


Abstract
Rad26p, a yeast homologue of human Cockayne syndrome B with an ATPase activity, plays a pivotal role in stimulating DNA repair at the coding sequences of active genes. On the other hand, DNA repair at inactive genes or silent areas of the genome is not regulated by Rad26p. However, how Rad26p recognizes DNA lesions at the actively transcribing genes to facilitate DNA repair is not clearly understood in vivo. Here, we show that Rad26p associates with the coding sequences of genes in a transcription-dependent manner, but independently of DNA lesions induced by 4-nitroquinoline-1-oxide in Saccharomyces cerevisiae. Further, histone H3 lysine 36 methylation that occurs at the active coding sequence stimulates the recruitment of Rad26p. Intriguingly, we find that Rad26p is recruited to the site of DNA lesion in an elongating RNA polymerase II-dependent manner. However, Rad26p does not recognize DNA lesions in the absence of active transcription. Together, these results provide an important insight as to how Rad26p is delivered to the damage sites at the active, but not inactive, genes to stimulate repair in vivo, shedding much light on the early steps of transcription-coupled repair in living eukaryotic cells.
INTRODUCTION
Genomic DNA is continuously challenged by a variety of damaging agents which can be of either exogenous or endogenous origin (1,2). The deficiencies in repairing DNA lesions are fundamental to the etiology of most cancers (2–11). However, a cell employs a number of repair pathways to maintain its genomic stability (7–30). One such repair mechanism utilized by the cell is nucleotide excision repair (NER). NER removes a wide variety of DNA lesions including UV-induced pyrimidine dimers (cyclobutane pyrimidine dimers and 6-4 photoproducts) and DNA-helix distorting bulky chemical adducts [e.g. 4-nitroquinoline-1-oxide (4NQO) adduct], and thus, is the most flexible DNA repair pathway. It is a complex process that requires stepwise action of many proteins for damage recognition, local opening of DNA duplex around the lesion, dual incision of the damaged DNA strand, gap repair synthesis and strand ligation (1,2,8,13,15–17,19,24,25).
Significant insight was gained in human NER through the study of two autosomal recessive disorders, namely xeroderma pigmentosum (XP) and Cockayne syndrome (CS). The patients with either one of these disorders show severe UV hypersensitivity. XP disorder is caused by mutation in one of the seven genes (i.e. XPA, XPB, XPC, XPD, XPE, XPF and XPG), while the defect in the CSA or CSB gene leads to CS disease. The XP gene products perform various functions during damage recognition, DNA unwinding and incision (11,15–19,25). For example, XPC (Rad4p in yeast)/HHR23B (Rad23p in yeast) initially detects DNA lesion (19), and subsequently, XPA (Rad14p in yeast) and heterotrimeric replication protein A (RPA) bind to the site of DNA lesion to further aid in damage recognition. The XPB (Rad25p in yeast) and XPD (Rad3p in yeast) helicases (components of transcription factor, TFIIH) then unwind duplex DNA in the vicinity of lesion followed by XPG (Rad2p in yeast) and ERCC1 (Rad10p in yeast)/XPF (Rad1p in yeast) endonuclease-mediated incision of one strand of DNA at positions 3′ and 5′ to the damage, respectively, generating ∼30 base oligonucleotide containing lesion. On the other hand, the CS gene products participate in the NER process specifically at the transcriptionally active genes (2,14,20–24,27,30). Thus, NER removes DNA lesion from the active and inactive genes. Accordingly, NER has been classified into two types, namely global genomic NER (GG-NER) and transcription-coupled NER (TC-NER). The DNA lesions in the transcriptionally inactive or silent areas of the genome are repaired by GG-NER pathway (17,19,24,25). The TC-NER process repairs DNA lesions at the coding sequence of the transcriptionally active gene (8,17,24,26–40).
Both the subpathways of NER are fundamentally identical except their mechanisms of damage recognition. Interestingly, TC-NER functions at a much faster rate than GG-NER (8,17,24,26–40) as damage is promptly recognized in TC-NER. In GG-NER, the XPC/HHR23B participates in initial recognition of DNA lesions (15,19,25,40). However, the recognition of the damaged DNA during TC-NER does not require XPC (14,17,24,33,37,40). In TC-NER, the transcription elongation machinery stalls at the site of DNA lesion (24,31,32,36,38,41–46), and subsequently, elongating RNA polymerase II is disassembled through specific degradation of its largest subunit (47). Such a disassembly of RNA polymerase II has been proposed to allow the TC-NER-specific proteins such as CSA and CSB to gain access to the damaged DNA (14,38,40,43,48,49). However, how TC-NER-specific factors are recruited to the site of DNA lesion at the active gene in eukaryotes remains unknown in vivo. Here, primarily using a formaldehyde-based in vivo cross-linking and chromatin immunoprecipitation (ChIP) assay, we show that Rad26p, a yeast homologue of human CSB with a DNA-dependent ATPase activity, is recruited to the sites of 4NQO-induced DNA lesions at the coding sequences of the active genes with the help of RNA polymerase II and histone H3 lysine 36 (H3 K36) methylation in Saccharomyces cerevisae. Such targeting of Rad26p confers it to be a TC-NER-specific factor, and stimulates its recognition of DNA lesions at the active genes, eventually leading to a faster TC-NER as compared to relatively slow GG-NER.
MATERIALS AND METHODS
Plasmids
The plasmids, pFA6a-13Myc-KanMX6 and pFA6a-3HA-His3MX6 (50) were used for genomic tagging of the proteins of interest by myc and HA epitopes, respectively. The plasmid pRS416 was used in the PCR-based gene disruption.
Yeast strains
Yeast strain harboring temperature-sensitive (ts) mutation in RPB1 was obtained from the Young laboratory (Richard A. Young, MIT). Multiple myc-epitope tags were added to the original chromosomal loci of RPB1, RAD26 and RAD3 in W303a to generate ZDY4 (Rpb1p-myc, KAN), ZDY2 (Rad26p-myc, KAN) and ASY41 (Rad3p-myc, KAN) respectively. The strain, ZDY17 (Rad26p-myc, KAN; rpb1-ts), was generated by adding multiple myc-epitope tags at the C-terminal of Rad26p in the rpb1-ts strain. Three HA-epitope tags were added to the chromosomal locus of RPB3 in ZDY2 and ZDY17 to generate SMY8 (Rad26p-myc, KAN; Rpb3p-HA, HIS3) and SMY9 (Rad26p-myc, KAN; Rpb3p-HA, HIS3; rpb1-ts), respectively. The strains, SLY6A (Rad26p-myc, KAN; Δset1, URA3) and SLY7 (Rad26p-myc, KAN; Δset2, URA3), were generated by deleting the SET1 and SET2 genes, respectively, in ZDY2.
Growth media
For the studies at the GAL genes, the wild-type yeast strain carrying myc epitope-tagged Rad26p or Rpb1p was grown in YPR (yeast extract-peptone plus 2% raffinose) up to an OD600 (optical density at 600 nm) of 0.8 at 30°C, and then transferred to YPG (yeast extract–peptone plus 2% galactose) for 90 min to induce GAL1 prior to 4NQO treatment. The 4NQO-treated cells were grown in YPG for 20 min at 30°C prior to formaldehyde cross-linking. For rpb1-ts mutant and its wild-type equivalent, yeast cells were grown in YPG up to an OD600 of 0.85 at 23°C, and then transferred to 37°C for 1 h before cross-linking. For the induction of INO1, yeast cells were initially grown in synthetic complete medium (yeast nitrogen base and complete amino acid mixture plus 2% dextrose) containing 100 µM inositol at 30°C up to an OD600 of 0.45, and then switched to the same medium without inositol for 4 h prior to 4NQO treatment or formaldehyde-based in vivo cross-linking. For the studies at RPS5, the yeast strain carrying myc epitope-tagged Rad26p was grown in YPD (yeast extract, peptone plus 2% dextrose) up to an OD600 of 1.0.
