

Abstract
The synthesis of prodrugs targeted to the SH2 domain of Stat3 is reported. Using a convergent strategy, the pivaloyloxymethyl phosphonodiester of pentachlorophenyl 4-phosphonodifluoromethylcinnamate, a phosphotyrosine surrogate, was synthesized and used to acylate peptidomimetic fragments that were prepared on solid supports. Two prodrugs described here inhibited the phosphorylation of Stat3 in breast tumor cells.
Signal transducer and activator of transcription 3 (Stat3) is a cytosolic transcription factor that has elevated, constitutive activity in several cancer types.1,2 Via its SH2 domain, Stat3 is recruited to phosphotyrosine residues on activated growth factor or cytokine receptors and is then phosphorylated on tyrosine 705. Phosphorylated Stat3 (pStat3) dimerizes by reciprocal interactions between phosphotyrosine 705 and SH2 domains and the dimer is translocated to the nucleus where it binds to promoters and initiates transcription of genes involved in unchecked cell division, invasion, and angiogenesis, characteristic of cancer.1,2 Inhibitors targeting the SH2 domain would split dimers and would prevent receptor docking, thereby preventing phosphorylation on Tyr705 and subsequent gene expression.
SH2 domains recognize phosphotyrosine residues and are integral components of intracellular signaling complexes. The development of inhibitors targeting these domains is complicated because the negatively charged phosphate impedes crossing of cell membranes. In addition, phosphate esters of tyrosine are susceptible to cleavage by phosphatases thereby destroying the ability of the inhibitor to bind to the SH2 domain.3 Phosphonates, difluoromethylphosphonates, malonates, and heterocycles have been employed as phosphate surrogates to overcome this deficit.4,5 Although a large number of inhibitors of the SH2 domains of Grb2, Src, Lck, p85PI3K, and others have been reported, relatively few have demonstrated ability to cross cell membranes and inhibit their targets.3 In several examples, passive diffusion into cells was accomplished by capping phosphate or phosphonate oxygen atoms with enzyme-cleavable groups. S-acyl-2-thioethyl (SATE),6,7 amino acid phosphoramidate,8,9 nitrofurfuryl-phosphoramidate,10 and pivaloyloxymethyl (POM)11,12,13 prodrug approaches have been reported.
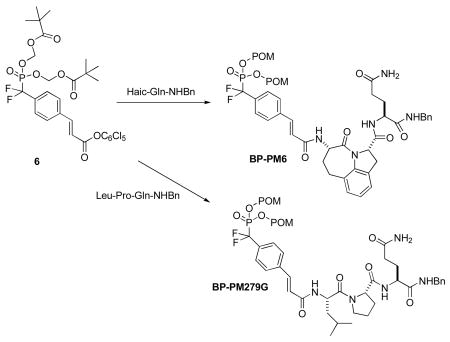
With the use of conformational constraints, we have converted our lead peptide inhibitor of Stat3, Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2, into high affinity peptidomimetics.14,15 Of these, pCinn-Haic-Gln-NHBn (PM-6) and pCinn-Leu-Pro-Gln-NHBn (PM-66F) (Figure 1) exhibited IC50 values of 162 and 138 nM, respectively, in a fluorescence polarization assay, as compared to 290 nM for the starting peptide (pCinn = 4-phosphoryloxycinnamate, Haic = 5-amino-1,2,4,5,6,7-hexahydro-4-oxo-azepino[3,2,1-hi]indole-2-carboxylic acid). Key to increased affinity was replacement of phosphotyrosine with pCinn. To stabilize against phosphatase inactivation, the phosphate group was replaced with the phosphonodifluoromethyl group16 (F2PmCinn). To impart cell permeability the POM prodrug method was employed. This approach required the development of a POM phosphonodiester of F2PmCinn. We report here the synthesis of a F2Pm(POM2)Cinn building block and its use in the preparation of cell-permeable prodrug analogues of PM-6 (BP-PM6) and PM-66F (BP-PM279G) (Fig 1). We demonstrate that they can indeed enter cells and inhibit the phosphorylation of Stat3.
Figure 1.

