of disease activity, and then declined (77). In a comparison of two models of EAE: SJL mice with relapsing-remitting EAE, and B6 mice with chronic EAE without complete remission, both SOCS-1 and SOCS-3 mRNA were elevated during active disease in both strains, although the B6 mice expressed less SOCS-3 (30). The failure of B6 mice to completely recover may be due to the lower levels of SOCS-3. Monocytes from relapsing remitting (RR) MS patients constitutively express higher levels of activated STAT-3 than cells from MS patients in remission. This was associated with decreased levels of SOCS-3 in RR patients compared to those in remission (31), suggesting an association of decreased SOCS-3 expression with MS relapses. In this regard, a recent study demonstrated that Simvastatin increased SOCS-3 expression in monocytes from RR MS patients, which was associated with decreased STAT-3 phosphorylation, and inhibition of IL-6 and IL-23 expression (78). Thus, the induction of SOCS-3 expression by statins may be of therapeutic efficacy in MS. SOCS-1 exerts a beneficial effect in the EAE model (32). Treatment with a mimetic of SOCS-1 (TKip) inhibited STAT-1 activation, and had a protective effect on EAE disease. This was associated with lower Myelin Basic Protein (MBP) antibody titers, suppression of MBP-induced splenocyte proliferation, and reduction of TNF-α production. Therefore, SOCS-1 and SOCS-3 proteins are possible therapeutic targets for ameliorating pathological inflammation within the CNS.
Cell-type specific expression of SOCS-1 or SOCS-3 reveals complexity in SOCS function. Targeted expression of SOCS-1 in developing oligodendrocytes protects against the detrimental effects of IFN-γ (79). In a transgenic mouse line, PLP/SOCS-1, in which SOCS-1 is expressed in oligodendrocytes, EAE development occurs with an accelerated onset associated with early inflammation and increased oligodendrocyte apoptosis (80). In this model, IFN-γ exerts a protective effect on mature oligodendrocytes, which is abrogated upon SOCS-1 expression. Thus, SOCS-1 has differing effects on oligodendrocytes, dependent on their state of maturation. SOCS-1 inhibits inflammatory responses in macrophages/microglia (81, 82), and as shown in this study, inhibits chemokine expression in astrocytes. SOCS-3 also has beneficial and detrimental functions in cells of the CNS. Conditional deletion of SOCS-3 in astrocytes, in the context of a spinal cord injury, enhanced migration of astrocytes and formation of glial scars (83). This resulted in less infiltration of inflammatory cells, sparing of myelin and oligodendrocytes, and improved functional recovery (83). These results imply that SOCS-3 represses the ability of reactive astrocytes to promote healing after spinal cord injury. Mice with conditional deletion of SOCS-3 in oligodendrocytes are protected against cuprizone-induced oligodendrocyte loss (84). SOCS-3 deletion allows LIF to inhibit myelin loss, thus modulating SOCS-3 levels may serve as a therapeutic strategy for demyelinating diseases. However, in the context of stroke, SOCS-3 has a neuroprotective effect, as antisense knockdown of SOCS-3 increased the severity of stroke symptoms (85). Previous findings from our laboratory indicated that SOCS-3 attenuates inflammatory events in macrophages/microglia (35), and this study revealed that SOCS-3 in astrocytes exerts an inhibitory effect on chemokine expression. Because both SOCS-1 and SOCS-3 modify responses in microglia, astrocytes and oligodendrocytes, understanding how SOCS expression is regulated in different CNS cell types will be clinically important. Studies to manipulate levels of SOCS expression in the CNS will aid in developing therapeutic strategies against neuroinflammatory diseases.
Acknowledgments
We thank Dr. Casey T. Weaver and James Oliver (University of Alabama at Birmingham, Birmingham, AL) for providing the STAT-1α deficient mice. We also thank Dr. Rebecca Goldstein for editorial assistance.
This work was supported by NIH grants NS45290 and NS36765 to E.N.B., a National Multiple Sclerosis Society (NMSS) grant RG 3892-A-12 to E.N.B. and H.Q., and a NMSS pilot grant PP1475 to H.Q.. B.J.B. is supported by NIH Training Grant T32-NS48039. We acknowledge the support of the University of Alabama at Birmingham Flow Cytometry Core Facility (AM-20614).