ChIP assay
The ChIP assay was performed as described previously (51–53). Briefly, yeast cells were treated with 1% formaldehyde, collected and resuspended in lysis buffer. Following sonication, cell lysate (400 µl lysate from 50 ml of yeast culture) was precleared by centrifugation, and then 100 µl lysate was used for each immunoprecipitation. Immunoprecipitated protein–DNA complexes were treated with protease K, the cross-links were reversed, and DNA was purified. Immunoprecipitated DNA was dissolved in 20 µl TE 8.0 (10 mM Tris–HCl pH 8.0 and 1 mM EDTA), and 1 µl of immunoprecipitated DNA was analyzed by PCR. PCR reactions contained [α-32P]dATP (2.5 µCi for each 25 µl reaction), and the PCR products were detected by autoradiography after separation on a 6% polyacrylamide gel. As a control, ‘input’ DNA was isolated from 5 µl lysate without going through the immunoprecipitation step, and dissolved in 100 µl TE 8.0. To compare PCR signal arising from the immunoprecipitated DNA with the input DNA, 1 µl of input DNA was used in the PCR analysis. Serial dilutions of input and IP DNAs were used to assess the linear range of PCR amplification as described previously (51). All the PCR data presented in this article are within the linear range of the PCR analysis.
For analysis of Rad26p and Rad3p recruitment, we modified the above ChIP protocol as follows. A total of 800 µl lysate was prepared from 100 ml of yeast culture. The 400 µl lysate was used for each immunoprecipitation (using 10 µl of anti-HA or anti-myc antibody and 100 µl of protein A/G plus agarose beads from Santa Cruz Biotechnology, Inc.), and immunoprecipitated DNA sample was dissolved in 10 µl TE 8.0 of which 1 µl was used for the PCR analysis. In parallel, the PCR analysis for ‘input’ DNA was performed using 1 µl DNA that was prepared by dissolving purified DNA from 5 µl lysate in 100 µl TE 8.0.
All ChIP experiments were repeated multiple times, and consistent results were obtained. The primer pairs used for PCR analysis are presented in Table 1. Autoradiograms were scanned and quantitated by the National Institutes of Health image 1.62 program. Immunoprecipitated (IP) DNAs were quantitated as the ratio of IP to input. The input DNA was isolated from 5% of the lysate that was used for immunoprecipitation in the normal ChIP assay. In case of modified ChIP assay for analysis of the recruitment of Rad26p to the gene loci, the input DNA was prepared from 1.25% of the lysate that was used for immunoprecipitation. A 1/100th of the input DNA was used for the PCR analysis. A 1/20th and 1/10th of the immunoprecipitated DNAs were used for the PCR analysis in the normal and modified ChIP assays, respectively.
Table 1.
The primers used in this study
| GAL1 (UAS): | 5′-CGCTTAACTGCTCATTGCTATATTG-3′ |
| 5′-TTGTTCGGAGCAGTGCGGCGC-3′ | |
| GAL1 (Core): | 5′-ATAGGATGATAATGCGATTAGTTTTT TAGCCTT-3′ |
| 5′-GAAAATGTTGAAAGTATTAGTTAAAG TGGTTATGCA-3′ | |
| GAL1 (ORF1): | 5′-CAGTGGATTGTCTTCTTCGGCCGC-3′ |
| 5′-GGCAGCCTGATCCATACCGCCATT-3′ | |
| GAL1 (ORF2): | 5′-CAGAGGGCTAAGCATGTGTATTCT-3′ |
| 5′-GTCAATCTCTGGACAAGAACATTC-3′ | |
| GAL7 (Core): | 5′-CTATGTTCAGTTAGTTTGGCTAGC-3′ |
| 5′-TTGATGCTCTGCATAATAATGCCC-3′ | |
| GAL7 (ORF): | 5′-TGAGACCTTGGTCATTTCAAAGAAG-3′ |
| 5′-ATGGATACCCATTGAGTATGGGAAA-3′ | |
| GAL10 (Core): | 5′-GCTAAGATAATGGGGCTCTTTACAT-3′ |
| 5′-TTTCACTTTGTAACTGAGCTGTCAT-3′ | |
| GAL10 (ORF): | 5′-TTAATGCGAATCATAGTAGTATCGG-3′ |
| 5′-TTACCAATAGATCACCTGGAAATTC-3′ | |
| INO1 (UAS): | 5′-TATGAAATACGTGCCGGTGTTCCGG-3′ |
| 5′-TCAATATTCTGGGAAAGAAGGATGA-3′ | |
| INO1 (Core): | 5′-TTCACATGGAGCAGAGAAAGCGCA-3′ |
| 5′-GGATAAAACTAACATTAGGAAC CCGAC-3′ | |
| INO1 (ORF1): | 5′-TGCCCATGGTTAGCCCAAACGACTT-3′ |
| 5′-AAGGAAGAGGCTTCACCAAGGACA-3′ | |
| INO1 (ORF2): | 5′-GTTCTGTCATAGATGACATCATCG-3′ |
| 5′-GGTTATGGCCACCTAACATCAACTC-3′ | |
| RPS5 (UAS): | 5′-AGAAACAATGAACAGCCTTGAG TTCTC-3′ |
| 5′-GCAGGGCCATTCTCATCTGA-3′ | |
| RPS5 (Core): | 5′-GGCCAACTTCTACGCTCACGTTAG-3′ |
| 5′-CGGTGTCAGACATCTTTGGAATGGTC-3′ | |
| RPS5 (ORF): | 5′-AGGCTCAATGTCCAATCATTGAAAG-3′ |
| 5′-CAACAACTTGGATTGGGTTTTGGTC-3′ | |
| GAL1 (whole gene): | 5′-CGCTTAACTGCTCATTGCTATATTG-3′ |
| 5′-TTTTGTCCCTGTGTTTTAAAGTTT GTGG-3′ | |
| GAL7 (whole gene): | 5′-TCATACATTCTTAAATTGCTTTGCCT CTCC-3′ |
| 5′-CTTACAAGCTGCATTGTATTCCTAA-3′ | |
| GAL10 (whole gene): | 5′-TTGTTCGGAGCAGTGCGGCGC-3′ |
| 5′-AATTTTTGATGTCTCCATGGTGGTA-3′ | |
| INO1 (whole gene): | 5′-TATGAAATACGTGCCGGTGTTCCGG-3′ |
| 5′-GGTTATGGCCACCTAACATCAACTC-3′ |
ORF, open reading frame; ORF1, towards the 5′-end of the ORF; ORF2, towards the 3′-end of the ORF; Core, core promoter; and UAS, upstream activating sequence.
DNA damage protocol
Yeast cells were grown to a desired OD600 as mentioned above in growth media, and then a concentrated solution of 4NQO in ethanol (0.4 mg/ml) was added to the growing yeast culture to a final concentration of 4 µg/ml. The 4NQO-treated cells were allowed to grow under inducible conditions at 30°C for 20 min, and then these cells were processed for the ChIP analysis. To ensure maximum damage, yeast cells were also treated with a final concentration of 16 µg/ml 4NQO in YPR, and then allowed to grow for 10 min at 30°C.
