

Abstract
Cyanylated cysteine, or β-thiocyanatoalanine, is an artificial amino acid that can be introduced into peptides and proteins by post-translational chemical modification of solvent-exposed cysteine side chains, and thus it can be used in any protein with a suitable expression and mutagenesis system. In this study, cyanylated cysteine is introduced at selected sites in two model peptides that have been shown to bind to membrane interfaces: a membrane-binding sequence of the human myelin basic protein and the antimicrobial peptide CM15. Far-UV circular dichroism indicates that the secondary structures of the bound peptides are not influenced by introduction of the artificial side chain. Infrared spectra of both systems in buffer and exposed to dodecylphosphocholine micelles indicate that the CN stretching absorption band of cyanylated cysteine can clearly distinguish between membrane burial and solvent exposure of the artificial side chain. Since infrared spectroscopy can be applied in a wide variety of lipid systems, and since cyanylated cysteine can be introduced into proteins of arbitrary size via mutagenesis and post-translational modification, this new probe could see wide use in characterizing the protein−lipid interactions of membrane proteins.
The dynamic, atom-level interactions that occur between membrane-bound proteins and their native lipid systems are typically not well-characterized by experiment, despite many recent experimental advances in the isolation and structural characterization of membrane proteins. A site-specific probe of such interactions with fast time resolution could be used broadly to characterize the interplay between membrane proteins and their often heterogeneous environments. X-ray crystallography has successfully assigned the structure of many membrane proteins crystallized with help from added surfactants and mesophase-based crystallization techniques,(1) but these excellent high-resolution structures are static and limited to their artificial crystalline environments. Techniques for characterizing membrane−protein interfacial geometry and interactions include nuclear magnetic resonance (NMR), electron paramagnetic resonance site-directed spin labeling (EPR-SDSL), fluorescent probes such as the naturally occurring tryptophan side chain, and several recently popularized vibrational probes. New model lipid and surfactant systems, as well as techniques for orienting samples preferentially versus the applied field, have led to many advances in membrane protein structure determination via NMR;(2) a quite promising development is the recent extension of solid-state NMR techniques to membrane proteins bound to larger and slower-moving lipid particles than the artificial micelles and bicelles needed for conventional solution NMR experiments.(3) In particular, for short membrane-bound peptides and proteins, aligned bilayer samples spun at the magic angle have provided excellent structural resolution of antimicrobial peptide sequences in contact with lipids of varying composition, with solution exposure or burial depth left to either paramagnetic relaxation measurements or through-space magnetic interactions with magnetically active lipid nuclei.(4) However, there remains no single NMR experiment that is universally applicable to membrane proteins, with each particular system requiring a different sample preparation and usually multiple instrumental approaches.
Natural lipid systems often form very large (100s of nanometers or larger) objects with complicated and heterogeneous phase behavior: site-specific optical probes that report on variations in either the peptide or lipid structure at their interface might find more general use in determining the role that proteins play in modulating and organizing this heterogeneous phase behavior. Tryptophan fluorescence(5) has been used widely to note the extent of membrane burial of single native Trp side chains. However, in membrane-bound proteins with multiple or no tryptophans in their native sequence, the protein−membrane interface may be significantly disrupted by mutations required to add or remove Trp side chains or other artificial fluorophores. Since there are often no wavelength-dependent fluorescence spectral properties that vary with membrane burial, the main fluorescence quantity that is typically used to calculate the depth of burial is the extent of quenching,(6) which must be modulated by introducing or excluding third-party quenching agents in either aqueous or lipid phases. EPR-SDSL is also able to measure the extent of membrane burial of a covalently attached spin label, similarly by its interaction with third-party magnetically active species introduced artificially in either phase.(7) The nanosecond-time scale sensitivity of EPR spin labels is a widely used probe of site-specific protein folding dynamics;(8) however, the dynamics of the lipid interface can range over many other orders of magnitude, including the faster picosecond time scale directly accessible to vibrational spectroscopy.
Nitrile-containing artificial amino acids such as p-cyano-phenylalanine (PheCN)(9) and 5-cyano-tryptophan(10) have been shown to be quite sensitive probes of site-specific protein−lipid contacts using infrared spectroscopy. However, their use is restricted (with limited exceptions(11)) to synthetically tractable peptides containing suitable mutation sites for the larger phenylalanine and tryptophan side chains. For C≡N-containing side chains, the CN stretching peak occurs in a relatively isolated region of the IR spectrum (usually between 2100 and 2300 cm−1), so a more universally incorporable amino acid containing this vibrational chromophore might see wide use in membrane proteins.