References
- 1.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 2.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong Y, Benveniste EN. Immune function of astrocytes. GLIA. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 4.Rostene W, Kitabgi P, Parsadaniantz SM. Chemokines: a new class of neuromodulator? Nat Rev Neurosci. 2007;8:895–903. doi: 10.1038/nrn2255. [DOI] [PubMed] [Google Scholar]
- 5.Williams A, Piaton G, Lubetzki C. Astrocytes-friends or foes in multiple sclerosis? Glia. 2007;55:1300–1312. doi: 10.1002/glia.20546. [DOI] [PubMed] [Google Scholar]
- 6.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 7.Rebenko-Moll NM, Liu L, Cardona A, Ransohoff RM. Chemokines, mononuclear cells and the nervous system: heaven (or hell) is in the details. Curr Opin Immunol. 2006;18:683–689. doi: 10.1016/j.coi.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 9.Benveniste EN, Qin H. Type I interferons as anti-inflammatory mediators. Sci STKE. 2007;2007:70–73. doi: 10.1126/stke.4162007pe70. [DOI] [PubMed] [Google Scholar]
- 10.Noppert SJ, Fitzgerald KA, Hertzog PJ. The role of type I interferons in TLR responses. Immunol Cell Biol. 2007;85:446–457. doi: 10.1038/sj.icb.7100099. [DOI] [PubMed] [Google Scholar]
- 11.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 12.Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T, Holmdahl R, Issazadeh-Navikas S. IFN-β gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol. 2003;170:4776–4784. doi: 10.4049/jimmunol.170.9.4776. [DOI] [PubMed] [Google Scholar]
- 13.Teige I, Liu Y, Issazadeh-Navikas S. IFN-β inhibits T cell activation capacity of central nervous system APCs. J Immunol. 2006;177:3542–3553. doi: 10.4049/jimmunol.177.6.3542. [DOI] [PubMed] [Google Scholar]
- 14.Floris S, Ruuls SR, Wierinckx A, van der Pol SMA, Dopp E, van der Meide PH, Dijkstra CD, De Vries HE. Interferon-β directly influences monocyte infiltration into the central nervous system. J Neuroimmunol. 2002;127:69–79. doi: 10.1016/s0165-5728(02)00098-x. [DOI] [PubMed] [Google Scholar]
- 15.Okada K, Kuroda E, Yoshida Y, Yamashita U, Suzumura A, Tsuji S. Effects of interferon-β on the cytokine production of astrocytes. J Neuroimmunol. 2005;159:48–54. doi: 10.1016/j.jneuroim.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F, Coccia EM. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol. 2005;64:706–715. doi: 10.1097/01.jnen.0000173893.01929.fc. [DOI] [PubMed] [Google Scholar]
- 17.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 18.Belmadani A, Tran PB, Ren D, Miller RJ. Chemokines regulate the migration of neural progenitors to sites of neuroinflammation. J Neurosci. 2006;26:3182–3191. doi: 10.1523/JNEUROSCI.0156-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sorensen TL. Targeting the chemokine receptor CXCR3 and its ligand CXCL10 in the central nervous system: potential therapy for inflammatory demyelinating disease? Curr Neurovasc Res. 2004;1:183–190. doi: 10.2174/1567202043480143. [DOI] [PubMed] [Google Scholar]
- 20.Rivieccio MA, John GR, Song X, Suh HS, Zhao Y, Lee SC, Brosnan CF. The cytokine IL-1{beta} activates IFN response factor 3 in human fetal astrocytes in culture. J Immunol. 2005;174:3719–3726. doi: 10.4049/jimmunol.174.6.3719. [DOI] [PubMed] [Google Scholar]
- 21.Kim MO, Suh HS, Brosnan CF, Lee SC. Regulation of RANTES/CCL5 expression in human astrocytes by interleukin-1 and interferon-β. J Neurochem. 2004;90:297–308. doi: 10.1111/j.1471-4159.2004.02487.x. [DOI] [PubMed] [Google Scholar]
- 22.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 23.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 24.Campbell IL. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res Brain Res Rev. 2005;48:166–177. doi: 10.1016/j.brainresrev.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 25.Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- 26.Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 27.Wong PK, Egan PJ, Croker BA, O’Donnell K, Sims NA, Drake S, Kiu H, McManus EJ, Alexander WS, Roberts AW, Wicks IP. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest. 2006;116:1571–1581. doi: 10.1172/JCI25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, Nagata K, Yoshimura A. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest. 2001;108:1781–1788. doi: 10.1172/JCI13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med. 2005;11:892–898. doi: 10.1038/nm1269. [DOI] [PubMed] [Google Scholar]