Structure of Stat3 inhibitors and their corresponding phosphatase-stable, cell-permeable prodrugs
Stankovic et al.11 synthesized POM prodrugs of phosphonodifluoromethylphenylalanine-containing inhibitors of the Src SH2 domain. It was reported that only one POM group could be installed on their peptidomimetics. For inhibitors of the SH2 domains of Stat4 and Stat6, McKinney et al. also employed F2Pm(POM2)Cinn.12,13 Their synthesis of this unit employed Arbuzov phosphonylation of 4-iodobenzoate followed by DAST treatment to install a diethyl phosphonodifluoromethyl group and subsequent palladium-mediated cross coupling to add the acryloyl group. After deprotection and coupling to a peptide mimetic, POM groups were installed using silver salts of the phosphonate and esterification with pivaloyloxymethyl iodide (POM-I) in toluene. It is unclear how successful this approach was as no yield data or biological testing were reported for the final prodrug.
BP-PM6 was used as the target to develop synthesis methodology. The first attempts involved esterification of the phosphonate oxygens of PM6 incorporating F2PmCinn. F2PmCinn was prepared as an active pentachlorophenyl carboxyester (5) for use in solid phase synthesis, as shown in Scheme 1. Starting with iodobenzaldehyde, Horner-Emmons coupling with tert-butyl (diethylphosphono)acetate gave the iodocinnamate, 2. Copper-cadmium cross coupling of 2 with diethyl bromodifluoromethyl-phosphonate17 provided the diethyl phosphonate 3. Since phenolic esters are stable to trimethylsilyliodide (TMSI)-mediated dealkylation of phosphate and phosphonate esters.,18 we employed the pentachlorophenyl ester to serve as both protection and activation of the carboxyl group. The tert-butyl ester of 3 was deprotected with TFA and the carboxyl group was esterified with pentachlorophenol and DCC to give intermediate 4. As expected, the pentachlorophenyl carboxy ester moiety was stable to TMSI, which was used to cleave the ethyl protecting groups to yield 5.19
Scheme 1.

Synthesis of F2PmCinn-OPcp (5) and F2Pm(POM2)Cinn-OPcp (6)
To synthesize the starting mimetic, F2PmCinn-Haic-Gln(R)-NHBn, Fmoc-Glu-NHBn20 was coupled to Rink amide resin via the side chain using diisopropylcarbodiimide and 1-hydroxybenzotriazole (HOBt) (R = Resin). After removal of the Fmoc group, Fmoc-Haic-OH was coupled in the same manner. Tyrosine mimic 5 was then coupled in the presence of HOBt and diisopropylethylamine (DIEA). Note that both Fmoc-Haic and 5 were coupled in 1.5 fold excess, which often required overnight reaction.
The first attempt to prepare BP-PM6 involved POM ester formation on the solid support. F2PmCinn-Haic-Gln(R)-NHBn was treated with excess POM-I in presence of DIEA in DMF for 24 h. After cleavage from the resin, the product was a mixture of the mono-POM ester (MP-PM6), the unprotected phosphonate (NP-PM6), and none of the POM diester. In a second approach, F2PmCinn-Haic-Gln-NHBn was cleaved from the resin, purified by HPLC to >98% purity, and subsequently treated with POM-I/DIEA in DMF for 24 h at room temperature. The product of this reaction was also a mixture of unprotected phosphonate and mono-POM ester. The di-silver salt of of F2PmCinn-Haic-Gln-NHBn was nearly insoluble in toluene, the optimum solvent for this reaction,21 precluding this method of POM esterification.
These unsatisfactory results led to the development of a preformed POM diester of F2PmCinn-OPcp (6, Scheme 1) that can be coupled to peptides or peptidomimetics in solution. To install the POM groups, the phosphonate oxygens of 5 were neutralized with two equivalents of NaOH and the sodium salt was treated with AgNO3 to give an insoluble silver salt. After thorough drying and pulverizing, the phosphonate oxygens were alkylated with three equivalents of pivaloyloxymethyl iodide in toluene (rt, 3 h) to give the prodrug building block 6.