β-Thiocyanato-alanine, or cyanylated cysteine (C*) is a C≡N-containing amino acid that can be introduced using site-selective chemical modification at cysteine, thus it could be more easily incorporated into larger and more diverse proteins than the previously mentioned artificial amino acids. The CN stretching band of aliphatic thiocyanate has a qualitatively similar, yet quantitatively quite different, response to its environment12,13 compared to the CCN-containing probes mentioned above. Although the −SCN group is apparently able to accept very weak and nonspecific hydrogen bonds at the N atom according to density functional theory (DFT) calculations,(14) solvent-dependent studies have shown that aliphatic SCN does not form well-defined 1:1 hydrogen-bonded complexes with any functional groups that naturally occur in proteins or lipids, thus making C* a relatively passive probe of its environment. Compared to PheCN, the frequency variation of C* in response to environmental changes is less dramatic, but this is balanced by its smaller size and its possible incorporation into a much wider variety of proteins via post-translational modification rather than peptide synthesis.
The incorporation of C* in proteins follows a similar strategy used in EPR-SDSL, namely, site-directed mutagenesis of a single-cysteine mutant to a protein of interest followed by covalent chemical addition of the probe moiety at the free cysteine thiol group.(15) However, in the context of membrane proteins, cyanylated cysteine has two distinct advantages over other covalently attached EPR or fluorescent probes: its remarkably small size versus other probes and its ability to report membrane exposure of specific sites without its presence of third-party quenching or paramagnetic species. This study was designed to demonstrate using two different well-characterized systems that the CN stretching band of C* is in fact sensitive to the formation of site-specific membrane contacts in a diagnostic way.
To examine the sensitivity of C* to membrane-interface binding, two model peptides with single-site mutations to C* were chosen (see Table 1 for sequences). The first peptide is amino acids 81−95 of human myelin basic protein (MBP),(16) a membrane-binding domain whose bound orientation has been previously characterized by EPR-SDSL(17) and NMR(18) studies and whose release from the membrane presents an antigenic target. MBP(16) is one example of a protein associated with disease pathogenesis that has important membrane-binding and mechanical activity. The second peptide is CM15, whose bound orientation versus bacterially derived membranes and detergent micelles has also been studied previously by EPR-SDSL and NMR techniques.(19) CM15, an artificial hybrid of the N-termini of cecropin A and melittin, is the shortest potent antimicrobial peptide sequence. It disrupts and reorganizes membranes through an unknown mechanism that begins with its binding to the membrane surface.(19) Each of these two peptides is unstructured in solution and helical when bound to lipids. In each peptide, we have chosen two centrally located residues to replace with C*, one of which is known to be solvent-exposed, and one of which is buried in the lipid interface. For best agreement with the previous studies that employed NMR and EPR techniques, the lipid system used here is dodecylphosphocholine (DPC) micelles. It is important to keep in mind, however, that future use of C* as a probe of membrane binding will not be limited to micelles or other such “artificial” lipid systems. Indeed, given its ability to avoid complexation with natural hydrogen bond donors, it is expected that C* should be useful in complex lipid systems containing hydrogen bond donors such as cholesterol and sphingolipids without strong perturbation of the lipid media by the probe and possible spectral discrimination of phase segregation by the probe’s IR spectrum. The local structure and dynamics of more complex lipid media will be the subject of future work; the purpose of the present study is to evaluate the sensitivity of C* to membrane binding in an otherwise well-understood context. Due to their short length, the peptides studied here were constructed via solid-phase peptide synthesis; but in a general sense, it is important to note that the C* probe is not in any way restricted to use in short, synthetic peptides.
