

Abstract
High resolution melting is a new method of genotyping and variant scanning that can be seamlessly appended to PCR amplification. Limitations of genotyping by amplicon melting can be addressed by unlabeled probe or snapback primer analysis, all performed without labeled probes. High resolution melting can also be used to scan for rare sequence variants in large genes with multiple exons and is the focus of this article. With the simple addition of a heteroduplex-detecting dye before PCR, high resolution melting is performed without any additions, processing or separation steps. Heterozygous variants are identified by atypical melting curves of a different shape compared to wild type homozygotes. Homozygous or hemizygous variants are detected by prior mixing with wild type DNA. Design, optimization, and performance considerations for high resolution scanning assays are presented for rapid turnaround of gene scanning. Design concerns include primer selection and predicting melting profiles in silico. Optimization includes temperature gradient selection of the annealing temperature, random population screening for common variants, and batch preparation of primer plates with robotically deposited and dried primer pairs. Performance includes rapid DNA preparation, PCR, and scanning by high resolution melting that require, in total, only 3 hours when no variants are present. When variants are detected, they can be identified in an additional 3 hours by rapid cycle sequencing and capillary electrophoresis. For each step in the protocol, a general overview of principles is provided, followed by an in depth analysis of one example, scanning of CYBB, the gene that is mutated in X-linked chronic granulomatous disease.
Keywords: High resolution melting, heteroduplex scanning, genotyping, LCGreen Plus, LightScanner
Introduction
Melting is a fundamental property of DNA. As the double helix is heated, its strands separate. If the duplex region is short, as for most synthesized probes, melting occurs in one transition (“all” or “none”) without intermediate states and the melting temperature (Tm) is defined as the temperature at which half of the duplexes have dissociated. The Tm of short duplexes can usually be estimated to within 2°C by considering the thermodynamics of neighboring bases [1]. In contrast, longer PCR products may melt in multiple stages or “domains”, with AT-rich regions melting at lower temperatures than GC-rich regions. Typically, melting domains range from 50–500 bps [2]. When more than one domain exists the Tm is not defined and melting curve prediction requires more complex recursive calculations [3].
Thermal melting of DNA is conventionally monitored by UV absorbance. For high quality melting curves, μg amounts of DNA and rates of 0.1–1.0 °C/min are typically employed. In contrast to absorbance, monitoring DNA melting with fluorescence is more sensitive and only ngs are required, conveniently provided by PCR amplification. Fluorescent DNA melting in the context of real time PCR was introduced with the LightCycler® in 1997 [4]. Small sample volumes and enhanced heat transfer allowed much faster melting rates of 0.1–1.0°C/sec. Both probe melting for genotyping [5] and product melting with SYBR® Green I [6] are widely used on real-time instruments today.
Because DNA melting is such a simple process that requires no more than PCR and a generic DNA dye, efforts to increase its information content eventually led to high resolution melting curve analysis, sometimes abbreviated as HRM, HRMA, or HRMCA, among others. High resolution melting was made possible by progress along three fronts, dye chemistry, instrument resolution and data analysis. Although SYBR Green I can differentiate many homozygous variants that differ in Tm, e.g., large deletions [7], complex repeat regions [8] and methylation analysis [9], it is difficult to detect heteroduplexes with SYBR Green I. Most high resolution melting applications depend on saturating DNA dyes that detect heteroduplexes, a new functional class of dyes either synthesized de novo or identified from existing dyes [10]. Some saturating dyes that are available outside of commercial master mixes reveal differences in their ability to detect heteroduplexes [11]. Similarly, melting instruments vary in their resolution, as shown in a series of studies [12–14]. Finally, targeted software is necessary to identify the small melting curve differences that identify a variant or genotype [15]. The sensitivity and specificity of high resolution melting depends on the dye, instrument and software used. Like high fidelity music, compromise in one component can spoil the end result.
The two major applications of high resolution amplicon melting are targeted genotyping [16] and gene scanning [17]. Most single base variants can be genotyped by high resolution melting because many homozygotes differ in Tm. However, the homozygotes of some single base variants and many insertions and deletions have similar or identical Tms and cannot be differentiated. Specifically, the homozygous genotypes of type 3 and 4 single base variants [16], also known as “base pair neutral” variants [18], are difficult to distinguish by Tm. In the human genome, 84% of single base variants are type 1 or 2 with homozygous Tm differences around 1°C that are easy to differentiate. Type 3 and 4 variants have Tm differences around 0.25°C (12% of human single base variants), except when nearest neighbor symmetry predicts no difference at all (4%). Even under the best conditions and with the best instruments, some loci cannot be genotyped by simple amplicon melting [18–20].
Problematic loci for amplicon genotyping should be identified in silico by Tm calculations and alternative methods employed. Robust solutions can be found in related melting methods that, although not quite as simple, use the same dyes and instruments. For example, quantitative heteroduplex analysis mixes the unknown with a known genotype [21]. Alternatively, melting analysis can be performed on smaller duplexes formed with unlabeled probes [22, 23] or the hairpin formed after PCR using snapback primers [24]. Unlabeled probe and snapback primer genotyping detect all single base variants within the duplex and allow many genotypes to be distinguished [25]. Both probe genotype (using unlabeled probes or snapback primers) and amplicon scanning can be performed in the same reaction simultaneously [26]. In such cases with a broad range of analyzed temperatures, software with exponential background removal is necessary [15]. With such software, single base genotyping in up to four amplicons with two temperature controls is possible in a single color multiplex PCR [27, 28].
High resolution melting is the method of choice for gene scanning of multiple exons based on simplicity, cost, sensitivity and specificity [29, 30]. Over 60 genes have been analyzed by this method, and for one (BRCA1/2), six reports have been published [31]. The protocols detailed below are based on the most common instrument (LightScanner®) and dye (LCGreen Plus) reported for genetic scanning in the literature [31]. At each step, different procedures and/or instruments are possible with the caveat that gene scanning results strongly depend on the dye, instrument and software used. For additional information on high resolution melting applications, including gene scanning, small amplicon genotyping, unlabeled probe genotyping, snapback primer genotyping, methylation analysis, sequence matching, and repeat typing, several recent reviews are available.[11, 25, 31–36].
Description of Method
High resolution melting analysis for gene scanning is an attractive option for laboratories with time and resource constraints. Careful design and optimization enable performance of gene scanning in less than an 8 hour workday and most sequencing is eliminated by identifying normal samples and normal coding regions of variant samples. Because scanning accuracy depends on high quality PCR, optimization is critical. Optimization success often depends on design, and informed design requires knowledge of the gene to be scanned. The following outline lists steps in the design, optimization and performance of scanning assays. Web sites useful in naming and cataloging genes and variants are listed below in 1.1–1.2, but these preliminary steps are not further detailed here.