Genomic DNA preparation
The genomic DNA was extracted from 5 ml of 4NQO-treated yeast cells. Briefly, the harvested cells were suspended in 200 µl lysis buffer (50 mM HEPES, pH 7.5; 140 mM NaCl; 1 mM EDTA; 1% Triton X-100; and 0.1% Na-deoxycholate) with 200 µl volume-equivalent of glass beads, and then were vortexed for 30 min at 4°C using a Tommy vortexer. The whole-cell extract (WCE) was collected by punching a hole at the bottom of the eppendorf tube, and then was partitioned with 200 µl of phenol:chloroform:isoamylalcohol. The aqueous phase following phenol:chloroform extraction was treated with ethanol to precipitate genomic DNA.
The genomic DNA was analyzed for 4NQO-induced DNA damage at GAL1, GAL7, GAL10 and INO1 loci, using the primer pairs listed for the whole genes in Table 1. The PCR products were analyzed by agarose gel electrophoresis and ethidium bromide staining. The PCR signals were quantitated by National Institutes of Health image 1.62 program.
WCE preparation and western blot analysis
For analysis of global level of Rad26p in the SET2 deletion mutant and its isogenic wild-type equivalent, the yeast strains expressing myc epitope-tagged Rad26p were grown in 20 ml YPD up to an OD600 of 1.0, and then harvested cells were lysed to prepare the WCE following the protocol as described previously for the ChIP assay (51–53). To analyze the global level of Rad26p in the rpb1-ts mutant and wild-type strains, yeast cells were grown in YPD up to an OD600 of 0.85 at 23°C, and then shifted to 37°C for 1 h before harvesting to prepare WCE. The WCE was run on SDS–polyacrylamide gel, and then analyzed by western blot assay. The anti-myc antibody (9E10; Santa Cruz Biotechnology, Inc.) was used for western blot analysis.
Co-immunoprecipitation assay
The co-immunoprecipitation assay was performed as described previously (54). Briefly, the yeast strain carrying HA-tagged Rpb3p and myc-tagged Rad26p was grown in YPD up to an OD600 of 1.0, and then cross-linked by formaldehyde. The WCE was prepared by lysing and sonicating the cross-linked yeast cells as described in the ChIP assay. Immunoprecipitation was performed using an anti-HA antibody and protein A/G plus agarose beads. An anti-FLAG was used as a non-specific antibody. After immunoprecipitation, the agarose beads were washed as in the ChIP assay. The washed A/G plus agarose beads were boiled in the SDS–PAGE loading buffer, and then the supernatant was used for SDS–PAGE and western blot analysis. The anti-myc antibody was used in the western blot analysis. For DNAase treatment in the co-immunoprecipitation assay, the WCE was treated with 50 U of DNAase I for 30 min at 37°C in the presence of protease inhibitors prior to the addition of antibody. For co-immunoprecipitation assay in the rpb1-ts mutant strain, both the wild-type and mutant strains were grown in YPD up to an OD600 of 0.85 at 23°C, and then switched to 37°C for 1 h before harvesting.
RESULTS
Rad26p associates with the coding sequences of active, but not inactive, genes
To determine whether Rad26p is recruited to the active gene in a DNA lesion-dependent or -independent manner, we analyzed its association with an inducible yeast gene, GAL1, in the absence of DNA lesions, using a ChIP assay. The GAL1 expression is driven by the activator, Gal4p. In raffinose-containing (non-inducing) growth medium, Gal80p binds to the activation domain of Gal4p (55–57), and hence prevents the formation of the PIC assembly. Thus, the expression of GAL1 is switched off in raffinose-containing growth medium. When the yeast cells grown in raffinose-containing growth medium is transferred to galactose-containing (inducing) growth medium, Gal80p does not remain bound to the Gal4p activation domain (56,57), and hence PIC is assembled at the GAL1 core promoter (55,58,59), leading to the expression of GAL1.
To perform the ChIP assay at GAL1, we tagged Rad26p by myc-epitope at its chromosomal locus in the wild-type strain, W303a. The functionality of myc-tagged Rad26p was analyzed by the association of RNA polymerase II with the GAL1 coding sequence as previous studies (60,61) have implicated Rad26p in transcriptional elongation of GAL genes. We find that the association of RNA polymerase II with the GAL1 coding sequence is significantly decreased in Δrad26 (Supplementary Figure S1), consistent with previous studies (60,61). However, myc-epitope tagging of Rad26p does not alter the association of RNA polymerase II with the GAL1 coding sequence (Supplementary Figure S1). Further, the initial growth rate of the yeast strain following UV-irradiation was not decreased by myc-epitope tagging of Rad26p as compared to the strain containing untagged Rad26p (Supplementary Figure S2). However, the deletion of RAD26 significantly slowed down the initial growth of the yeast strain following UV-irradiation as compared to the wild type equivalent (Supplementary Figure S3). Together, these results support that myc-tagged Rad26p is functional in transcription as well as in DNA repair. In fact, epitope-tagged Rad26p has also been used previously in several studies (62–67).
Using the strain bearing myc epitope-tagged Rad26p, we performed the ChIP assay at the upstream activating sequence (UAS), core promoter, and two different regions (ORF1, towards the 5′-end; and ORF2, towards the 3′-end) within the ORF of GAL1 (Figure 1A, bottom panel), following the protocol as described in the Materials and Methods section. Figure 1A (top panel) shows that myc-tagged Rad26p was predominantly associated with the coding sequence (ORF1 and ORF2) of GAL1 under inducible conditions. An anti-HA was used as a non-specific antibody, and thus, provided background signal in the ChIP assay. It is clear from the Figure 1A that the ChIP signal arising from myc-tagged Rad26 at the GAL1 coding sequence is much higher than that of the anti-HA. Thus, Rad26p is specifically associated with the coding sequence, but not promoter, of the active GAL1 gene, even in the absence of the DNA lesions.
Figure 1.

Rad26p is associated with the coding sequence but not promoter of the GAL1 gene in a transcription-dependent manner. (A) Top panel: analysis of the recruitment of Rad26p at the UAS, core promoter, ORF of the GAL1 gene. The yeast strain expressing myc-tagged Rad26p was grown in YPG up to an OD600 of 1.0 prior to cross-linking. The ChIP assay was performed as described in the Materials and Methods section. Immunoprecipitation was performed using a mouse monoclonal antibody against the c-myc epitope-tag (9E10; Santa Cruz Biotechnology, Inc.). An anti-HA (Santa Cruz Biotechnology, Inc.) was used as a non-specific antibody. Both the input and immunoprecipitated DNA samples were analyzed by PCR. The dilution factors for input and immunoprecipitated DNA samples are mentioned in the modified ChIP protocol (see ‘Materials and Methods’ section). The immunoprecipitated DNA was quantitated as the ratio of immunoprecipitate over the input in the autoradiogram. The fold enrichment of Rad26p at the GAL1 coding sequence with respect to UAS is presented. IP, immunoprecipitate. Bottom panel: the PCR primer pairs located at the UAS, core promoter, and towards the 5′-(ORF1) and 3′-(ORF2) ends of the ORF of the GAL1 gene. (B) RNA polymerase II is associated with the coding sequence of the active GAL1 gene. The yeast strain was grown in raffinose (YPR)- or galactose (YPG)-containing growth medium up to an OD600 of 1.0 prior to crosslinking. Immunoprecipitations were performed using a mouse monoclonal antibody 8WG16 (Covance) against the carboxy terminal domain of the largest subunit (Rpb1p) of RNA polymerase II. (C) Rad26p is associated with the ORF of the active GAL1 gene. The fold enrichment of Rad26p at the GAL1 coding sequence in galactose-containing growth medium in comparison to raffinose-containing growth medium is presented. (D) Rpb1p is essential for recruitment of Rad26p to the GAL1 coding sequence. (E) Analysis of the global levels of Rad26p in the rpb1-ts and wild-type strains at the non-permissive temperature. The yeast strain expressing myc epitope-tagged Rad26p were grown in YPD medium at permissive (23°C) and non-permissive (37°C) temperatures as discussed in the ‘Materials and Methods’ section. The WCE was run on SDS–polyacrylamide gel, and then analyzed by western blot assay. (F) Rad26p interacts with RNA polymerase II as revealed by the co-immunoprecipitation assay. (G) Analysis of Rad26p association with RNA polymerase II in the rpb1-ts mutant using a co-immunoprecipitation assay. (H) The co-immunoprecipitation assay as in panel G in the presence of DNAase. (I) Analysis of DNA digestion as a control for the experiments presented in (H). PCR-amplified DNA was treated with DNAase for 30 min at 37°C, and then was analyzed by agarose gel electrophoresis.