Table 1. Sequences of Model Peptides, Where C* = Cyanylated Cysteine.
| CM15 | Ac-KWKLFKKIGAVLKVL-NH2 | |
| CM15 A10C* | Ac-KWKLFKKIGC*VLKVL-NH2 | C* solvent-exposed |
| CM15 I8C* | Ac-KWKLFKKC*GAVLKVL-NH2 | C* buried |
| MBP 81−95 | Ac-NPVVHFFKNIVTPRT-NH2 | |
| MBP N89C* | Ac-NPVVHFFKC*IVTPRT-NH2 | C* solvent-exposed |
| MBP F87C* | Ac-NPVVHFC*KNIVTPRT-NH2 | C* buried |
The probe CN group was introduced into the CM15 and MBP single-cysteine mutants by standard modification chemistry (see below) with near-unity yields in all cases. Since these are partially amphipathic peptides, their aqueous solubilities are limited to ∼1 mM or less. Each of these peptides is also strongly cationic at neutral pH, so substantial buffer concentrations were used to prevent the concentrated peptides from raising their own local pH outside the window in which the probe group is stable (pH 6.0−8.0 according to previous work as well as ours).
Far-UV circular dichroism (CD) was used to verify that mutation and cyanylation did not perturb the helical secondary structure of the peptides’ DPC-bound states. Figure 1 presents far-UV CD spectra in aqueous buffer solution and in the presence of 100 mM DPC for all six peptides. For each mutant peptide, substitution of C* led to no observable changes in the global secondary structure of the DPC-bound peptides as compared to the C*-free peptide. Interestingly, the aqueous structures of the peptides did change to some extent, indicating that the mutation to C* did introduce steric changes that biased the unstructured ensemble toward a less random-coil-like average structure. However, the helical propensity of each peptide in its bound form is unaltered with the addition of C*.
Figure 1.

Far-UV CD spectra of all peptides in phosphate buffer and DPC-bound conditions: (a) native-sequence MBP 81−95, (b) MBP N89C*, (c) MBP F87C*, (d) unlabeled CM15, (e) CM15 A10C*, (f) CM15 I8C*. Note: since the MBP sequence does not contain a good UV chromophore for concentration determination, scaled raw ellipticities of samples are presented, and shape comparisons are most useful.
Previous studies on the solvent dependence of aliphatic thiocyanate’s CN stretching band indicate that its frequency depends on the dipolar character and hydrogen bonding ability of the solvent.(12) The CN stretching band of methyl thiocyanate in solvents relevant to this work (most of which were not included in the original solvent study) is shown in Figure 2. The probe vibration exhibits a red shift of 4−5 cm−1 versus its aqueous position in less-hydrogen bonding solvents with the exception of alkanes, which are not able to stabilize the charge distribution of the non-H-bonded −SCN moiety. The CN line width narrows considerably in tetrahydrofuran (THF) and alkane solvents compared to other more dipolar solvents, likely due to a smaller inhomogeneous frequency distribution imposed by the dipoles of surrounding solvent molecules. If THF is the best solvent model of the membrane interface (largely alkane but with substantial dipole variations), then the expectation is that a C* side chain’s stretching vibration should red-shift by 4−5 cm−1 when buried in the membrane interface versus its frequency when solvent-exposed. The band might also narrow due to dipolar and/or dynamical differences between aqueous and lipid environments. The bandwidth of this particular vibration was shown previously to be particularly sensitive to picosecond time scale solvent dynamics.(12)
Figure 2.

Solvent dependence of the CN stretching infrared absorption band of methyl thiocyanate in solvents relevant to membrane binding studies.
The CN stretching band of each cyanylated peptide was recorded under the same aqueous and DPC-exposed conditions as the CD spectra. Peptide concentrations were limited by solubility for the aqueous samples and by maintaining a suitably large lipid:peptide ratio for the DPC samples, and since the CN stretching band of SCN is weaker than for CCN, the approximate optical density of the CN band is approximately 100 μOD for each cyanylated sample. Figure 3 compares the infrared spectrum in the CN stretching region for each peptide in aqueous buffer versus DPC micelle solutions.
Figure 3.

CN stretching absorption bands for cyanylated peptides in aqueous solution and DPC micelles: (a) MBP N89C*, (b) MBP F87C*, (c) CM15 A10C*, (d) CM15 I8C*. Intensities are scaled to the peak intensity for comparison; all optical densities are between 200 and 400 μOD.