-
1. Design
-
1.1 Identify genes and coding regions of interest
1.1.1 Use HUGO gene nomenclature (http://www.genenames.org/)
1.1.2 Scan the literature for prior studies (http://www.ncbi.nlm.nih.gov/pubmed)
-
1.2 Search for known variants
1.2.1 Use HGVS variant (c.) nomenclature (http://www.hgvs.org/mutnomen/)
-
1.2.2 Database extraction
Gene or disease specific database
Human mutation database (http://www.hgmd.cf.ac.uk/ac/)
dbSNP (http://www.ncbi.nlm.nih.gov/SNP/)
Genome browser (http://genome.ucsc.edu/)
Pseudogenes and copy number variants
1.2.3 Catalog known mutations, benign variants and frequencies
1.3 Design primers
1.4 Predict melting profiles
-
-
2. Optimization
2.1 Prepare DNA
2.2 PCR optimization
2.3 Random population screening
2.4 Prepare primer plates
-
3. Performance
3.1 PCR
3.2 Scanning
3.3 Cycle sequencing
3.4 Capillary electrophoresis
1. Design
Gene scanning assays usually need to cover multiple exons and/or genes. For simultaneous analysis, all primer pairs need to be amplified under identical conditions, preferably on one plate with a layout that is straightforward and convenient. Following the above outline, the gene(s) of interest (1.1) and known variants (1.2) are first identified. Subsequent steps, beginning with primer design and melting profile prediction are detailed below.
1.3 Design primers
Primers are designed to bracket each region of interest. Usually, this includes exonic coding segments and adjacent intronic splicing regions. Based on the consensus sequence associated with splicing [37] at least 15 bases upstream from the 5′-end of the exon and 6 bases downstream from the 3′-end of the exon should be included (Fig. 1). Probable splice branching sites can also be identified 10–50 bases upstream from the 5′-end of each exon. Regulatory and/or promoter regions may be included if warranted by mutation frequency.
Fig. 1.

Consensus sequences for splicing and branch sites of human exons. The branch site is typically 20–50 bases upstream of the exon. Splice sites bracket each exon with consensus bases reaching at least 15 bases upstream and 6 bases downstream. The colored bars indicate the proportion of each nucleotide at the position given (A , C
, C , T
, T , G■). Primers should be designed to avoid consensus bases where variation is likely to effect splicing.
, G■). Primers should be designed to avoid consensus bases where variation is likely to effect splicing.
Both freely available (http://frodo.wi.mit.edu/primer3/) and commercial software are useful in primer design. Design criteria should include a similar melting temperature (Tms within 4°C of each other) for all primers. Primers with significant hairpins, homodimers, or heterodimers should be excluded. Although the optimal amplicon length and number of domains are controversial and may depend on the system, amplicons are typically kept below 400 base pairs with 3 or fewer melting domains. Large exons may require multiple amplicons for full coverage.
Detailed example
Primers are designed to amplify the 13 exons of CYBB, the gene responsible for the X-linked form of chronic granulomatous disease (OMIM 300481, accessible from http://www.ncbi.nlm.nih.gov/). The reference sequence gene of CYBB is formatted and downloaded from the UCSC Genome Browser. Then, LightScanner primer design software (Idaho Technology) is used to simultaneously design primers for multiple exons. This software automatically breaks up large exons into multiple amplicons based on a maximum amplicon size. Finally, to simplify sequencing, primers are tailed with M13 sequencing primers.
Protocol
Open the UCSC genome browser (http://genome.ucsc.edu/).
Click, “Genomes,” on the top toolbar.
Enter, “CYBB,” as the, “position or search term,” and click, “submit.”
Click on the CYBB RefSeq Gene.
In the browser window, click on the, “CYBB,” label of the RefSeq Gene track.
Click on, “Genomic Sequence,” from assembly.
-
Check boxes for only:
CDS Exons
One FASTA record per region with 200 bases on each side
CDS in upper case, UTR in lower case
Click, “Submit.”
Copy the FASTA text file into notepad.
Open LightScanner primer design software and click on, “Scanning Primers.”
Import the notepad FASTA file and convert uppercase regions to exons.
-
Open, “Common Settings,” under the, “Settings,” menu and adjust as follows:
Max Amplicon Size = 400 bps
Primer Tm between 62–66°C
Primer size between 18–35 bases
5′ exclusion buffer = 15 bases
3′ exclusion buffer = 6 bases
Minimum overlap = 5 bases
Primer concentrations = 0.5 μM
Press, “OK,” and click, “Search All.”
Inspect the primers by clicking on the tabs for each exon. Alternative primers may be selected if current ones overlap known variants.
Select, “Export to CSV,” in the, “Sequence,” menu.
Append the appropriate M13 sequencing primers onto the 5′-end of each primer to facilitate sequencing and have the primers synthesized.
The complete amplicons with M13 tails should be considered when evaluating length and melting characteristics. The primer sequences and sizes of the resulting PCR products are shown in Table 1.
Table 1.
Primers and product sizes for the 13 exon gene CYBB.
| Primer Name | Sequencea (5′-3′) | Product Size (bps) |
|---|---|---|
| Ex1F | AGAAGCATAGTATAGAAGAAAGGC | 180 |
| Ex1R | CCCGAGAAGTCAGAGAATTTATAAC | |
| Ex2F | CTACTGTGGAAATGCGGA | 214 |
| Ex2R | AGCCAATATTGCATGGGAT | |
| Ex3F | GGACAGGGCATATTCTGTG | 249 |
| Ex3R | GCCTTTGAAAATTAGAGGAACTTAGTA | |
| Ex4F | CTTTCCTGTTAACAATTACTATTCCAT | 202 |
| Ex4R | TCCCTGGTTCCAAGTTTTCTTTAATA | |
| Ex5F | TCATACCCTTCATTCTCTTTGTTT | 281 |
| Ex5R | AGTCCTCAATTGTAATGGCCTA | |
| Ex6F | TGTGTGTGTGTGTGTTTATATTTTAC | 309 |
| Ex6R | CTGCCTAGAAATTGAGGGAC | |
| Ex7F | TTAATTTCCTATTACTAAATGATCTGGACTT | 255 |
| Ex7R | TGTCAGTAATGAAACTGTAATAACAAC | |
| Ex8F | CCTCTGAATATTTTGTTATCTATTACCACTTA | 252 |
| Ex8R | ACTTGTCCATGATATAGTTAGACAC | |
| Ex9F | GGCAAGTATTTAGGAAAAATGTCAT | 401 |
| Ex9R | GCTATTTAGTGCCATTTTTCCTG | |
| Ex10F | GAGCAAGACATCTCTGTAACT | 276 |
| Ex10R | CTCTAAGGCCCTCCGAT | |
| Ex11F | AGGGCCTGCCAAATATAAT | 274 |
| Ex11R | CTGTACACTATGGGAAGGACC | |
| Ex12F | GTATGTGCTTTTACAGAATGTCTC | 260 |
| Ex12R | GCAGATGCAAGCCTCAA | |
| Ex13F | ATCCCAAAGCTTGAAATTGTC | 251 |
| Ex13R | CATTTGGCAGCACAACC | |
| amel-F | CCCTGGGCTCTGTAAAGAATAGTG | 106 (X) |
| amel-R | ATCAGAGCTTAAACTGGGAAGCTG | 112 (Y) |
The following M13 sequencing tails were added: 5′-ACGACGTTGTAAAACGAC-3′ to each forward and 5′-CAGGAAACAGCTATGACC-3′ to each reverse primer.
Primers to the amelogenin gene that amplify both the X and Y chromosomes are included to provide an internal amplification control and to identify male vs female DNA. All primers are prepared by standard phosphoramidite synthesis and resuspended in 10 mM Tris, pH 8.0, 0.1 mM EDTA at 100 μM (A260 = 2.0).