To synthesize BP-PM6, H-Haic-Gln-NHBn, 7, was synthesized on Rink Resin, cleaved with trifluoroacetic acid:triisopropylsilane:H2O (TFA:TIS:H2O)22 and was purified by reverse phase HPLC. Active ester 6 was coupled in 1.1 fold excess to the peptide (Scheme 2). Initial experiments using HOBt as a catalyst and DIEA as the base in DMF resulted in 15-25% coupling yields, based on the amount of starting peptide, after HPLC purification. Addition of a solution of 1.1 equivalents of 6 in CH2Cl2 to the peptide substrate in N-methylpyrrolidone (NMP) in the presence of HOBt and DIEA increased the yields to 50% (6 was sparingly soluble in NMP). However, the replacement of HOBt with 0.5 equivalents of dimethylaminopyridine (DMAP) and DIEA with N-methylmorpholine (NMM) increased the yield to 72% after HPLC purification. Under these conditions, coupling was complete in ca 2 h, as judged by analytical HPLC. In these experiments, BP-PM6 was purified to >98% purity by reverse phase HPLC using gradients of MeCN in H2O. Note: addition of 0.1% TFA to the HPLC mobile phase results in premature hydrolysis of one of the POM groups.
Scheme 2.
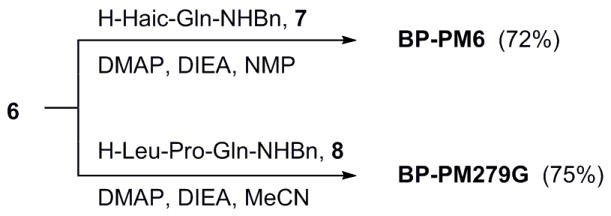
Synthesis of prodrugs
The versatility of the active ester approach was demonstrated in the synthesis of BP-PM279G (Scheme 2). As above, H-Leu-Pro-Gln-NHBn, 8, was synthesized by solid phase methods. In this case 6 (in CH2Cl2) was added to the peptide, DMAP, and NMM in MeCN. Based on starting tri-peptide, the yield of BP-PM279G was 75% after HPLC purification.
BP-PM6 and BP-PM279G were added to cultured MDA-MB-468 breast tumor cells that possess constitutively phosphorylated Stat3. Cells were treated with the inhibitors for two h, lysed, and pStat3 was detected by Western blotting. As shown in Figure 2A, both prodrugs inhibited Stat3 phosphorylation in a dose dependent manner. BP-PM6, the less peptidic prodrug, is significantly more potent than BP-PM279G, which has a higher proportion of natural amino acids. These results suggest that the prodrugs enter the cells where the POM groups are cleaved by carboxyesterases, exposing the aryl difluoromethylphosphonate for binding to the SH2 domain of Stat3. This breaks up preformed Stat3 dimers exposing the phosphotyrosine to cleavage by phosphatases and blocks the binding of Stat3 to receptors leading to reduced phosphorylation. NP-PM6 and MP-PM6 do not inhibit Stat3 phosphorylation at 25 μM (Figure 2B). Therefore two POM groups are required for efficient cell penetration and inhibitory activity.
Figure 2.
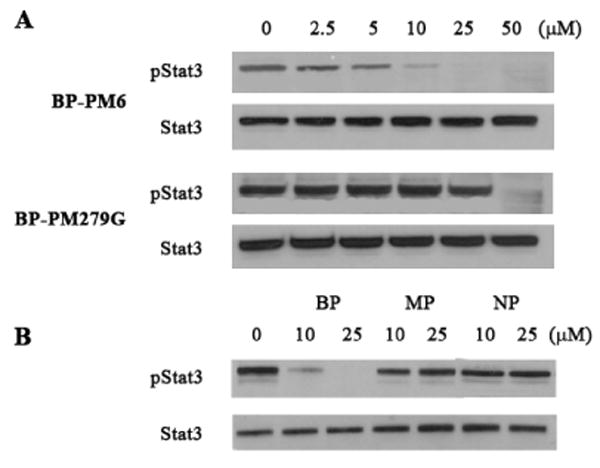
A. Inhibition of Stat3 phosphorylation in MD-MBA-468 breast tumor cells by prodrugs BP-PM6 and BP-PM279G.
B. MP-PM6 (MP) and NP-PM6 (NP) do not inhibit Stat3 phosphorylation (BP = BP-PM6).
In conclusion, we have developed a bis-POM phosphonodiester building block that can be used to synthesize prodrug inhibitors of Stat3. Evaluation of BP-PM6 and BP-PM279G indicates that the POM groups lead to cell permeability and that the compounds bind to Stat3 and prevent its phosphorylation. Full biological evaluation will be reported under separate cover.