The fact that Rad26p is associated with the GAL1 coding sequence under inducible conditions indicates the requirement of active transcription for its association. To test this possibility, we next analyzed the association of Rad26p with the GAL1 coding sequence under non-inducible conditions. We find that, like RNA polymerase II, Rad26p was also not associated with the GAL1 coding sequence when GAL1 transcription was switched off under non-inducible conditions (Figures 1B and C). Consistent with this observation, we also find that the association of Rad26p with the GAL1 coding sequence was significantly impaired in the rpb1-ts mutant strain (Figure 1D). However, such an impaired association of Rad26p could be due to the loss of its stability in the rpb1-ts mutant strain at non-permissive temperature. To test this possibility, we analyzed the global level of Rad26p in the RPB1 wild-type and ts mutant strains at permissive (23°C) as well as non-permissive (37°C) temperatures. We find that the stability of Rad26p was not altered in the rpb1-ts mutant strain while Rpb1p was significantly degraded at non-permissive temperature (Figure 1E). The level of TBP was monitored as a loading control, and it was unchanged (Figure 1E). Thus, RNA polymerase II or active transcription plays an important role in association of Rad26p with the GAL1 coding sequence.
Since Rad26p and RNA polymerase II were associated with the GAL1 coding sequence, it seems likely that Rad26p might be associated with RNA polymerase II either directly or indirectly during transcriptional elongation. To test this possibility, we analyzed the association of Rad26p with RNA polymerase II using a cross-linking-based co-immunoprecipitation assay as described previously (54). In this direction, we generated a strain carrying myc-tagged Rad26p and HA-tagged Rpb3p subunit of RNA polymerase II. Using this strain, we performed the co-immunoprecipitation assay as schematically shown in Figure 1F (top panel). An anti-Flag was used as a non-specific antibody in this assay. Figure 1F (bottom panel) shows the association of Rad26p with RNA polymerase II. Such an association was significantly impaired in the rpb1-ts mutant strain (Figure 1G), thus implying the association of Rad26p with structurally and/or functionally intact RNA polymerase II, since Rpb1p maintains the structural/functional integrity of RNA polymerase II (47,68–72). Further, this association is not mediated by DNA as the treatment of WCE with DNAase did not alter the interaction of Rad26p with RNA polymerase II (Figures 1H and I). However, we cannot rule out the possibility that DNA might be playing an important role to bring Rad26p and RNA polymerase II in proximity prior to cross-linking. Nonetheless, our data demonstrate that Rad26p is associated with RNA polymerase II. However, whether this association is either direct or indirect remains to be elucidated. Consistent with our observations, Li and Smerdon (73) have also demonstrated the genetic interaction between Rad26p and RNA polymerase II.
Next, we asked whether Rad26p is associated with other genes in a transcription-dependent manner. To address this question, we analyzed the association of Rad26p with the promoters and coding sequences of two other inducible GAL genes, namely GAL7 and GAL10 (Figure 2A, right panel), under both inducible and non-inducible conditions. We find that Rad26p was associated with the coding sequences but not promoters of GAL7 and GAL10 under inducible conditions (Figures 2A and B). Thus, like GAL1, the GAL7 and GAL10 genes recruit Rad26p at their coding sequences in a transcription-dependent manner. Likewise, Rad26p was also found to be associated with the coding sequence, but not promoter, of a different class of inducible gene, INO1, in a transcription-dependent manner (Figure 2C). Similar association of Rad26p was also observed at the constitutively active gene, RPS5 (Figure 2D). Taken together, our data presented in Figures 1 and 2 demonstrate that Rad26p associates with the coding sequences of the active genes, indicating a possible role of Rad26p in transcriptional elongation. Indeed, Lee et al. (60,61) have implicated Rad26p in the positive regulation of transcriptional elongation.
Figure 2.

Rad26p is associated with the coding sequences but not promoters of the genes in a transcription-dependent manner. (A) Rad26p is associated with the coding sequences of the active GAL7 and GAL10 genes. The ChIP assay was performed as in Figure 1A. The fold enrichment of Rad26p at the coding sequence in comparison to the core promoter is presented. (B) Rad26p is associated with the ORFs of GAL7 and GAL10 in a transcription-dependent manner. The fold enrichment of Rad26p at the coding sequence in galactose-containing growth medium in comparison to raffinose-containing growth medium is presented. (C) Rad26p is associated with the coding sequence of the INO1 gene in a transcription-dependent manner. The yeast strain expressing myc-tagged Rad26p was induced for INO1 as described in the ‘Materials and Methods’ section. Primer pairs located at the UAS, core promoter, two different locations (ORF1 and ORF2) of ORF of INO1 (bottom panel) were used for the PCR analysis of the immunoprecipitated DNA samples. +, induced stated; and −, repressed state. The ChIP assay was performed as in Figure 1A. The fold enrichment of Rad26p at different locations of INO1 under transcriptionally active conditions in comparison to inactive conditions is presented. (D) Rad26p is associated with the coding sequence, but not promoter, of a constitutively active gene, RPS5. The yeast strain carrying myc epitope-tagged Rad26p was grown in YPD up to an OD600 of 1.0 prior to cross-linking. The ChIP assay was performed as in Figure 1A. The fold enrichment of Rad26p at the RPS5 coding sequence with respect to UAS is presented.
The DNA lesion alone does not target the recruitment of Rad26p in the absence of active transcription
In addition to its role in transcriptional elongation, Rad26p has been implicated to recognize DNA lesions in TC-NER for signaling recruitment of other NER factors (45,74–82). However, it is not known whether DNA lesion alone can be sufficient enough to recruit Rad26p in the absence of active transcription. With this view, we introduced DNA lesions by 4NQO at GAL1 when its transcription was switched off in raffinose-containing growth medium. 4NQO interacts with DNA, and forms bulky adducts primarily with guanine bases and to a lesser extent with adenine bases. These bulky adducts are removed by NER. Figure 3A shows the presence of 4NQO-induced DNA lesions at the GAL1 gene based on significant loss of the PCR signal, consistent with our previous studies (47). We next asked whether Rad26p is recruited to DNA lesions at GAL1 in the absence of active transcription. In this direction, we analyzed the recruitment of Rad26p at GAL1 in raffinose-containing medium following 4NQO-treatment. Figure 3B shows that the recruitment of Rad26p at the promoter as well as coding sequence of GAL1 was not induced by DNA lesions. Further, in raffinose-containing growth medium, RNA polymerase II was not associated with GAL1 (Figure 1B). Thus, DNA lesion alone does not induce the recruitment of Rad26p to GAL1 in the absence of active transcription or RNA polymerase II.
Figure 3.