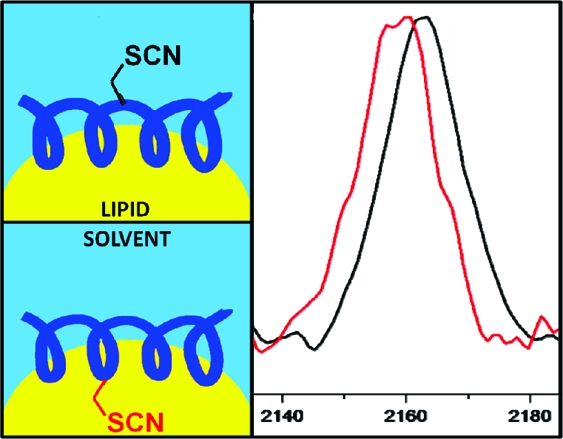
CN stretching bands for the MBP mutants (Figure 3a,b) show that there is a distinct difference between the membrane-buried versus the solvent-exposed C*. As shown in Figure 3a, there are no significant changes to the line width or central peak frequency between the solvent-exposed N89C* probe in buffer and that in DPC micelles. However, the membrane-buried F87C*, shown in Figure 2d, exhibits a red shift of ∼4 cm−1 (from 2163 to 2159 cm−1) as well as a substantial narrowing in the presence of the lipid.
For CM15, in Figure 3c, there is no significant change in the line width or central peak frequency between the solvent-exposed A10C* probe in buffer versus DPC. However, the CN stretching band of the membrane-buried I8C* probe redshifts by ∼4 cm−1 with DPC (Figure 3d) from 2163 to 2159 cm−1.
The observed CN stretching red shift of about 4 cm−1 for the presumably micelle-buried probe groups is in quantitative agreement with a THF-like environment when compared to the spectra of MeSCN. A recent study of C* inserted into the aqueous, water-soluble alanine peptides (AAAAK)5 observed a red shift of approximately 1.5 cm−1 upon the formation of helical secondary structure.(20) However, this red shift was from 2164.5 to 2163.0 cm−1 and accompanied a decrease in the temperature of the sample; a similar temperature-dependent shift was also observed for MeSCN over the same temperature range. In the aqueous helical peptides, formation of helical structure did correspond with a change in the observed CN stretching bandwidth. However, in the case of the membrane-bound peptides investigated here, the CN stretching band of C* is observed well outside the frequency window of any CN stretching band observed for water-exposed C*, thus we can say that the red shift is unmistakably due to burial in a nonaqueous and non-H-bonding environment.
In two different peptide systems with different neighboring side chains and different physiological effects on natural membranes, the C* probe is shown here to be sensitive to lipid burial and reports the orientation of the peptide at the membrane interface. The difference in line width between the two buried side chains (Figure 3b,d) suggests the possibility of depth and/or dynamic differences at the two peptide−micelle interfaces. This difference is interesting, given the contrasting natural roles of these two peptide sequences: MBP serves to stabilize and solidify the myelin sheath, at least partly due to lipid interactions, and CM15’s toxicity derives from its ability to permeabilize bacterial membranes. Recent detailed NMR experiments by Zangger et al.(21) suggest that CM15 is nearly completely buried in DPC micelles; this is not quite what is suggested by our results for the A10C* mutant of CM15, in which the CN band of the C* probe is nearly indistinguishable from that of A10C* in aqueous solution. This may be due to fact that the CN stretching band is sensitive simply to the presence of water in its local environment, rather than to the artificially introduced solution-phase paramagnetic species that are needed to reveal solvent exposure in NMR experiments. Solid-state NMR measurements of oriented samples that explicitly take into account membrane−peptide interactions via peptide−lipid nuclear Overhauser effects (NOEs) have recently been able to quantify membrane burial depth dependence in some antimicrobial peptides better than paramagnetic exposure.(22) It is our hope that our approach might in time also lead to clear quantitation of burial depth in membrane-bound proteins of arbitrary size.
Future experiments will address each peptide in the context of its natural lipid system, with variability in the line width and frequency of buried labels expected to report on local membrane dynamics and speciation near the peptides. The prior observation that aliphatic SCN is weakly sensitive to H-bonding groups presents the intriguing possibility that buried, covalently attached SCN groups could be used to document the presence of H-bonding physiological bilayer components such as sterols near membrane proteins. For now, these results indicate that the cyanylated cysteine side chain should be a sensitive and widely incorporable vibrational probe of membrane binding in membrane proteins.