Supplementary Material
Acknowledgments
We are grateful to the National Cancer Institute (CA096652) and the M. D. Anderson Cancer Center University Cancer Fund for support of this work. We also acknowledge the NCI Cancer Center Support Grant CA016672 for the support of our NMR facility and Chiyi Zhong of the Department of Experimental Diagnostic Imaging for Mass Spectrometry.
Footnotes
References
- 1.Darnell JE., Jr Nat Rev Cancer. 2002;2:740–9. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 2.Yu H, Jove R. Ibid. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Echeverria C. Protein Tyrosine Kinases. 2006:31–52. [Google Scholar]
- 4.Burke TR, Jr, Yao ZJ, Liu DG, Voigt J, Gao Y. Biopolymers. 2001;60:32–44. doi: 10.1002/1097-0282(2001)60:1<32::AID-BIP1002>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 5.Burke TR, Jr, Lee K. Acc Chem Res. 2003;36:426–33. doi: 10.1021/ar020127o. [DOI] [PubMed] [Google Scholar]
- 6.Liu WQ, Vidal M, Mathe C, Perigaud C, Garbay C. Bioorg Med Chem Lett. 2000;10:669–72. doi: 10.1016/s0960-894x(00)00077-9. [DOI] [PubMed] [Google Scholar]
- 7.Liu WQ, Vidal M, Olszowy C, Million E, Lenoir C, Dhotel H, Garbay C. J Med Chem. 2004;47:1223–33. doi: 10.1021/jm031005k. [DOI] [PubMed] [Google Scholar]
- 8.Gay B, Suarez S, Caravatti G, Furet P, Meyer T, Schoepfer J. Int J Cancer. 1999;83:235–241. doi: 10.1002/(sici)1097-0215(19991008)83:2<235::aid-ijc15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 9.Gay B, Suarez S, Weber C, Rahuel J, Fabbro D, Furet P, Caravatti G, Schoepfer J. J Biol Chem. 1999;274:23311–23315. doi: 10.1074/jbc.274.33.23311. [DOI] [PubMed] [Google Scholar]
- 10.Garrido-Hernandez H, Moon KD, Geahlen RL, Borch RF. J Med Chem. 2006;49:3368–76. doi: 10.1021/jm060142p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stankovic CJ, Surendran N, Lunney EA, Plummer MS, Para KS, Shahripour A, Fergus JH, Marks JS, Herrera R, Hubbell SE, Humblet C, Saltiel AR, Stewart BH, Sawyer TK. Bioorg Med Chem Lett. 1997;7:1909–1914. doi: 10.1016/s0968-0896(96)00200-3. [DOI] [PubMed] [Google Scholar]
- 12.McKinney J, Raimundo BC, Cushing TD, Yoshimura H, Ohuchi Y, Hiratate A, Fukushima H, Xu F, Peto C. Application. WO: 2001. p. 85. [Google Scholar]
- 13.McKinney J, Raimundo BC, Cushing TD, Yoshimura H, Ohuchi Y, Hiratate A, Fukushima H. Application. US: 2002. p. 31. [Google Scholar]
- 14.Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Bioorg Med Chem Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandal PK, Limbrick D, Coleman DR, Dyer GA, Ren Z, Birtwistle JS, Xiong C, Chen X, Briggs JM, McMurray JS. J Med Chem. 2009;52:2429–42. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke TR, Jr, Smyth MS, Otaka A, Nomizu M, Roller PP, Wolf G, Case R, Shoelson SE. Biochemistry. 1994;33:6490–4. doi: 10.1021/bi00187a015. [DOI] [PubMed] [Google Scholar]
- 17.Qabar MN, Urban J, Kahn M. Tetrahedron. 1997;53:11171–11178. [Google Scholar]
- 18.Blackburn GM, Ingleson D. J Chem Soc., Perk T. 1980;1:1150–1153. [Google Scholar]
- 19.Gordeev MF, Patel DV, Barker PL, Gordon EM. Tetrahedron Lett. 1994;35:7585–7588. [Google Scholar]
- 20.Coleman DRt, Ren Z, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Campbell M, Chen X, McMurray JS. J Med Chem. 2005;48:6661–70. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- 21.Farquhar D, Khan S, Srivastva DN, Saunders PP. J Med Chem. 1994;37:3902–9. doi: 10.1021/jm00049a009. [DOI] [PubMed] [Google Scholar]
- 22.Pearson DA, Blanchette M, Baker ML, Guindon CA. Tetrahedron Lett. 1989;30:2739–2742. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.