1.4 Predict Melting Profiles
Before any PCR is performed, predict all amplicon melting curves in silico. Although the mathematics is complex, Poland’s algorithm [38] as modified by Fixman and Freire [39] and implemented by Steger [3] is freely available on public web sites. Knowing what the melting curves should look like aids PCR optimization. Although the absolute temperatures are seldom accurately predicted, the shape and number of domains are usually correct. The number of predicted domains is less important than matching the overall shape of the predicted curve to observation.
Detailed example
Access http://www.biophys.uni-duesseldorf.de/local/POLAND/. Cut and paste an amplicon sequence (including the M13 tails) into the Sequence box on the website. For thermodynamic parameters select, “DNA (75 mM NaCl, Blake and Delcourt)” [40]. Enter in temperature limits of 65 and 90°C with a step size of 0.2°C and leave all other parameters at their defaults. Select melting curve graphics and click, “Submit.” The predicted absorbance melting curve is displayed. Click on “Bunch of numbers (x,y values)” and find the melting curve data (temperature and A260 hypochromicity) near the end of the data file. This data gives the melting curve (x) as increasing absorbance with temperature and can be inverted (1-x) to directly simulate fluorescent melting curve data. The observed melting curve shapes (using the reagents and conditions given below) closely follow the predicted curves, although they are shifted 5–6°C higher than predicted.
2. Optimization
High resolution melting analysis depends on comparing the melting curves of multiple samples. Sequence differences are identified by subtle deviations in the melting profiles. It is therefore critical to optimize the process by: 1) reducing sample to sample variation in DNA preparation, 2) developing robust, specific PCR amplification of all products, 3) demonstrating tight clustering of wild type samples, and 4) obtaining consistent primer plates by automation.
2.1 Prepare DNA
DNA extraction and purification should be standardized so that all samples are prepared in the same buffers to minimize ionic strength differences that affect melting curves. Additionally, spectrophotometric quantification and dilution of sample DNA to a standard concentration for input into PCR is recommended. Although advanced instruments with curve overlay (temperature shifting) software options can mask some sample differences, it is best to minimize these differences before melting analysis. Gene scanning depends on detecting heterozygotes by shape differences of the melting curves, not on homozygous differences that primarily affect Tm. Accuracy of variant detection is increased by overlaying the curves to focus only on shape differences, not on Tm differences that are influenced by sample and instrument variance. Homozygous (or hemizygous X-linked) variants are best detected by mixing normal and unknown DNA so that any homozygous variant in the unknown will be detected as a heterozygote in the mixture. Scanning of X-linked targets in males is performed by mixing with wild-type male DNA to generate heterozygotes for detection of base substitutions and small insertions/deletions. Large deletions encompassing entire exons of X-linked genes can also be detected in male samples by directly analyzing samples that are not mixed. Mixing is not necessary to detect heterozygous variants in females.
Detailed example
Several rapid, automated methods of DNA extraction from whole blood are now available that can provide high quality DNA in less than 1 hour. The specific method used is less important than consistency within the chosen method.
Materials
Whole blood anti-coagulated with sodium heparin.
MagNA Pure Compact Instrument (Roche Applied Science).
MagNA Pure Compact Nucleic Acid Isolation Kit I (Roche Applied Science).
NanoDrop 1000 or 8000 Spectrophotometer (Thermo Scientific).
Procedure
Mix the specimen well by inversion in the original tube.
Transfer 500 μL of whole blood to a 1.5 mL conical tube and place the tube in the sample rack.
Load the MagNA Pure Compact with the sample rack and appropriate cartridges and disposables.
Program the instrument to process 400 μL of specimen and elute in 100 μL of elution buffer.
Proceed with the extraction according to the automated protocol.
When the process is complete, remove the tubes containing the extracted nucleic acid and shut down instrument.
Measure the A260 of the sample nucleic acid on the NanoDrop 1000.
Dilute the nucleic acid to 50 ng/μL (A260 = 1.0) using 10 mM Tris, pH 7.5. Store short term at 4°C or long term at −20°C.
For male DNA, prepare both mixed and unmixed samples for CYBB (X-linked) analysis. To generate the mixed sample, mix the unknown test DNA with known wild type male DNA (1:1 v/v), resulting in a 50 ng/μL (A260 = 1.0) mixture in 10 mM Tris, pH 7.5. Female DNA is processed without mixing.
2.2 PCR Optimization
There are many methods for PCR optimization. If a gradient thermal cycler is available, PCR optimization of multiple primer sets is easily accomplished with an annealing temperature gradient. The predicted Tm of each primer pair is bracketed with an annealing temperature range extending 10–15°C above and 0–5°C below the Tm. The PCR products are evaluated by melting analysis as detailed below in section 3.2 and compared to predicted melting curves. Each primer pair is analyzed across the gradient and the annealing temperature range over which the product is pure is determined. Most PCR products <200 bp will melt completely in one transition between 76°C and 94°C. Some products, especially those >300 bps melt in more than one transition (multiple domains). If nonspecific products are present, they usually appear on derivative plots as small melting peaks with low Tms. Gel electrophoresis is only performed if the observed melting curves do not match the predicted profiles. A final common annealing temperature is selected that is within the acceptable temperature windows of all primer pairs to produce specific PCR products. Wide temperature windows indicate more robust designs. Optimization of parameters beyond the annealing temperature, including primer and Mg++ concentrations can be performed, although it is often easier to replace difficult primer sets. If a product melts at greater than 92°C, additives such as 5–10% DMSO and/or 1–2 M betaine can be included to lower its melting temperature. Evaporation during cycling and potential contamination are prevented by both an oil overlay and sealing tape.
Detailed example
Annealing temperature optimization for the 13 exons of CYBB and a control gene are performed on a gradient cycler. Results are compared to predicted melting curves and optimal temperature zones for the annealing temperature of each target are determined. A common annealing temperature for all products must be found for co-amplification on the same plate.
Materials
Primers with M13 tails for the 13 CYBB exons and the amelogenin control target (100 μM).
PCR master mix with heteroduplex detecting dye (LightScanner Master Mix, Idaho Technology) containing Taq polymerase, anti-Taq antibody, dNTPs, magnesium chloride, and LCGreen Plus dye.
Hard-shell, thin-walled 96-well microplates with white wells and a black shell (BioRad, HSP-9665)
Light mineral oil (Sigma M5904).
Optically clear (real time) sealing tape for plates (Bio-Rad 223-9444 or equivalent).
Gradient PCR instrument – 96-well (BioRad C1000).
Plate centrifuge (Eppendorf 5430).
High resolution melting instrument (Idaho Technology, LightScanner 96).
Procedure
Prepare 5X (2.5 μM) primer pair solutions for each target by combining 5 μl of one primer stock with 5 μl of its paired primer stock (each at 100 μM) and diluting with 190 μl of water.
Program the thermal cycler with an annealing temperature gradient 10–15°C above and 0–5°C below the predicted Tm of the primers (use the Tms were previously calculated by the LightScanner primer design program without the M13 tails). After an initial denaturation at 95°C for 10 sec, use 40 cycles of 94°C for 10 sec, the annealing gradient for 10 sec, and 75°C for 10 sec, followed with 1 cycle at 95°C for 10 sec and a final hold at 15°C.