The DNA lesion alone does not target the recruitment of Rad26p in the absence of active transcription. (A) Analysis of DNA damage at the GAL1, GAL7 and GAL10 loci within the first 20 min of 4NQO treatment. The yeast cells were grown in YPR, and then treated with 4NQO for 20 min. The genomic DNA was prepared and was analyzed by PCR. (B) The DNA lesions at the GAL1 gene do not target the recruitment of Rad26p in the absence of active transcription. The yeast strain carrying myc-tagged Rad26p was grown in YPR up to an OD600 of 1.0, and then treated with 4NQO for 20 min prior to cross-linking. The ChIP assay was carried out as in Figure 1A. The fold change of Rad26p ChIP signal at GAL1 in 4NQO-treated cells in comparison to untreated cells under transcriptionally inactive conditions. The DNA lesions at the GAL10 (C) and GAL7 (D) genes also do not target the recruitment of Rad26p in the absence of active transcription. (E) Analysis of DNA damage at the INO1 gene within the first 20 min of 4NQO treatment. Yeast cells were grown in synthetic complete medium containing 100 µM inositol at 30°C up to an OD600 of 1.0, and then treated with 4NQO for 20 min. The whole INO1 gene was amplified by PCR, using the specific primer pairs as mentioned in Table 1. (F) The DNA lesions at the INO1 gene do not target the recruitment of Rad26p in the absence of active transcription. The yeast strain expressing myc-tagged Rad26p was grown in the synthetic complete medium containing 100 µM inositol at 30°C up to an OD600 of 1.0, and then treated with 4NQO for 20 min prior to cross-linking. (G) Analysis of DNA damage at the GAL1 locus within the first 10 min of 4NQO treatment at a concentration of 16 µg/ml. The yeast cells were grown in YPR and treated with 4NQO as in (A). The whole GAL1 locus, core promoter, ORF1 and ORF2 regions were amplified by PCR, using the specific primer pairs as mentioned in Table 1. (H) Analysis of the association of Rad26p with GAL1 following 4NQO treatment at the concentration of 16 µg/ml in the absence of transcription. Yeast cells were grown in YPR. The ChIP assay was performed as in Figure 1A. The fold change of Rad26p ChIP signal at GAL1 in 4NQO-treated cells in comparison to untreated cells under transcriptionally inactive conditions.
To determine whether DNA lesions present at other inactive genes can target Rad26p, we analyzed the recruitment of Rad26p to GAL7 and GAL10 in raffinose-containing growth medium following 4NQO-treatment. Figure 3A shows the presence of 4NQO-induced DNA lesions at GAL7 and GAL10 based on the loss of PCR signal, consistent with our previous studies (47). However, 4NQO-induced DNA lesions at GAL7 and GAL10 do not induce the recruitment of Rad26p in raffinose-containing growth medium (Figures 3C and 3D). Similarly, 4NQO-induced lesions at INO1 (Figure 3E) do not trigger the recruitment of Rad26p in the absence of active transcription (Figure 3F). Collectively, our data presented in Figures 3A–F demonstrate that DNA lesions alone do not induce the recruitment of Rad26p in the absence of active transcription.
To determine whether severe DNA damage at higher 4NQO concentration can induce the recruitment of Rad26p at the inactive coding sequence, we treated yeast cells carrying myc-tagged Rad26p in raffinose-containing growth medium with 4NQO at a final concentration of 16 µg/ml for 10 min. Then, we analyzed the recruitment of Rad26p to GAL1. Figure 3G shows that the whole GAL1 gene was severely damaged following 4NQO treatment. However, the smaller regions (ChIP PCR regions such as GAL1-Core, GAL1-ORF1 and GAL1-ORF2 with an average size of ∼150 bp) of GAL1 were not significantly damaged, thus allowing the ChIP PCR as schematically shown in Figure 1A (bottom panel). Figure 3H shows that even severe damage at GAL1 does not induce the recruitment of Rad26p in the absence of active transcription.
Although our data demonstrate that DNA lesions present at the inactive gene cannot induce the recruitment of Rad26p, it is quite possible that 4NQO might be generating lesions at or around the ChIP PCR region (∼150 bp) in a small fraction of cells. Such a possibility could lead to our inability to detect the recruitment of Rad26p at the inactive coding sequence in response to 4NQO-induced DNA damage. Furthermore, the presence of DNA lesion within the ChIP PCR region will inhibit PCR, and thus, the recruitment of Rad26p to the lesion within the ChIP PCR region would not be detected. However, our data in Figure 4B show that PCR signal at the ChIP PCR region (i.e. GAL1-ORF1) is not significantly altered, thus indicating insufficient damage at the ChIP PCR region. This is consistent with our previous study (47). However, the recruitment of Rad26p at the DNA lesions around the ChIP PCR region can be detected in the ChIP analysis (Figure 4C), since DNA fragments of ∼500 bp or less are generated by sonication (Figure 4A) and significant fractions (∼50%) of cells have DNA lesions within 300–500 bp fragments of the GAL1 coding sequence (Figure 4B). Thus, if Rad26p is recruited within ∼500 bp fragment around the ChIP PCR region of the GAL1 ORF (i.e. GAL1-ORF1), we would detect it in the ChIP assay as schematically shown in Figure 4C. However, we do not observe the recruitment of Rad26p to the GAL1-ORF1 in response to 4NQO-induced DNA damage in raffinose-containing growth medium (Figure 3B and H). Thus, DNA lesions alone do not appear to recruit Rad26p in the absence of transcription. However, it may be quite possible that the recruitment of Rad26 to the site of DNA lesion may make the site more vulnerable to sonication, which in turn could lead to our inability to observe the Rad26p ChIP signal as schematically shown in Figure 4C. In fact, we observe the recruitment of Rad26p to the GAL1 coding sequence following 4NQO-induced DNA damage in a transcription-dependent manner (see below; Figure 5B and C). Further, we observe the recruitment of Rad3p to the active GAL1 coding sequence in response to 4NQO-induced DNA damage (see below; Figure 5D). Therefore, the DNA lesion sites associated with Rad26p do not seem to be more readily broken during chromatin shearing than those not associated with Rad26p. This is further supported by the fact that the association of Gal4p with the GAL1 UAS was not altered following 4NQO-induced DNA damage in our ChIP analysis (Supplementary Figure S4).
Figure 4.

Analysis of DNA lesion in the GAL1 coding sequence. (A) Analysis of the size of DNA fragments in the ChIP assay. Yeast cells were grown in YPR prior to cross-linking as in Figure 3B. The genomic DNA was isolated following sonication (7 times, 10 s each) of WCE and was analyzed by agarose gel electrophoresis. (B) Analysis of DNA damage at GAL1 with varying lengths of coding sequence. Yeast cells were grown, cross-linked and sonicated as in (A). Cells were treated with 4NQO at a final concentration of 4 µg/ml as described in Figure 3A. DNA was isolated and analyzed by PCR using specific primer pairs. PCR products with different sizes were analyzed by 2% agarose gel electrophoresis. (C) The schematic diagram for the analysis of Rad26p recruitment at the site of DNA lesion in the ChIP assay. The ‘star’ represents the site of DNA lesion.
Figure 5.

RNA polymerase II promotes the recruitment of Rad26p to the DNA lesions at the coding sequences of the active GAL1 and GAL10 genes. (A) Analysis of the association of RNA polymerase II with the GAL1 coding sequence in the presence and absence of 4NQO-induced DNA damage. The yeast strain was grown in YPR up to an OD600 of 0.8 at 30°C, and then transferred to YPG for 90 min to induce GAL1 prior to 4NQO treatment for 20 min. As a control, yeast cells were also grown under similar growth conditions without 4NQO treatment. These cells were used for the ChIP assay to analyze the level of Rpb1p at the GAL1 ORF in 4NQO-treated (+) and untreated (−) cells. The percentage of DNA immunoprecipitated in the 4NQO-treated cells relative to that of the untreated cells is represented as ‘%IP of -4NQO’. (B) Analysis of Rad26p recruitment towards the 5′- and 3′-ends of the GAL1 ORF in the presence and absence of 4NQO-induced DNA damage. The yeast strain carrying myc-tagged Rad26p was grown and crosslinked as in (A). Immunoprecipitation was performed as in Figure 1A. (C) The ChIP experiments at the GAL1 coding sequence using the strain that bears untagged Rad26p. An anti-myc antibody was used in the ChIP assay. (D) Analysis of Rad3p recruitment at the GAL1 coding sequence following 4 µg/ml 4NQO treatment. Immunoprecipitation was performed using the modified ChIP protocol as described in the ‘Materials and Methods’ section. (E) Analysis of the association of Rad26p and RNA polymerase II with the GAL10 ORF.