Experimental Methods
Materials
Fmoc-labeled amino acids and all peptide synthesis reagents were purchased from Advanced Chem Tech, except for N,N-diisopropylethylamine (DIEA; Pharmco-AAPER) and acetic anhydride (Aldrich). All peptide cleavage reagents were purchased from Aldrich, and high-performance liquid chromatography (HPLC) solvents were purchased from Pharmco-AAPER. 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) was purchased from Acros and d,l-dithiothreitol (DTT) and NaCN were purchased form Aldrich. Methyl thiocyanate was purchased from Acros. All purchased items above were used as received. DPC was purchased from Avanti Polar Lipids dissolved in chloroform. All aqueous samples were constituted using doubly deionized, Milli-Q quality water.
Peptide Synthesis and Analysis
All peptides were synthesized on an Applied Biosystems API 443A synthesizer using standard fmoc chemistry with an HBTU/HOBt activation cocktail. All branched amino acids were double-coupled. The PAL resin was used to furnish a C-terminal carboxamide on cleavage; treatment of the resin bound peptides with acetic anhydride was used for N-acetylation. Purity and identity of peptides was verified by reverse-phase HPLC using an analytical-scale Dynamax C18 column and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS; performed at the Wistar Institute, Philadelphia, PA). Since all peptides were nearly (95% or greater) pure following peptide cleavage according to analytical HPLC and MALDI analysis, preparative-scale HPLC was not performed.
Cyanylation of Peptides
Lyophilized cysteine-containing peptide from cleavage was treated for 20 min in 0.01 M HCl solution and relyophilized to remove residual TFA. The peptide was weighed after lyophilization, dissolved in 50 mM phosphate buffer, pH 7.0−7.5, and treated with 100× DTT to ensure the free thiol. Residual DTT and any other small impurities were separated from the peptide using a 28 cm column of Sephadex G-10, and the reduced peptide was lyophilized. The peptide was redissolved in 50 mM phosphate buffer (CM15 peptides) or 20 mM phosphate (MBP peptides), pH 6.5−7.0, and treated with 5× DTNB dissolved in 200 mM phosphate buffer, pH 6.5−7.0, to form a mixed disulfide at cysteine. In limited cases, absorbance of the sample was monitored at 412 nm to observe release of the thionitrobenzoate anion and quantify the yield of the mixed disulfide; yield for all cyanylated peptides was near unity when determined. The DTNB−adduct sample was then treated with 50× NaCN, and the cyanylated peptide was isolated using the same 28 cm Sephadex G-10 column equilibrated with 50 mM (CM15 peptides) or 20 mM (MBP peptides) sodium phosphate buffer, pH 6.5−7.0. Peptide-containing fractions were concentrated about 5× using a Speed Vac centrifugal vacuum device, and the presence of the C≡N label was verified using infrared spectroscopy. The final concentrated sample concentration was 1−2 mM peptide in 100−200 mM phosphate buffer, pH 6.5−7.0.
Lipid-Containing Samples
DPC was dried from chloroform by evaporating the solvent under a steady stream of nitrogen. Dynamic light scattering using a DynaPro (Wyatt Technologies, Santa Barbara, CA) in a 1.5 mm quartz cuvette (Helma) indicated a uniform micelle size distribution of ∼2 nm. To obtain uniform micelle size, samples were extruded 15 times using a mini-extruder (Avanti Polar Lipids, Alabaster, AL). Speed-vac’ed peptide samples from above were mixed with the dried lipid to yield samples that were approximately 2 mM in peptide and 100 mM in DPC in a 200 mM phosphate buffer.
Far-UV CD
CD spectra were collected from 185 to 260 nm using an Aviv model 410 spectropolarimeter. Peptide samples at 1−2 mM peptide concentration (and 100−200 mM sodium phosphate buffer) were analyzed in a 0.1 mm quartz demountable cell (Starna Cells).
Infrared Spectroscopy
Cyanylated peptide samples (in buffer or 100 mM DPC) were placed between the windows of a 22 μm CaF2 BioCell (BioTools, Jupiter, FL) placed inside a BioJack temperature-circulating jacket held at 25 °C. All spectra were collected at 2 cm−1 resolution using a Bruker Optics Vertex 70 FTIR spectrometer with a photovoltaic HgCdTe detector. A spectrum of buffer solution or 100 mM DPC in buffer solution was subtracted from the raw spectrum, and further baseline correction was accomplished by fitting the baseline (outside the region from 2145 to 2185 cm−1) to a polynomial and subtracting the fit.