Prepare PCR solutions with 1X master mix, 0.5 μM each primer and 5 ng/μl wild type DNA. On the BioRad C1000, the annealing gradients are down microplate columns (8 wells), so an appropriate mixture would be 40 ul of 2.5X LightScanner master, 20 μl of 5X primer pair solution, 10 μl of 50 ng/μl DNA and 30 ul of water.
Dispense 10 μl of the PCR solution into each well of a column on the 96-well microplate. Up to 12 amplicons can be run on one plate at the same time.
Add 12 μL mineral oil to each well, seal the plate with optically clear sealing tape and spin for 30 seconds at 1600 g (4650 rpm on the Eppendorf 5430).
Place the plate in the gradient cycler and run the gradient PCR.
Following PCR, remove the plate from the thermal cycler and spin for 30 seconds at 1600 g.
Plates may be scanned immediately, stored at room temperature for less than a day, or stored at 4 °C for up to a month before analysis. If plates have been refrigerated, spin for 30 s at 1600 g before analysis.
Obtain melting curves on the LightScanner as detailed below in section 3.2.
Use the predicted melting curve profiles as a guide to analyze the melting curves generated from each primer pair on LightScanner software. Determine the annealing temperature range for specific amplification of each product.
If the observed melting curves do not fit the predicted curves, perform gel electrophoresis on the PCR products. Standard 1.5% agarose slab gels in 0.5X TBE (45 mM Tris-borate, 1 mM EDTA, 0.5 μg/ml ethidium bromide) are adequate. Alternatively, an automated microelectrophoresis system can be used. Assess the amount and purity of the PCR products and determine the annealing temperature range over which each PCR product is pure.
If extraneous products are present, increase the specificity by decreasing the concentration of primers to 0.10–0.25 μM, or add 5–10% DMSO and/or 1 M betaine to lower primer Tms. If the yield is low or no PCR products are obtained, decrease the specificity by increasing the Mg++ concentration. Repeat the optimization. It may be easier to choose alternative primers than to perform extensive optimization with difficult primer sets.
2.3 Random population screening
Once PCR optimization of all primer pairs is accomplished and a common annealing temperature is selected, analyze random DNA samples from healthy individuals to screen for common variants. Set up one 96-well plate for each primer pair and analyze 95 individual DNA samples and one no template control. Any variants in healthy people detected by high-resolution melting analysis are typically identified by sequencing. The most common variant is assumed to be wild type and does not need confirmation by sequencing unless more than one common cluster is present.
Detailed example
Melting curves are generated from the DNA of 95 healthy females for each CYBB primers pair. DNA plates containing 95 different DNA samples and a single no template control are first prepared. Then, PCR master mix containing one primer pair and all PCR reagents is added to all wells. After PCR amplification, the samples are melted and analyzed. Results for CYBB exon 1 using 95 different DNA samples are shown in Fig. 2, both as a normalized/overlaid melting plot and as a difference plot. When no variants are present, all curves cluster tightly on the normalized plot as shown. Difference plots magnify any variance between samples, showing the single wild type cluster spread out over a fluorescence difference of about +/− 1%. A fluorescence difference of 5% has been suggested as a cutoff for identifying variant samples [41].
Fig. 2.
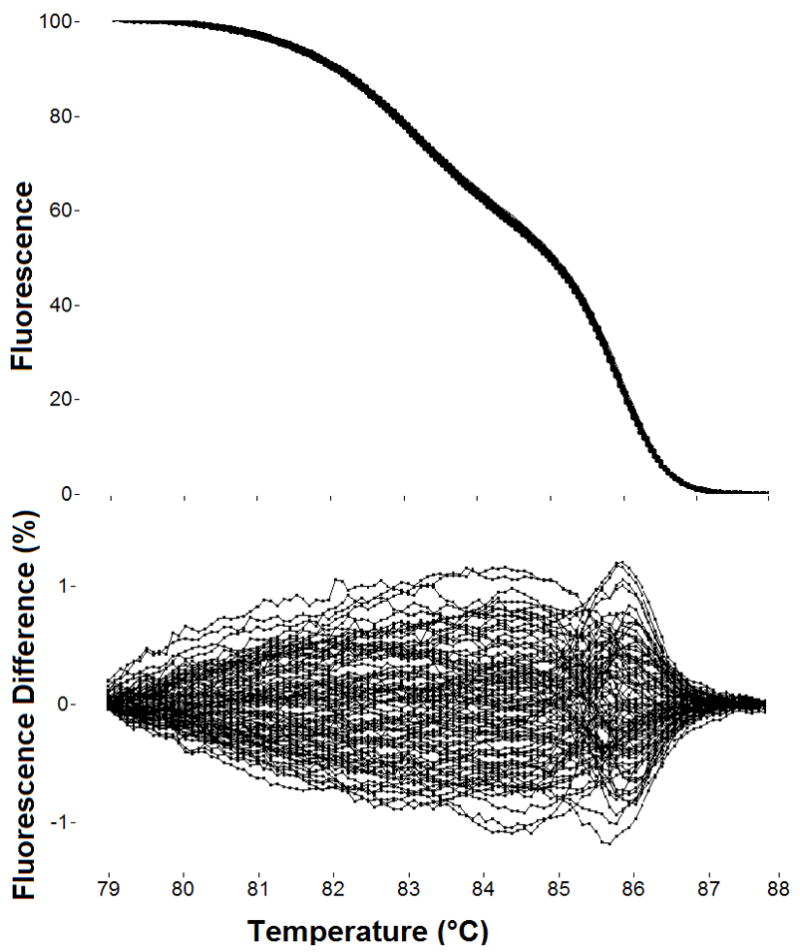
Ninety-five melting curves of CYBB exon 1 after PCR from the DNA of 95 healthy individuals. After exponential background subtraction, melting curves are shown normalized and overlaid (top) and as a difference from the average (bottom). No sequence variations were detected in the 146 bp product with two domains, typical of the low variation found in CYBB. Melting curves were acquired on the LightScanner in the presence of the saturating DNA dye, LCGreen Plus. The overall reproducibility of PCR and melting analysis can be assessed from the tightness of such clusters, typically +/− 1% on difference plots.
Additional Materials
DNA isolated from 96 random healthy females at 50 ng/μl (see 2.1)).
5X (2.5 μM) primer pair solutions for the 13 CYBB exons with M13 tails.
Procedure
Prepare DNA microplates by adding 1 uL of DNA (50 ng/uL) to each well from 95 different healthy females. Let the DNA solutions evaporate.
For each target, combine 400 ul of 2.5X LightScanner Master Mix, 200 μl of 5X primer pair solution and 400 μl water.
Add 10 μL of the PCR solution to each well of the DNA microplate.
Add 12 μL of mineral oil to each well.
Seal the plate with optically clear sealing tape and centrifuge for 30 seconds at 1600 g.
Place the plate in the C1000 thermal cycler and cycle using the following program: 1 cycle at 95°C for 10 sec, 40 cycles of 94°C for 10 sec, 64°C for 10 sec, 75°C for 10 sec, followed by 1 cycle at 95°C for 10 sec and a final hold at 15°C.
Following PCR, remove the plate from the thermal cycler and spin for 30 seconds at 1600 g.
Plates may be scanned immediately, stored at room temperature for less than a day, or stored at 4 °C for up to a month before analysis. If plates are refrigerated, spin for 30 s at 1600 g before analysis.