- 30.Stark JL, Cross AH. Differential expression of suppressors of cytokine signaling-1 and -3 and related cytokines in central nervous system during remitting versus non-remitting forms of experimental autoimmune encephalomyelitis. Int Immunol. 2006;18:347–353. doi: 10.1093/intimm/dxh373. [DOI] [PubMed] [Google Scholar]
- 31.Frisullo G, Mirabella M, Angelucci F, Caggiula M, Morosetti R, Sancricca C, Patanella AK, Nociti V, Iorio R, Bianco A, Tomassini V, Pozzilli C, Tonali PA, Matarese G, Batocchi AP. The effect of disease activity on leptin, leptin receptor and suppressor of cytokine signalling-3 expression in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2007;192:174–183. doi: 10.1016/j.jneuroim.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Mujtaba MG, Flowers LO, Patel CB, Patel RA, Haider MI, Johnson HM. Treatment of mice with the suppressor of cytokine signaling-1 mimetic Peptide, tyrosine kinase inhibitor Peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J Immunol. 2005;175:5077–5086. doi: 10.4049/jimmunol.175.8.5077. [DOI] [PubMed] [Google Scholar]
- 33.Qin H, Wilson C, Lee SJ, Benveniste EN. IFN-β induced SOCS-1 negatively regulates CD40 gene expression in macrophages and microglia. FASEB J. 2006;20:985–987. doi: 10.1096/fj.05-5493fje. [DOI] [PubMed] [Google Scholar]
- 34.Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-Induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–5976. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- 35.Qin H, Wilson C, Roberts K, Baker B, Zhao X, Benveniste E. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006;177:7761–7771. doi: 10.4049/jimmunol.177.11.7761. [DOI] [PubMed] [Google Scholar]
- 36.Dong Y, Rohn WM, Benveniste EN. IFN-γ regulation of the type IV class II transactivator promoter in astrocytes. J Immunol. 1999;162:4731–4739. [PubMed] [Google Scholar]
- 37.Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 38.Sakata D, Taniguchi H, Yasuda S, Adachi-Morishima A, Hamazaki Y, Nakayama R, Miki T, Minato N, Narumiya S. Impaired T lymphocyte trafficking in mice deficient in an actin-nucleating protein, mDia1. J Exp Med. 2007;204:2031–2038. doi: 10.1084/jem.20062647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang GX, Baker CM, Kolson DL, Rostami AM. Chemokines and chemokine receptors in the pathogenesis of multiple sclerosis. Mult Scler. 2000;6:3–13. doi: 10.1177/135245850000600103. [DOI] [PubMed] [Google Scholar]
- 40.Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4:590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Y, Ivashkiv LB. Costimulation of chemokine receptor signaling by matrix metalloproteinase-9 mediates enhanced migration of IFN-α dendritic cells. J Immunol. 2006;176:6022–6033. doi: 10.4049/jimmunol.176.10.6022. [DOI] [PubMed] [Google Scholar]
- 42.Weinstock-Guttman B, Ransohoff RM, Kinkel RP, Rudick RA. The interferons: biological effects, mechanisms of action, and use in multiple sclerosis. Ann Neurol. 1995;37:7–13. doi: 10.1002/ana.410370105. [DOI] [PubMed] [Google Scholar]
- 43.Dhib-Jalbut S. Mechanisms of interferon β action in multiple sclerosis. Multiple Sclerosis. 1997;3:397–401. doi: 10.1177/135245859700300609. [DOI] [PubMed] [Google Scholar]
- 44.Hohlfeld R, Wekerle H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14599–14606. doi: 10.1073/pnas.0404874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veldhuis WB, Floris S, van der Meide PH, Vos IM, de Vries HE, Dijkstra CD, Bar PR, Nicolay K. Interferon-β prevents cytokine-induced neutrophil infiltration and attenuates blood-brain barrier disruption. J Cereb Blood Flow Metab. 2003;23:1060–1069. doi: 10.1097/01.WCB.0000080701.47016.24. [DOI] [PubMed] [Google Scholar]
- 46.Lu HT, Riley JL, Babcock GT, Huston M, Stark GR, Boss JM, Ransohoff RM. Interferon (IFN) β acts downstream of IFN-γ-induced class II transactivator messenger RNA accumulation to block major histocompatibility complex class II gene expression and requires the 48-kD DNA-binding protein, ISGF3-γ. J Exp Med. 1995;182:1517–1525. doi: 10.1084/jem.182.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Chen M, Wandinger KP, Williams G, Dhib-Jalbut S. IFN-β–1b inhibits IL-12 production in peripheral blood mononuclear cells in an IL-10-dependent mechanism: relevance to IFN-β–1b therapeutic effects in multiple sclerosis. J Immunol. 2000;165:548–557. doi: 10.4049/jimmunol.165.1.548. [DOI] [PubMed] [Google Scholar]