Acknowledgments
We gratefully acknowledge the groups of Karin Akerfeldt and Robert Fairman at Haverford College for help with peptide synthesis and circular dichroism. C.H.L. acknowledges the Camille and Henry Dreyfus Foundation for a New Faculty Start-Up Award, the Research Corporation for a Cottrell College Science Award, and the National Institute of General Medical Sciences for AREA Grant R15GM088749. K.N.A. thanks the Howard Hughes Medical Institute for a summer fellowship. This content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies above.
Funding Statement
National Institutes of Health, United States
References
- Caffrey M. Crystallizing Membrane Proteins for Structure Determination: Use of Lipidic Mesophases. Annu. Rev. Biophys. 2009, 38, 29–51. [DOI] [PubMed] [Google Scholar]
- Opella S. J.; Marassi F. M. Structure Determination of Membrane Proteins by NMR Spectroscopy. Chem. Rev. 2004, 104, 3587–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott A. Structure and Dynamics of Membrane Proteins by Magic Angle Spinning Solid-State NMR. Annu. Rev. Biophys. 2009, 38, 385–403. [DOI] [PubMed] [Google Scholar]; Opella S. J.; Zeri A. C.; Park S. H. Structure, Dynamics, and Assembly of Filamentous Bacteriophages by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Phys. Chem. 2008, 59, 635–657. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy A. Beyond NMR Spectra of Antimicrobial Peptides: Dynamical Images at Atomic Resolution and Functional Insights. Solid State Nucl. Magn. Reson. 2009, 35, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loura L. M. S.; de Almeida R. F. M.; Coutinho A.; Prieto M. Interaction of Peptides with Binary Phospholipid Membranes: Application of Fluorescence Methodologies. Chem. Phys. Lipids 2003, 122, 77–96. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay A.; London E. Parallax Method for Direct Measurement of Membrane Penetration Depth Utilizing Fluorescence Quenching by Spin-Labeled Phospholipids. Biochemistry 1987, 26 (1), 39–45. [DOI] [PubMed] [Google Scholar]
- Klug C. S.; Feix J. B.. Methods and Applications of Site-Directed Spin Labeling EPR Spectroscopy. In Biophysical Tools for Biologists: In Vitro Techniques; Elsevier Academic Press Inc: San Diego, CA, 2008; Vol. 84, pp 617−658. [DOI] [PubMed] [Google Scholar]
- Columbus L.; Hubbell W. L. A New Spin on Protein Dynamics. Trends Biochem. Sci. 2002, 27, 288–295. [DOI] [PubMed] [Google Scholar]
- Tucker M. J.; Getahun Z.; Nanda V.; DeGrado W. F.; Gai F. A New Method for Determining the Local Environment and Orientation of Individual Side Chains of Membrane-Binding Peptides. J. Am. Chem. Soc. 2004, 126, 5078–5079. [DOI] [PubMed] [Google Scholar]; Getahun Z.; Huang C. Y.; Wang T.; De Leon B.; DeGrado W. F.; Gai F. Using Nitrile-Derivatized Amino Acids As Infrared Probes of Local Environment. J. Am. Chem. Soc. 2003, 125, 405–411. [DOI] [PubMed] [Google Scholar]
- Waegele M. M.; Tucker M. J.; Gai F. 5-Cyanotryptophan As an Infrared Probe of Local Hydration Status of Proteins. Chem. Phys. Lett. 2009, 478, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz K. C.; Supekova L.; Ryu Y. H.; Xie J. M.; Perera R.; Schultz P. G. A Genetically Encoded Infrared Probe. J. Am. Chem. Soc. 2006, 128, 13984–13985. [DOI] [PubMed] [Google Scholar]; Taskent-Sezgin H.; Chung J.; Patsalo V.; Miyake-Stoner S. J.; Miller A. M.; Brewer S. H.; Mehl R. A.; Green D. F.; Raleigh D. P.; Carrico I. Interpretation of p-Cyanophenylalanine Fluorescence in Proteins in Terms of Solvent Exposure and Contribution of Side-Chain Quenchers: A Combined Fluorescence, IR and Molecular Dynamics Study. Biochemistry 2009, 48, 9040–9046. [DOI] [PubMed] [Google Scholar]