Perform melting analysis on the LightScanner as detailed below in section 3.2. Analyze the melting curves by normalization, curve overlay (temperature shifting) and difference plots to identify heterozygous variants. Common variants will cluster together and representative samples within any cluster can be sequenced as described below in section 3.3–3.4).
2.4 Prepare primer plates
The plate layout should maximize the number of samples that can be analyzed per plate. Multiple targets on a single DNA sample can be analyzed in one or more columns or in one or more rows. For ease of use and simplicity of design, some wells may be left empty. Table 2 provides options for formatting plates based on the number of samples and targets. An example plate design for CYBB is shown in Fig. 3.
Table 2.
Format options for a 96-well plate.
| Number of Samples | Number of Amplicons | Format Options |
|
|---|---|---|---|
| Each amplicon goes into: | Each sample goes into: | ||
| 49–96 | 1 | The entire plate | - |
| 33–48 | 2 | 4 rows or 6 columns | - |
| 25–32 | 3 | 4 columns | - |
| 17–24 | 4 | 2 rows or 3 columns | - |
| 13–16 | 5–6 | 2 columns | - |
| 9–12 | 7–8 | 1 row | 1 column |
| 7–8 | 9–12 | 1 column | 1 row |
| 5–6 | 13–16 | - | 2 columns |
| 4 | 17–24 | - | 2 rows or 3 columns |
| 3 | 25–32 | - | 4 columns |
| 2 | 33–48 | - | 4 rows or 6 columns |
| 1 | 49–96 | - | The entire plate |
Fig 3.

Workflow diagram for CYBB mutation scanning by high resolution melting analysis. Wild type samples containing no variants can be reported within 3 hours. Rare samples with aberrant melting profiles are sequenced for variant identification and can be reported within 6 hours. (A) DNA preparation from whole blood in 60 min (Roche MagNA Pure Compact) with quantification and dilution in 15 min (NanoDrop ND-8000 Spectrophotometer), (B) PCR preparation in 15 min with PCR cycling in 45 min (BioRad C1000), (C) Mutation scanning in 15 min (Idaho Techonology LightScanner 96), (D) Scanning data analysis in 30 min → if no variants detected, report within 3 hours, (E) Cycle sequence preparation (PCR product dilution, master mix preparation, and plate loading) in 30 min followed by cycle sequencing in 45 min (BioRad C1000), (F) Capillary electrophoresis preparation (ABI 3130xl) and product clean-up in 30 min with capillary electrophoresis in 45 min, (G) Sequencing data analysis in 45 min → report variant(s) within 6 hours. The hatched area indicates overlap of cycle sequencing and capillary electrophoresis preparation.
When multiple targets are analyzed, the complexity of primer placement can be simplified by preparing, “primer plates,” with all primer sets robotically dispensed and dried into appropriate wells. Automated liquid handling instruments can be used to add primers to the appropriate wells. After dispensing, the plates are air dried in an oven and then stored at room temperature for as long as 8 months. Alternatively, the plates may be dried during centrifugation in a Speed-Vac concentrator. PCR setup is greatly simplified because the primer matrix is prepared before the assay is performed. The PCR master mix with sample DNA is added directly to the wells containing primers and mixed before amplification.
Detailed example
The plate layout for CYBB is shown in Table 3 and allows the analysis of six samples per plate, two columns per sample. After primer deposition, the plates are sealed, centrifuged, uncovered, and then dried in a 37°C oven. When reconstituted with 10 μL of PCR master mix and DNA the final concentration of each primer is 0.5 μM.
Table 3.
Location of CYBB and amelogenin primers on X-linked chronic granulomatous disease primer plates.
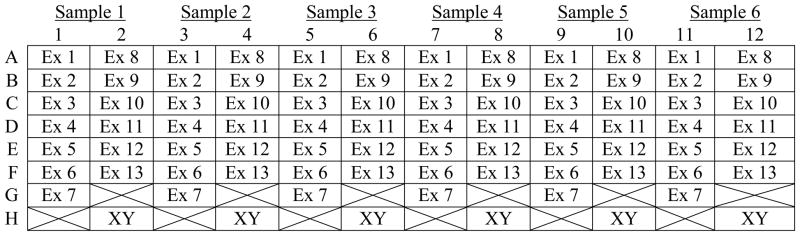 |
Additional Materials
Liquid handling instrument (Nanodrop Express, Innovadyne Technologies).
Aluminum sealing tape (VWR 82028-086).
Procedure
Program the Nanodrop Express to dispense 2000 nL (2 μL) of 2.5 μM primer pair solutions according to the plate pattern shown in Table 3.
Label 14 sterile, 1.5 mL microfuge tubes with the target gene and exon.
Add 380 μL of water to each tube.
Add 10 μL of 100 μM stock forward primer and 10 μL of 100 μM stock reverse primer to the tubes for a final primer concentration of 2.5 μM each. One plate requires 15 μL of primer pair solution plus 10 μL dead volume. The Innovadyne 8-well dispensing strips hold up to 250 μL/well so up to 10 plates can be dispensed in one run.
Dispense the appropriate volume of 2.5 μM primer pair solution into two 8-well strips as shown in Table 4. Add water for any empty wells.
Place the 8-well strips on the Nanodrop Express.
Place a 96-well microplate on the instrument.
Run the Nanodrop Express to dispense each primer pair solution into the appropriate wells.
When the dispensing is complete, remove the first plate and repeat with any subsequent plates.
Cover the plates and centrifuge them for 30 s at 1600 g to get the primers to the bottom of the wells.
Dry the plates, uncovered, in a single layer in a 37 °C oven.
Seal the plates with aluminum sealing tape and place them in individual zip-lock bags, removing excess air.
Store at room temperature for up to 8 months.
Table 4.
Location of the 2.5 μM working CYBB primer pair solutions in 8-well strips for robotic dispensing.
| H2O | exon 7 | exon 6 | exon 5 | exon 4 | exon 3 | exon 2 | exon 1 |
| AMEL X/Y | H2O | exon 13 | exon 12 | exon 11 | exon 10 | exon 9 | exon 8 |
3 Performance
Although high resolution melting for gene scanning is often used for high throughput research studies, it can also be used for rapid clinical diagnostics, targeting only a few patient samples at one time. If primer plates are available it is completely feasible, starting from whole blood, to complete screening of negative samples in 3 hours and to identify any variants by sequencing is less than 6 hours (Fig. 3). As more and more rare genetic diseases are found, efficient testing of such orphan genes can be rapidly performed by high resolution melting.
3.1 PCR
Using previously prepared primer plates, bulk master mix containing sample DNA but without primers is added to each well. When all wells are filled, the plate is covered with sealing tape and mixed to dissolve the primers. Each well is then overlaid with mineral oil, re-sealed securely and centrifuged. PCR is performed using conditions determined during PCR optimization (section 2.2) and validated on healthy controls (section 2.3).
Detailed example
PCR is performed using the CYBB primer plates prepared in section 2.4). Each plate can analyze six samples, typically one wild type control, one variant control, and two unknown male samples, analyzed both with and without mixing. Mixing with a known wild type male sample allows sensitive detection of hemizygous small variants, including single base variants and small deletions and insertions. Analyzing the samples without mixing will detect large deletions.
Additional Materials
Purified wild type and unknown DNA adjusted to 50 ng/μl (section 2.1).
CYBB primer plates (section 2.4).