- 48.Comabella M, Imitola J, Weiner HL, Khoury SJ. Interferon-β treatment alters peripheral blood monocytes chemokine production in MS patients. J Neuroimmunol. 2002;126:205–212. doi: 10.1016/s0165-5728(02)00064-4. [DOI] [PubMed] [Google Scholar]
- 49.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (α/β) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 50.Baker BJ, Qin H, Benveniste EN. Regulation of SOCS-3 gene expression in astrocytes by Oncostatin M. Glia. 2008 doi: 10.1002/glia.20694. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stark JL, Lyons JA, Cross AH. Interferon-γ produced by encephalitogenic cells induces suppressors of cytokine signaling in primary murine astrocytes. J Neuroimmunol. 2004;151:195–200. doi: 10.1016/j.jneuroim.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 52.Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J Biol Chem. 2003;278:14747–14752. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- 53.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TWH, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 54.Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, Alexander WS. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. 1998;95:14395–14399. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, Saito H, Nagasawa T, Uchiyama Y, Kishimoto T. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc Natl Acad Sci USA. 1998;95:15577–17782. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diehl S, Anguita J, Hoffmeyer A, Zapton T, Ihle JN, Fikrig E, Rincon M. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity. 2000;13:805–815. doi: 10.1016/s1074-7613(00)00078-9. [DOI] [PubMed] [Google Scholar]
- 57.Kawazoe Y, Naka T, Fujimoto M, Kohzaki H, Morita Y, Narazaki M, Okumura K, Saitoh H, Nakagawa R, Uchiyama Y, Akira S, Kishimoto T. Signal transducer and activation of transcription (STAT)-induced STAT inhibitor 1 (SSI-1)/suppressor of cytokine signaling 1 (SOCS1) inhibits insulin signal transduction pathway through modulating insulin receptor substrate 1 (IRS-1) phosphorylation. J Exp Med. 2001;193:263–269. doi: 10.1084/jem.193.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 59.Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ. JAB/SOCS1/SSI-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood. 2001;97:221–226. doi: 10.1182/blood.v97.1.221. [DOI] [PubMed] [Google Scholar]
- 60.Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, Metcalf D, Hilton DJ, Alexander WS. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci USA. 2001;98:9324–9329. doi: 10.1073/pnas.161271798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 62.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 63.Canfield S, Lee Y, Schroder A, Rothman P. Cutting Edge: IL-4 induces suppressor of cytokine signaling-3 expression in B Cells by a mechanism dependent on activation of p38 MAPK. J Immunol. 2005;174:2494–2498. doi: 10.4049/jimmunol.174.5.2494. [DOI] [PubMed] [Google Scholar]
- 64.Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: Characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci USA. 1999;96:6964–6969. doi: 10.1073/pnas.96.12.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emery B, Merson TD, Snell C, Young KM, Ernst M, Kilpatrick TJ. SOCS3 negatively regulates LIF signaling in neural precursor cells. Mol Cell Neurosci. 2006;31:739–747. doi: 10.1016/j.mcn.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 66.Yadav A, Kalita A, Dhillon S, Banerjee K. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem. 2005;280:31830–31840. doi: 10.1074/jbc.M501316200. [DOI] [PubMed] [Google Scholar]
- 67.Owaki T, Asakawa M, Kamiya S, Takeda K, Fukai F, Mizuguchi J, Yoshimoto T. IL-27 suppresses CD28-medicated IL-2 production through suppressor of cytokine signaling 3. J Immunol. 2006;176:2773–2780. doi: 10.4049/jimmunol.176.5.2773. [DOI] [PubMed] [Google Scholar]
- 68.Sands WA, Woolson HD, Milne GR, Rutherford C, Palmer TM. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol Cell Biol. 2006;26:6333–6346. doi: 10.1128/MCB.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strengell M, Lehtonen A, Matikainen S, Julkunen I. IL-21 enhances SOCS gene expression and inhibits LPS-induced cytokine production in human monocyte-derived dendritic cells. J Leukoc Biol. 2006;79:279–285. doi: 10.1189/jlb.0905503. [DOI] [PubMed] [Google Scholar]