- Maienschein-Cline M. G.; Londergan C. H. The CN Stretching Band of Aliphatic Thiocyanate Is Sensitive to Solvent Dynamics and Specific Solvation. J. Phys. Chem. A 2007, 111, 10020–10025. [DOI] [PubMed] [Google Scholar]
- Oh K. I.; Choi J. H.; Lee J. H.; Han J. B.; Lee H.; Cho M. Nitrile and Thiocyanate IR Probes: Molecular Dynamics Simulation Studies. J. Chem. Phys. 2008, 128, 154504. [DOI] [PubMed] [Google Scholar]
- Choi J. H.; Oh K. I.; Lee H.; Lee C.; Cho M. Nitrile and Thiocyanate IR Probes: Quantum Chemistry Calculation Studies and Multivariate Least-Square Fitting Analysis. J. Chem. Phys. 2008, 128, 134506. [DOI] [PubMed] [Google Scholar]
- Degani Y.; Neumann H.; Patchornik A. Selective Cyanylation of Sulfhydryl Groups. J. Am. Chem. Soc. 1970, 92, 6969–6971. [DOI] [PubMed] [Google Scholar]; Degani Y.; Patchornik A. Cyanylation of Sulfhydryl groups by 2-Nitro-5-thiocyanobenzoic Acid. High-Yield Modification and Cleavage of Peptides at Cysteine Residues. Biochemistry 1974, 13, 1–11. [DOI] [PubMed] [Google Scholar]; Fafarman A. T.; Webb L. J.; Chuang J. I.; Boxer S. G. Site-Specific Conversion of Cysteine Thiols into Thiocyanate Creates an IR Probe for Electric Fields in Proteins. J. Am. Chem. Soc. 2006, 128, 13356–13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G.; Ladizhansky V.; Boggs J. M. Structural Polymorphism and Multifunctionality of Myelin Basic Protein. Biochemistry 2009, 48, 8094–8104. [DOI] [PubMed] [Google Scholar]
- Musse A. A.; Boggs J. M.; Harauz G. Deimination of Membrane-Bound Myelin Basic Protein in Multiple Sclerosis Exposes an Immunodominant Epitope. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4422–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares C.; Libich D. S.; Harauz G. Solution NMR Structure of an Immunodominant Epitope of Myelin Basic Protein - Conformational Dependence on Environment of an Intrinsically Unstructured Protein. FEBS J. 2006, 273, 601–614. [DOI] [PubMed] [Google Scholar]
- Bhargava K.; Feix J. B. Membrane Binding, Structure, and Localization of Cecropin−Mellitin Hybrid Peptides: A Site-Directed Spin-Labeling Study. Biophys. J. 2004, 86, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pistolesi S.; Pogni R.; Feix J. B. Membrane Insertion and Bilayer Perturbation by Antimicrobial Peptide CM15. Biophys. J. 2007, 93, 1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein L.; Stetz M. A.; McMahon H. A.; Londergan C. H., unpublished results.
- Zangger K.; Respondek M.; Goebl C.; Hohlweg W.; Rasmussen K.; Grampp G.; Madl T. Positioning of Micelle-Bound Peptides by Paramagnetic Relaxation Enhancements. J. Phys. Chem. B 2009, 113, 4400–4406. [DOI] [PubMed] [Google Scholar]
- Porcelli F.; Buck-Koehntop B. A.; Thennarasu S.; Ramamoorthy A.; Veglia G. Structures of the Dimeric and Monomeric Variants of Magainin Antimicrobial Peptides (MSI-78 and MSI-594) in Micelles and Bilayers, Determined by NMR Spectroscopy. Biochemistry 2006, 45, 5793–5799. [DOI] [PubMed] [Google Scholar]; Porcelli F.; Verardi R.; Shi L.; Henzler-Wildman K. A.; Ramamoorthy A.; Veglia G. NMR Structure of the Cathelicidin-Derived Human Antimicrobial Peptide LL-37 in Dodecylphosphocholine Micelles. Biochemistry 2008, 47, 5565–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]