Rad26p is recruited to the sites of DNA lesions at the coding sequences of the active genes with the help of RNA polymerase II
How does Rad26p get recruited to the site of DNA lesion at the coding sequence of the active gene in TC-NER? We show here that Rad26p is co-associated with RNA polymerase II at the coding sequences of the active genes (Figures 1 and 2). RNA polymerase II stalls when it encounters the DNA lesion (24,31,32,36,38,41–46). Further, we have recently demonstrated that elongating RNA polymerase II is disassembled through the degradation of its largest subunit at the coding sequences of several active genes in response to DNA damage (47). Based on these observations, we hypothesize that elongating RNA polymerase II promotes the recruitment of Rad26p to the site of damaged DNA followed by its disassembly through degradation of its largest subunit. According to this hypothesis, if elongating RNA polymerase II delivers Rad26p to DNA lesion, Rad26p would remain associated with the coding sequence even after the disassembly of elongating RNA polymerase II from the site of DNA lesion. To test this hypothesis, we analyzed the association of RNA polymerase II (Rpb1p) and Rad26p with the GAL1 coding sequence at 20 min following 4NQO-treatment in galactose-containing growth medium. The 4NQO-treatment time was chosen to be 20 min based on our recent studies (47). Within this 4NQO-treatment time, the association of RNA polymerase II with the GAL1 ORF was significantly lost due to the disassembly of RNA polymerase II (47). As a control, the association of RNA polymerase II and Rad26p with the GAL1 coding sequence was also analyzed without 4NQO-treatment. Figure 5A (left panel) shows significant loss of RNA polymerase II association with the 5′-end of the GAL1 coding sequence (ORF1) in 4NQO-treated cells as compared to that of the untreated cells. Such a loss of RNA polymerase II association with the GAL1 coding sequence has been attributed in our recent studies (47) to the disassembly of elongating RNA polymerase II through the degradation of its largest subunit at the site of DNA lesion. Interestingly, even when association of RNA polymerase II with the GAL1 ORF1 was significantly lost in 4NQO-treated cells, Rad26p was retained at the GAL1 ORF1 (Figure 5B, left panel). In contrast, Rad26p did not remain significantly associated with the GAL1 ORF in the absence of RNA polymerase II or active transcription when DNA was not damaged by 4NQO (Figure 1C and D). Together, these observations support the retention of Rad26p at the GAL1 coding sequence following the disassembly of RNA polymerase II in response to 4NQO-induced DNA damage. As a negative control, we show the absence of the Rad26p ChIP signal in the strain bearing untagged Rad26p, using an anti-myc antibody in the ChIP assay (Figure 5C). To verify as a positive control whether DNA lesions in the active coding sequence can also induce the recruitment of other DNA repair factors, like Rad26p, we analyzed the recruitment of Rad3p, a DNA repair factor with a helicase activity that is involved in unwinding DNA duplex around the lesion, to the active GAL1 coding sequence in response to 4NQO-induced DNA damage. We find that Rad3p is recruited to the active GAL1 coding sequence in response to 4NQO-induced DNA damage (Figure 5D). Thus, like Rad26p, Rad3p is also recruited to the coding sequence of active gene following 4NQO-induced DNA damage.
Next, we analyzed the association of RNA polymerase II and Rad26p with the 3′-end of the GAL1 coding sequence following 20 min 4NQO-treatment. Similar to the 5′-end, association of RNA polymerase II with the 3′-end of the GAL1 coding sequence was lost in 4NQO-treated cells (Figure 5A, right panel). However, like RNA polymerase II, Rad26p was also not significantly associated with the 3′-end of the GAL1 coding sequence following 4NQO-treatment (Figure 5B, right panel). Such an observation can be attributed to the fact that RNA polymerase II did not reach the 3′-end of the GAL1 coding sequence due to the presence of multiple DNA lesions (within ∼1200 base pair fragment) ahead of the 3′-end in the continuously 4NQO-treated cells as shown in our previous studies (47). Thus, the association of RNA polymerase II with the 3′-end of the GAL1 coding sequence was not observed in 4NQO-treated cells (Figure 5A, right panel). Since RNA polymerase II did not reach the 3′-end of the GAL1 coding sequence, it did not carry Rad26p. Thus, the association of Rad26p with the 3′-end of the GAL1 ORF was not observed in 4NQO-treated cells (Figure 5B, right panel). Together, our data support the role of elongating RNA polymerase II in recruitment of Rad26p to the site of DNA lesion.
To determine whether RNA polymerase II also promotes the recruitment of Rad26p at the coding sequences of other active genes in response to DNA damage, we analyzed the association of Rad26p and RNA polymerase II with the coding sequence of another GAL gene, namely GAL10, in 4NQO-treated cells. Figure 5E shows that the association of RNA polymerase II with the GAL10 coding sequence was significantly decreased in 4NQO-treated cells, consistent with our recent studies (47). However, the association of Rad26p with the GAL10 coding sequence remained unchanged (Figure 5E) even when the association of RNA polymerase II was significantly decreased in 4NQO-treated cells in comparison to that of the untreated cells (Figure 5E). Thus, similar to the results obtained at GAL1, we find that Rad26p remained associated with the GAL10 coding sequence following the disassembly of elongating RNA polymerase II in response to 4NQO-induced DNA damage. Next, we extended this study to a different inducible gene, INO1. We find that the association of Rpb1p with the INO1 coding sequence was significantly decreased in response to 4NQO-induced DNA damage (Figure 6A), consistent with our previous studies at GAL1, GAL7 and GAL10 (47). However, the association of Rad26p with the 5′-end of the INO1 coding sequence remained unchanged (Figure 6B and C), similar to the results obtained at GAL1 and GAL10 (Figure 5B and E). The association of Rad26p with the 3′-end of the INO1 coding sequence was moderately decreased. Collectively, our data presented in Figures 5 and 6 demonstrate the retention of Rad26p at the coding sequences of the active genes following the degradation of Rpb1p in response to 4NQO-induced DNA damage.
Figure 6.

RNA polymerase II promotes the recruitment of Rad26p to the site of DNA lesion at the coding sequence of the active INO1 gene. (A) Analysis of association of RNA polymerase II with the INO1 coding sequence in the presence and absence of 4NQO-induced DNA damage. The INO1 gene was induced prior to 4NQO treatment (4 µg/ml) for 20 min as described in the ‘Materials and Methods’ section. As a control, yeast cells were also grown under similar growth conditions without 4NQO treatment. These cells were used for the ChIP assay to analyze the level of Rpb1p at the INO1 ORF in 4NQO-treated (+) and untreated (−) cells. (B) Analysis of the recruitment of Rad26p towards the 5′- and 3′-ends (ORF1 and ORF2, respectively) of the INO1 ORF in the presence and absence of the 4NQO-induced DNA damage. The yeast strain carrying myc-tagged Rad26p was grown and cross-linked as in (A). Immunoprecipitation was performed as in Figure 1A. (C) The data of the (A and B) were plotted in the form of a histogram.