Plate mixer (Eppendorf, MixMate 5353 000.014).
Procedure
Prepare sufficient master mix for a volume of 10 μL per well for the 13 CYBB exons and the control target.
Refer to Table 5 to determine the component volumes to add for master mix preparation. This table assumes samples are run in singlet and includes 1 well dead volume. Male DNA is analyzed both mixed 1:1 with wild type DNA and without mixing.
Add 10 μL per well of the master mix including DNA into the appropriate wells according to the plate map shown in Table 3.
Seal the plate lightly with sealing tape and place on the plate mixer for 30 seconds at 1650 rpm to dissolve the primers with the master mix.
Remove the tape and add 12 μL mineral oil to each well.
Seal the plate firmly with sealing tape and centrifuge for 30 s at 1600 g.
Place the plate in the C1000 thermal cycler and amplify using the following program: 1 cycle at 95°C for 10 sec, 40 cycles of 94°C for 10 sec, 64°C for 10 sec, 75°C for 10 sec, followed by 1 cycle at 95°C for 10 sec and a final hold at 15°C.
After PCR, remove the plate from the thermal cycler and spin for 30 seconds at 1600 g.
Plates may be scanned immediately, stored at room temperature for less than a day, or stored at 4 °C for up to a month before analysis. If plates are refrigerated, spin for 30 s at 1600 g before analysis.
Table 5.
Master mix components for gene scanning of X-linked chronic granulomatous disease.
| Component | Each well (μL) | Female sample not mixed (μL) | Male sample not mixed (μL) | Male sample mixed (μL) | Control Sample (μL) |
|---|---|---|---|---|---|
| Water | 5 | 75 | 75 | 75 | 75 |
| 2.5X LightScanner Master | 4 | 60 | 60 | 60 | 60 |
| Wild type DNA | 1 | --- | --- | 7.5 | 15 |
| Unknown DNA | 15 | 15 | 7.5 | --- | |
| Total | 10 | 150 | 150 | 150 | 150 |
3.2 Scanning
High resolution melting is the only scanning method that does not require physical processing or separation steps. Samples can be melted immediately after PCR and changes in melting curve shape identify samples that vary from wild type.
Detailed example
The frequency of CYBB variants is very low. The disease is rare, and benign variants are uncommon. Six samples were PCR amplified at all 13 exons and melted. The normalized and overlaid fluorescence curves are shown in Fig. 4. Within an exon, the different samples trace each other very closely, except for exon 11, where altered curve shape of an unknown male sample indicates a variant. Fig. 5 compares the individual curves for exon 11 in more detail, contrasting a normal sample to an unknown male specimen with a variant sequence.
Fig. 4.

Normalized and overlaid high resolution melting curves for all 13 CYBB exons. All traces were generated on a single 96-well plate using 3 samples run in duplicate. The samples analyzed were an unknown male, a wild type male (WT) and a mixed sample containing both unknown and WT. The traces for exon 11 (circled) suggested a heterozygous sequence in the mixed sample. The traces for the other 12 exons were normal. Additional analysis of exon 11 for this unknown male sample is shown in Fig. 5.
Fig. 5.

High resolution melting analysis of CYBB exon 11 in wild type DNA (left panels) and variant DNA (right panels). Melting curves are displayed normalized (top), normalized and overlaid (middle) and as difference curves (bottom). In X-linked disorders, variants are best detected after heteroduplex formation by mixing unknown DNA with wild type (WT) DNA. Less reliable is detecting the unknown sample without mixing, although in this case the unmixed hemizygous sample (Unknown) can also be distinguished from wild type by difference curves. The unknown male sample had a T duplication at c.1456 by sequencing.
Procedure
Program the LightScanner as follows: Hold Temp 67°C, Start Temp 70°C, End Temp 94°C, Exposure Auto.
Load the plate into the LightScanner, inserting the notched edges first and start the run.
When the run finishes (about 10 min), open the data folder for the scan just completed.
Observe the *.tif screen image for even, bright fluorescence. Some signal attenuation on the plate edges is normal, but very low or absent fluorescence in wells indicates likely deletion of entire exons.
Open the LightScanner software, click on “Scanning” Analysis.
-
Open the *.mlt Analysis File to analyze a run. Follow either a) or b):
If you have analyzed the same primer plate configuration before (same PCR products and plate layout), import the previously saved subsets and cursor settings by selecting, “Import”, and “Scanning Analysis”. Find and load your previously saved *.mat file with the desired subsets and cursor settings.
Build subsets for each exon and the XY amelogenein target according to the User Manual for the LightScanner instrument. These subsets can be stored at any time for future reference by saving (Save as) an Analysis (*.mat) file.
Choose subset XY, go to the Negative Filter Tab, and examine the melting peaks: 2 peaks, one at ~76°C and one at ~84°C, are present in male specimens. Only one peak at ~84 °C is present in females.
In the top window, select the Exon 1 subset, go to the Negative Filter tab, check that fluorescence levels are adequate for all wells, and that negative samples are excluded. Note any sharp drops or increases in fluorescence that can result from bubble or film artifacts. If present, spin and melt the plate again and resume at step 3 above.
Click on the Normalize tab and adjust the vertical cursors on the top graph to encompass all predicted melting domains (section 1.4).
Select the Curve Shift tab and adjust the horizontal cursor on the top graph to optimize clustering of the curves.
Enter the Grouping tab and click on Select Baseline. Choose one or more wild type control wells for the baseline and click on Finish Selection.
Record any aberrant results by sample and exon. An exon with an unexpected melting pattern should be genotyped or sequenced.
Repeat steps 8–12 for all exons (subsets).
Retain the cursor settings and subsets for future analysis by saving a *.mat file.
3.3 Cycle sequencing
Targeted sequencing is performed using dideoxy-terminator chemistry on any exon with an abnormal scan. The PCR product from the exon of interest is recovered from the plate and diluted 1:50 in water. Cycle sequencing is performed with both forward and reverse M13 primers.
Detailed Example
Additional Materials
M13 sequencing primers, 5 μM (forward: 5′-ACGACGTTGTAAAACGAC-3′, reverse: 5′-CAGGAAACAGCTATGACC-3′).
BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems).
Procedure
Dilute the PCR product from the exon of interest 1:50 by adding 2 μL of PCR product to 98 μL of water.
-
Prepare the cycle sequencing reactions with 1:6th of the recommended concentration of BigDye terminator and a primer concentration of 0.5 μM as follows:
Master Mix component 1X (μL) BigDye Terminator v1.1 2.0 5X sequencing buffer 5.0 1:50 diluted PCR product 3.0 Water 17.0 Dispense 9 μL of the master mix into two wells of a 96-well plate and add 1 μL of 5 μM forward M13 primer into one well and 5 μM reverse M13 primer into the other well.
Seal the plate, place it in the C1000 thermal cycler, and run the following program: 1 cycle at 96°C for 10 sec, 25 cycles of 96°C for 5 sec, 50°C for 10 sec, 60°C for 20 sec, followed by a final hold at 4°C.
3.4 Capillary electrophoresis
After cycle sequencing, unincorporated terminator dyes and salts are removed by gel filtration. Formamide is added and the solution denatured briefly at 95°C. Capillary electrophoresis is performed using a 36 cm capillary array and an ultrafast protocol for short PCR products.