- 70.Adams TE, Hansen JA, Starr R, Nicola NA, Hilton DJ, Billestrup N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J Biol Chem. 1998;273:1285–1287. doi: 10.1074/jbc.273.3.1285. [DOI] [PubMed] [Google Scholar]
- 71.Jegalian AG, Wu H. Regulation of Socs gene expression by the proto-oncoprotein GFI-1B: two routes for STAT5 target gene induction by erythropoietin. J Biol Chem. 2002;277:2345–2352. doi: 10.1074/jbc.M105575200. [DOI] [PubMed] [Google Scholar]
- 72.Hayashi T, Kaneda T, Toyama Y, Kumegawa M, Hakeda Y. Regulation of receptor activator of NF-κB ligand-induced osteoclastogenesis by endogenous interferon-β (INF-β) and suppressors of cytokine signaling (SOCS) J Biol Chem. 2002;277:27880–27886. doi: 10.1074/jbc.M203836200. [DOI] [PubMed] [Google Scholar]
- 73.Bellezza I, Neuwirt H, Nemes C, Cavarretta IT, Puhr M, Steiner H, Minelli A, Bartsch G, Offner F, Hobisch A, Doppler W, Culig Z. Suppressor of cytokine signaling-3 antagonizes cAMP effects on proliferation and apoptosis and is expressed in human prostate cancer. Am J Pathol. 2006;169:2199–2208. doi: 10.2353/ajpath.2006.060171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cacalano NA, Sanden D, Johnston JA. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 2001;3:460–465. doi: 10.1038/35074525. [DOI] [PubMed] [Google Scholar]
- 75.Soriano SF, Hernanz-Falcon P, Rodriguez-Frade JM, De Ana AM, Garzon R, Carvalho-Pinto C, Vila-Coro AJ, Zaballos A, Balomenos D, Martinez-A C, Mellado M. Functional inactivation of CXC chemokine receptor 4-mediated responses through SOCS3 up-regulation. J Exp Med. 2002;196:311–321. doi: 10.1084/jem.20012041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Polizzotto MN, Bartlett PF, Turnley AM. Expression of “suppressor of cytokine signalling: (SOCS) genes in the developing and adult mouse nervous system. J Comp Neuorol. 2000;423:348–358. [PubMed] [Google Scholar]
- 77.Maier J, Kincaid C, Pagenstecher A, Campbell IL. Regulation of signal transducer and activator of transcription and suppressor of cytokine-signaling gene expression in the brain of mice with astrocyte-targeted production of interleukin-12 or experimental autoimmune encephalomyelitis. Am J Pathol. 2002;160:271–288. doi: 10.1016/S0002-9440(10)64371-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Jin J, Peng X, Ramgolam VS, Markovic-Plese S. Simvastatin Inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol. 2008;180:6988–6996. doi: 10.4049/jimmunol.180.10.6988. [DOI] [PubMed] [Google Scholar]
- 79.Balabanov R, Strand K, Kemper A, Lee JY, Popko B. Suppressor of cytokine signaling 1 expression protects oligodendrocytes from the deleterious effects of interferon-γ. J Neurosci. 2006;26:5143–5152. doi: 10.1523/JNEUROSCI.0737-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller S, Popko B. Interferon-γ-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci. 2007;27:2013–2024. doi: 10.1523/JNEUROSCI.4689-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.O’Keefe GM, V, Nguyen T, Tang LP, Benveniste EN. IFN-γ regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J Immunol. 2001;166:2260–2269. doi: 10.4049/jimmunol.166.4.2260. [DOI] [PubMed] [Google Scholar]
- 82.Wesemann D, Dong Y, O’Keefe GM, Nguyen VT, Benveniste EN. Suppressor of cytokine signaling 1 inhibits cytokine induction of CD40 expression in macrophages. J Immunol. 2002;169:2354–2360. doi: 10.4049/jimmunol.169.5.2354. [DOI] [PubMed] [Google Scholar]
- 83.Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 84.Emery B, Cate HS, Marriott M, Merson T, Binder MD, Snell C, Soo PY, Murray S, Croker B, Zhang JG, Alexander WS, Cooper H, Butzkueven H, Kilpatrick TJ. Suppressor of cytokine signaling 3 limits protection of leukemia inhibitory factor receptor signaling against central demyelination. Proc Natl Acad Sci USA. 2006;103:7859–7864. doi: 10.1073/pnas.0602574103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carmichael ST. Gene expression changes after focal stroke, traumatic brain and spinal cord injuries. Curr Opin Neurol. 2003;16:699–704. doi: 10.1097/01.wco.0000102621.38669.77. [DOI] [PubMed] [Google Scholar]