Histone H3K36 methylation enhances the association of Rad26p with the active coding sequence
Our data demonstrate that RNA polymerase II or active transcription is essential for association of Rad26p with the coding sequences of the active genes either in the presence or absence of DNA lesions. Next, we asked whether RNA polymerase II plays a direct or indirect role in recruitment of Rad26p. There can be at least three different scenarios: (i) elongating RNA polymerase II evicts histones, and, subsequently, allows free DNA to interact with Rad26p; (ii) Rad26p is associated with the coding sequence in a RNA polymerase II-dependent manner; and (iii) RNA polymerase II covalently modifies nucleosomal histones at the coding sequence indirectly by recruiting histone modification enzymes, and such modifications may trigger association of Rad26p. We rule out the first possibility based on the fact that Rad26p was not associated with the relatively nucleosome-free promoter region of GAL1 (83) (Figure 1A). Our data support the second possibility, since the recruitment of Rad26p is dependent on RNA polymerase II or active transcription (Figure 1C–I). However, histone covalent modifications that are mediated by RNA polymerase II or active transcription at the coding sequence could also stimulate the association of Rad26p. Thus, to test this third possibility, we analyzed the roles of histone H3 K4 and K36 methylation in recruitment of Rad26p, since these covalent modifications are mediated by elongating RNA polymerase II or active transcription through Set1p and Set2p histone methyltransferases, respectively. We find that histone H3 K4 methylation or Set1p is dispensable for the recruitment of Rad26p (Figure 7A). Interestingly, the recruitment of Rad26p to GAL1 was significantly impaired in the absence of Set2p (Figure 7A) that is essential for histone H3 K36 methylation at the GAL1 coding sequence (Figure 7B and C). However, such an impaired recruitment of Rad26p in the absence of histone H3 K36 methylation could be due to the reduced association of elongating RNA polymerase II or the decreased stability of Rad26p. To test these possibilities, we analyzed the association of Rpb1p with the GAL1 coding sequence in the SET2 deletion mutant and its isogenic wild-type equivalent. Figure 7D shows that the association of Rpb1p with the GAL1 coding sequence was modestly altered in the absence of histone H3 K36 methylation or Set2p. In contrast, a dramatic decrease of Rad26p recruitment was observed in Δset2 (Figure 7A). Further, the stabilities of Rad26p and Rpb1p were not altered in Δset2 (Figure 7E). Thus, our data demonstrate that histone H3 K36 methylation stimulates the association of Rad26p with the coding sequence of the active gene, and hence the factors (e.g. Ctk1p, RNA polymerase II and general transcription factors) involved in histone H3 K36 methylation are likely to alter indirectly the recruitment of Rad26p.
Figure 7.

Methylation of K36 but not K4 on histone H3 stimulates the association of Rad26p with the coding sequence of the active gene. (A) Histone H3K36 methylation stimulates the recruitment of Rad26p to the coding sequence of the active GAL1 gene. The deletion mutants of SET1 and SET2, and wild-type strains were grown in YPR up to an OD600 of 0.8 at 30°C, and then transferred to YPG for 90 min to induce GAL1 prior to cross-linking. The immunoprecipitations were performed as described in Figure 1A. The normalized occupancies of Rad26p at the GAL1 ORF in the wild-type and deletion mutant strains were plotted in the form of a histogram. (B) Histone H3 associated with the coding sequence of the active GAL1 gene is methylated on K36. Yeast cells were grown and cross-linked as in (A). Immunoprecipitation was performed using an anti-H3K36me3 antibody (Abcam-ab9050) as described for normal ChIP assay (see ‘Materials and Methods’ section). (C) Set2 is essential for histone H3 K36 methylation. The yeast strains were grown in YPG up to an OD600 prior to cross-linking. (D) Analysis of the association of RNA polymerase II with the GAL1 coding sequence in the absence of histone H3 K4/36 methylation. The wild-type and deletion mutant strains were grown and crosslinked as in (A). The immunoprecipitations were performed as in Figure 1B. The normalized occupancies of RNA polymerase II (Rpb1p) at the GAL1 ORF in the presence and absence of histone H3 K4/36 methylation were plotted in the form of a histogram. (E) Analysis of the global levels of Rad26p and Rpb1p in the wild-type and SET2 deletion mutant strains. Both the wild-type and mutant strains were grown as in (A) for western blot analysis. An anti-myc antibody was used against myc-tagged Rad26 in the western blot analysis. A mouse monoclonal antibody 8WG16 (Covance) against the carboxy terminal domain of Rpb1p was used.
Several studies (84–88) have implicated chromo, tudor, PHD (plant homeodomain) finger, PWWP and MBT (malignant brain tumor) domains in the recognition of methylated-lysine residues on histones H3 and H4. Therefore, it is likely that the stimulation of Rad26p recruitment to the coding sequence of the active gene is mediated by one of the above methylated-lysine recognition domains. To test this possibility, we performed the bioinformatic analysis which revealed the absence of the methylated-lysine recognition domain in Rad26p. Thus, Rad26p seems to interact indirectly with methylated-K36 on histone H3. In support of this model, Krogan et al. (89) have demonstrated the genetic interaction of Rad26p with Sin3p which is a common component of Rpd3S (the smaller version of a multi-subunit complex with histone deacetylase activity) and Rpd3L (the larger version of a multi-subunit complex with histone deacetylase activity). Rpd3L is recruited to the promoter while Rpd3S associates with the coding sequence of the active gene to prevent cryptic transcriptional initiation (90–93). The association of Rpd3S with the coding sequence depends on histone H3K36 methylation (90–93). Further, Li et al. (94) have demonstrated the requirement of a combination of the PHD finger and chromo domain of the specific Rco1p and Eaf3p components of Rpd3S for its interaction with methylated-K36 on histone H3. Thus, histone H3 K36 methylation appears to exert its stimulatory role in recruitment of Rad26p via the interaction of Rad26p with the Sin3p component of Rpd3S. However, such a model remains to be elucidated.
DISCUSSION
TC-NER and GG-NER fundamentally follow similar mechanisms except the recognition step of DNA lesion. Such a recognition step has been implicated in differential kinetics of these two repair processes. Here, we show that a TCR-specific factor, Rad26p, is recruited to the coding sequence of the active genes in response to 4NQO-induced DNA damage with the help of RNA polymerase II. On the other hand, search for the damaged DNA at the inactive genes or silent areas of the genome by Rad4p/Rad23p (XPC/HHR23B in human) is not augmented by elongating RNA polymerase II in GG-NER, since elongating RNA polymerase II is not associated with the inactive genes or silent areas of genome. Thus, our study provides important information for the rapid rate of TC-NER as compared to the slower GG-NER.
How is Rad26p recruited to the site of DNA lesion in the presence of elongating RNA polymerase II? Our data support that Rad26p is co-recruited with elongating RNA polymerase II to the coding sequences of the active genes. When RNA polymerase II stalls at the damaged DNA site, it is disassembled through the degradation of its largest subunit (47), leaving behind Rad26p to the site of DNA lesion. This is consistent with previous biochemical studies (95) that demonstrated the formation of a stable complex of the human homologue of Rad26p with elongating RNA polymerase II at the DNA pause site. Here, we find that elongating RNA polymerase II-mediated histone H3K36 methylation also enhances the association of Rad26p with the coding sequence of the active gene. Further, following its pause at the 4NQO-induced damaged DNA site, elongating RNA polymerase II might be changing its conformation in vivo, and such a conformational change may help Rad26p to interact more stably with DNA lesion. Moreover, Rad26p might be recognizing DNA lesion at the active gene in the presence of other, yet unknown, factor(s) in vivo. Additionally, Rad26p might be efficiently binding to the site of DNA lesion that has an altered conformation via the modulation of local base-stacking as well as Watson–Crick hydrogen bonding as has been structurally shown for the recognization of DNA lesion by Rad4p (96). However, these possibilities of Rad26p recognition of the DNA lesion in TC-NER remain to be further investigated. Nonetheless, our study demonstrates in vivo that Rad26p is recruited to 4NQO-induced DNA lesion with the help of elongating RNA polymerase II, and subsequently, RNA polymerase II is disassembled through the degradation of its largest subunit. Once Rad26p is recruited to the DNA lesion, it becomes independent on the retention of RNA polymerase II. Similar results were also obtained in case of UV-induced DNA lesions (Zhen Duan and S.R.B., unpublished data). Thus, the mode of Rad26p recognition of the lesion at the active genes seems to be independent of the nature of the single strand DNA damaging agents.