Detailed Example
Additional Materials
Gel Filtration Cartridges (Edge Performa® DTR).
Formamide (HiDi, ABI).
Capillary sequencer (ABI 3130xl).
36 cm capillary array (ABI).
POP-7 Polymer (ABI).
Mutation Surveyor DNA Variant Analysis Software (SoftGenetics).
Procedure
Prepare the gel filtration cartridges by centrifuging for 2 min at 750 g to remove storage water.
Add each cycle sequencing reaction to a prepared gel filtration cartridge.
Spin 2 minutes at 750 g and collect the eluted material in a 96-well plate. Proceed to electrophoresis or store at 4°C.
Add 10 uL formamide to each product to be sequenced.
Cover plate with the ABI septum and spin for 30 s at 1600 g.
Denature the solutions for 2 min at 95°C on a thermal cycler.
Load the solution onto the ABI 3130xl loaded with a 36 cm capillary array and POP-7 polymer.
Perform electrophoresis using the UltraSeq_POP7_1 instrument protocol and the 3130POP7_BDTv1.1 analysis protocol.
Analyze the data with the Sequencing Analysis program according to the ABI protocol.
Continue analysis with Mutation Surveyor software (v. 3.4) as follows:
Open Mutation Surveyor and click on the Open Files icon.
Add the appropriate GenBank reference file in GBK format to the top window and the experimental data Sample Files in AB1 format to the bottom window.
Click OK to return to the main screen and click on the Run icon to initiate the analysis.
Use the Graphic Display of Mutations icon to check that the sequence covers the area of interest.
Return to the main window and manually check the calls that Mutation Surveyor has made. Double click on the Mutation Surveyor call to move the chromatogram view to the call site. Confirm or delete the variations called by the software.
Click on Reports on the main menu, then select Custom Report. Go to the Nomenclature tab, click on Custom and under Reference click on Relative to CDS and Intronic Mutations Relative to Nearest CDS. Click OK.
A Custom Report Table will be generated that lists any variants identified.
Concluding Remarks
High resolution melting analysis provides a sensitive, homogeneous scanning method using controlled heating at a fast rate and high data density. Heterozygous variants are easily identified because they distort the melting curve shape compared to wild type. When a PCR product scans as negative, there is no need for further analysis. Therefore, scanning is most useful when the frequency of variants is low. No sequencing is needed for wild type samples. For samples with variants, only the PCR products that scan positive need to be further analyzed. Common variants can often be recognized by characteristic melting patterns [42] or small amplicon genotyping [43], but definitive genotyping requires unlabeled probe [23] or snapback primer [24] analysis (convenient because they use the same platforms and reagents reported here) or other means of genotyping. Rare variants, whether associated with disease or not, are best identified by sequencing.
In order to streamline turnaround in a clinical laboratory, the following protocols were implemented:
Primers were designed with tools that automatically select primers bracketing each exon (including likely splice sites) with consideration of high-resolution melting (e.g., PCR product size).
Melting curves were predicted by recursive algorithms to provide the relative position and number of melting domains expected. These predicted curves were used to verify PCR specificity and to select the temperature region to analyze for each product.
Primer pairs were optimized on a gradient PCR instrument. Analysis by high resolution melting and optional agarose gel electrophoresis provides a window of acceptable annealing temperatures for each PCR product. The final annealing temperature selected must be within these windows for all PCR products on the plate. The wider the window, the more robust the reaction.
Microtiter plates with dried primer pairs were robotically prepared in batches. The primer plates can be used for at least 8 months after preparation without degradation of performance. Instead of manually pipetting 26 different primers into selected wells, the assay complexity is handled robotically, requiring only the addition of a PCR reagent/DNA mixture at the time of testing.
An initial screen for common polymorphisms is performed by analyzing 95 random individuals across all exons. Identifying variants as common polymorphisms can significantly reduce the need for sequencing. Common polymorphisms can be identified by melting curve identity [42], mixing [44], small amplicon genotyping [16], unlabeled probes [23], or snapback primers [24], either in separate reactions or simultaneously in the same reaction [26]. All of these methods are based on melting and use the same dyes and instrumentation as heterozygote scanning, avoiding labeled probes and sequencing.
Only rare heterozygous PCR products are sequenced. In the case of CYBB analysis, 1 out of 13 (8%) of the PCR products from positive samples requires sequencing. This percentage is further reduced proportionally by the percentage of samples that are negative. For example, if half of the samples are positive, only 4% of exons require sequencing. Furthermore, once a variant is identified within a family, inheritance can be determined by melting alone, further reducing the need for sequencing.
Turnaround time is minimized with rapid protocols. Negative samples are identified after DNA preparation (60 min), quantification (15 min), PCR (60 min) using previously prepared primer plates, high resolution melting (15 min) and analysis (30 min). When a positive product is identified by melting, targeted sequencing of only that product is performed. Common sequencing primers are used that were introduced during PCR as 5′-tails. Sequencing requires an additional 2 hours and 15 min of rapid cycle sequencing with 45 min allowed for analysis. Negative samples are easily completed in less than 3 hours and the variants in positive samples are identified in 6 hours.
Throughput can be increased by multiplying the number of inexpensive standard PCR machines while using a single dedicated high resolution melting instrument. For example, plates from up to 4 thermal cyclers can be analyzed with 1 dedicated melting instrument because it takes 60 min to amplify but only 15 min to melt. This avoids a linear increase in instrument cost with volume when using more expensive real-time instruments.
Footnotes
Disclosure Statement
Development of high resolution melting was supported by grants GM060063, GM072419, GM073396, and GM082116 from the NIH. High resolution melting analysis is licensed from the University of Utah to Idaho Technology and Idaho Technology has sublicensed Roche and Qiagen. CTW is an inventor on high resolution melting patents and holds equity interest in Idaho Technology. ME has nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Santalucia J, Jr, Hicks D. Annu Rev Biophys Biomol Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 2.Lerman LS, Silverstein K. In: Methods in Enzymology. Wu R, editor. Vol. 155. Academic Press; New York: 1987. pp. 482–501. [DOI] [PubMed] [Google Scholar]
- 3.Steger G. Nucleic Acids Res. 1994;22:2760–2768. doi: 10.1093/nar/22.14.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyon E, Wittwer CT. J Mol Diagn. 2009;11:93–101. doi: 10.2353/jmoldx.2009.080094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lay MJ, Wittwer CT. Clin Chem. 1997;43:2262–2267. [PubMed] [Google Scholar]
- 6.Ririe KM, Rasmussen RP, Wittwer CT. Anal Biochem. 1997;245:154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- 7.Pornprasert S, Phusua A, Suanta S, Saetung R, Sanguansermsri T. Eur J Haematol. 2008;80:510–514. doi: 10.1111/j.1600-0609.2008.01055.x. [DOI] [PubMed] [Google Scholar]
- 8.Price EP, Smith H, Huygens F, Giffard PM. Appl Environ Microbiol. 2007;73:3431–3436. doi: 10.1128/AEM.02702-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Worm J, Aggerholm A, Guldberg P. Clin Chem. 2001;47:1183–1189. [PubMed] [Google Scholar]
- 10.Dujols VE, Kusukawa N, Mckinney JT, Dobrowolski SF, Wittwer CT. In: Real-Time PCR. Dorak MT, editor. Garland Science; New York: 2006. pp. 157–171. [Google Scholar]
- 11.Farrar JS, Reed GH, Wittwer CT. In: Molecular Diagnostics. Patrinos GP, Ansorge WJ, editors. Elsevier; London, UK: 2010. pp. 229–245. [Google Scholar]
- 12.Herrmann MG, Durtschi JD, Bromley LK, Wittwer CT, Voelkerding KV. Clin Chem. 2006;52:494–503. doi: 10.1373/clinchem.2005.063438. [DOI] [PubMed] [Google Scholar]
- 13.Herrmann MG, Durtschi JD, Bromley LK, Wittwer CT, Voelkerding KV. Clin Chem. 2007;53:150–152. doi: 10.1373/clinchem.2006.081240. [DOI] [PubMed] [Google Scholar]
- 14.Herrmann MG, Durtschi JD, Wittwer CT, Voelkerding KV. Clin Chem. 2007;53:1544–1548. doi: 10.1373/clinchem.2007.088120. [DOI] [PubMed] [Google Scholar]
- 15.Palais R, Wittwer CT. In: Methods in Enzymology. Johnson ML, Brand L, editors. Vol. 454. Academic Press; New York: 2009. pp. 323–343. [DOI] [PubMed] [Google Scholar]
- 16.Liew M, Pryor R, Palais R, Meadows C, Erali M, Lyon E, Wittwer C. Clin Chem. 2004;50:1156–1164. doi: 10.1373/clinchem.2004.032136. [DOI] [PubMed] [Google Scholar]
- 17.Reed GH, Wittwer CT. Clin Chem. 2004;50:1748–1754. doi: 10.1373/clinchem.2003.029751. [DOI] [PubMed] [Google Scholar]
- 18.Gundry CN, Dobrowolski SF, Martin YR, Robbins TC, Nay LM, Boyd N, Coyne T, Wall MD, Wittwer CT, Teng DH. Nucleic Acids Res. 2008;36:3401–3408. doi: 10.1093/nar/gkn204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rigon C, Andrisani A, Forzan M, D’antona D, Bruson A, Cosmi E, Ambrosini G, Tiboni GM, Clementi M. Fertil Steril. 2009 doi: 10.1016/j.fertnstert.2009.05.025. in press. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery J, Wittwer CT, Kent JO, Zhou L. Clin Chem. 2007;53:1891–1898. doi: 10.1373/clinchem.2007.092361. [DOI] [PubMed] [Google Scholar]
- 21.Palais RA, Liew MA, Wittwer CT. Anal Biochem. 2005;346:167–175. doi: 10.1016/j.ab.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Erali M, Palais R, Wittwer CT. In: Molecular Beacons: Signalling Nucleic Acid Probes, Methods and Protocols. Seitz O, Marx A, editors. Vol. 429. Humana Press; Totowa: 2008. pp. 199–206. [Google Scholar]
- 23.Zhou L, Myers AN, Vandersteen JG, Wang L, Wittwer CT. Clin Chem. 2004;50:1328–1335. doi: 10.1373/clinchem.2004.034322. [DOI] [PubMed] [Google Scholar]
- 24.Zhou L, Errigo RJ, Lu H, Poritz MA, Seipp MT, Wittwer CT. Clin Chem. 2008;54:1648–1656. doi: 10.1373/clinchem.2008.107615. [DOI] [PubMed] [Google Scholar]
- 25.Vossen RH, Aten E, Roos A, Den Dunnen JT. Hum Mutat. 2009;30:860–866. doi: 10.1002/humu.21019. [DOI] [PubMed] [Google Scholar]
- 26.Montgomery J, Wittwer CT, Palais RA, Zhou L. Nature Prot. 2007;2:59–66. doi: 10.1038/nprot.2007.10. [DOI] [PubMed] [Google Scholar]
- 27.Seipp MT, Durtschi JD, Voelkerding KV, Wittwer CT. J Biomol Tech. 2009;20:160–164. [PMC free article] [PubMed] [Google Scholar]
- 28.Seipp MT, Pattison D, Durtschi JD, Jama M, Voelkerding KV, Wittwer CT. Clin Chem. 2008;54:108–115. doi: 10.1373/clinchem.2007.097121. [DOI] [PubMed] [Google Scholar]
- 29.Aguirre-Lamban J, Riveiro-Alvarez R, Garcia-Hoyos M, Cantalapiedra D, Avila-Fernandez A, Villaverde C, Trujillo-Tiebas MJ, Ramos C, Ayuso C. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-4518. in press. [DOI] [PubMed] [Google Scholar]
- 30.Chou LS, Gedge F, Lyon E. J Mol Diagn. 2005;7:111–120. doi: 10.1016/S1525-1578(10)60016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery JL, Sanford LN, Wittwer CT. Expert Rev Mol Diagn. 2010;10 doi: 10.1586/erm.09.84. in press. [DOI] [PubMed] [Google Scholar]
- 32.Erali M, Voelkerding KV, Wittwer CT. Exp Mol Pathol. 2008;85:50–58. doi: 10.1016/j.yexmp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackay JF, Wittwer CT. PCR Optimization and Troubleshooting. Horizon Scientific Press; Norwich: 2010. in press. [Google Scholar]
- 34.Mckinney JT, Nay LM, De Koeyer D, Reed GH, Wall M, Palais RA, Jarret RL, Wittwer CT. In: The Handbook of Plant Mutation Screening. Meksem K, Kahl G, editors. Wiley-VCH; Weinheim: 2010. in press. [Google Scholar]
- 35.Reed GH, Kent JO, Wittwer CT. Pharmacogenomics. 2007;8:597–608. doi: 10.2217/14622416.8.6.597. [DOI] [PubMed] [Google Scholar]
- 36.Wittwer CT. Hum Mutat. 2009;30:857–859. doi: 10.1002/humu.20951. [DOI] [PubMed] [Google Scholar]
- 37.Zhang MQ. Hum Mol Genet. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 38.Poland D. Biopolymers. 1974;13:1859–1871. doi: 10.1002/bip.1974.360130916. [DOI] [PubMed] [Google Scholar]
- 39.Fixman M, Freire JJ. Biopolymers. 1977;16:2693–2704. doi: 10.1002/bip.1977.360161209. [DOI] [PubMed] [Google Scholar]
- 40.Blake RD, Delcourt SG. Nucleic Acids Res. 1998;26:3323–3332. doi: 10.1093/nar/26.14.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laurie AD, Smith MP, George PM. Clin Chem. 2007;53:2211–2214. doi: 10.1373/clinchem.2007.093781. [DOI] [PubMed] [Google Scholar]
- 42.Vandersteen JG, Bayrak-Toydemir P, Palais RA, Wittwer CT. Clin Chem. 2007;53:1191–1198. doi: 10.1373/clinchem.2007.085407. [DOI] [PubMed] [Google Scholar]
- 43.Dobrowolski SF, Ellingson C, Coyne T, Grey J, Martin R, Naylor EW, Koch R, Levy HL. Mol Genet Metab. 2007;91:218–227. doi: 10.1016/j.ymgme.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Zhou L, Vandersteen J, Wang L, Fuller T, Taylor M, Palais B, Wittwer CT. Tissue Antigens. 2004;64:156–164. doi: 10.1111/j.1399-0039.2004.00248.x. [DOI] [PubMed] [Google Scholar]