We find the loss of RNA polymerase II at the coding sequences, but not promoters, of the active GAL1, GAL7, GAL10 and INO1 genes following 4NQO-induced DNA damage. Such a loss of RNA polymerase II has been attributed in our previous studies (47) to the disassembly of elongating RNA polymerase II through the degradation of its largest subunit in response to 4NQO-induced DNA damage in vivo. Consistent with our data in yeast, a recent study (97) has also reported similar results in living mammalian cells. Such disassembly of elongating RNA polymerase II makes the damaged DNA accessible to the DNA repair factors, since elongating RNA polymerase II masks DNA lesion (40,98,99). Following the disassembly of elongating RNA polymerase II, Rad26p is left behind at the site of DNA lesion in vivo. Is Rad26p recruited to the DNA lesion after the disassembly of RNA polymerase II? We rule out this possibility, based on the fact that the DNA lesion at the coding sequence of inactive gene fails to recruit Rad26p. Thus, Rad26p is recruited to the site of DNA lesion prior to the disassembly of elongating RNA polymerase II.
Following its recruitment, how does Rad26p participate in DNA repair? Rad26p has a DNA-dependent ATPase activity (100,101), and thus, upon its recruitment to the site of DNA lesion, Rad26p might be altering the chromatin structure around the damaged DNA, hence facilitating the access of the DNA repair factors to the site of DNA lesion. Indeed, a previous study (67) has demonstrated the requirement of the chromatin remodeling activity of Rad26p in DNA repair. Furthermore, Rad26p might be contributing to the DNA repair process by recruiting DNA repair factors through its direct physical interaction. In support of this model, several studies (79–81) have previously demonstrated the physical interaction of Rad26p with Rad3p. Rad3p, in turn, facilitates the incision of the DNA strand containing the lesion for subsequent steps of DNA repair (82). Thus, Rad26p appears to facilitate DNA repair at the coding sequences of the active genes by modulating the chromatin structure to enhance recruitment of the DNA repair factors and/or by directly interacting with the DNA repair factor(s). However, this model remains to be elucidated in vivo.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
American Heart Association - National Scientist Development Grant (0635008N); American Cancer Society - Research Scholar Grant (06-52); an Edward Mallinckrodt, Jr. Foundation award; a Southern Illinois University Cancer Institute - RFA Grant; several internal grants of Southern Illinois University; pre-doctoral fellowship (0710187Z) from American Heart Association (to A.S.). Funding for open access charge: Edward Mallinckrodt, Jr. Foundation award.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Shruti Bagla, Zhen Duan, Pratibha Bajwa, Kathy Yee and Gaurav Tuli for technical assistance; and Sarah Frankland-Searby for critical reading of the article.
REFERENCES
- 1.Sancar A, Lindsey-Boltz LA, Unsal-Kaccmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 2.Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. 2nd edn. New York: McGraw-Hill; 2002. pp. 211–237. [Google Scholar]
- 3.Cooper PK, Nouspikel T, Clarkson SG, Leadon SA. Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science. 1997;275:990–993. doi: 10.1126/science.275.5302.990. [DOI] [PubMed] [Google Scholar]
- 4.Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse. Science. 2003;299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 5.Sands AT, Abuin A, Sanchez A, Conti CJ, Bradley A. High susceptibility to ultraviolet-induced carcinogenesis in mice lacking XPC. Nature. 1995;377:162–165. doi: 10.1038/377162a0. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell JR, Hoeijmakers JH, Niedernhofer LJ. Divide and conquer: nucleotide excision repair battles cancer and ageing. Curr. Opin. Cell Biol. 2003;15:232–240. doi: 10.1016/s0955-0674(03)00018-8. [DOI] [PubMed] [Google Scholar]
- 7.Cromie GA, Connelly JC, Leach DR. Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol. Cell. 2001;8:1163–1174. doi: 10.1016/s1097-2765(01)00419-1. [DOI] [PubMed] [Google Scholar]
- 8.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. Washington, DC: ASM Press; 1995. [Google Scholar]
- 9.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 10.Hartwell LH. Role of yeast in cancer research. Cancer. 1992;69:2615–2621. doi: 10.1002/1097-0142(19920515)69:10<2615::aid-cncr2820691036>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 11.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 12.Kanaar R, Hoeijmakers JH, van Gent DC. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998;8:483–489. doi: 10.1016/s0962-8924(98)01383-x. [DOI] [PubMed] [Google Scholar]
- 13.Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- 14.Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne syndrome. Mol. Cell. 2005;20:187–198. doi: 10.1016/j.molcel.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 15.Batty DP, Wood RD. Damage recognition in nucleotide excision repair of DNA. Gene. 2000;241:193–204. doi: 10.1016/s0378-1119(99)00489-8. [DOI] [PubMed] [Google Scholar]
- 16.Friedberg EC. DNA damage and repair. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 17.Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene. 2002;21:8949–8956. doi: 10.1038/sj.onc.1206096. [DOI] [PubMed] [Google Scholar]
- 18.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 19.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedberg EC. Cockayne syndrome—a primary defect in DNA repair, transcription, both or neither? BioEssays. 1996;18:731–738. doi: 10.1002/bies.950180908. [DOI] [PubMed] [Google Scholar]
- 21.Frosina G. The current evidence for defective repair of oxidatively damaged DNA in Cockayne syndrome. Free Radic. Biol. Med. 2007;43:165–177. doi: 10.1016/j.freeradbiomed.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Licht CL, Stevnsner T, Bohr VA. Cockayne syndrome group B cellular and biochemical functions. Am. J. Hum. Genet. 2003;73:1217–1239. doi: 10.1086/380399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Gool AJ, van der Horst GT, Citterio E, Hoeijmakers JH. Cockayne syndrome: defective repair of transcription? EMBO J. 1997;16:4155–4162. doi: 10.1093/emboj/16.14.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 25.Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18:64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- 27.Leadon SA. Transcription-coupled repair: a multifunctional signaling pathway. Cold Spring Harb. Symp. Quant. Biol. 2000;65:561–566. doi: 10.1101/sqb.2000.65.561. [DOI] [PubMed] [Google Scholar]
- 28.Mellon I, Spivak G, Hanawalt PC. Selective removal of transcriptionblocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 29.Smerdon MJ, Thoma F. Site-specific DNA repair at the nucleosome level in a yeast minichromosome. Cell. 1990;61:675–684. doi: 10.1016/0092-8674(90)90479-x. [DOI] [PubMed] [Google Scholar]
- 30.Sweder KS, Hanawalt PC. Transcription-coupled DNA repair. Science. 1993;262:439–440. doi: 10.1126/science.8211165. [DOI] [PubMed] [Google Scholar]
- 31.Somesh BP, Sigurdsson S, Saeki H, Erdjument-Bromage H, Tempst P, Svejstrup JQ. Communication between distant sites in RNA polymerase II through ubiquitylation factors and the polymerase CTD. Cell. 2007;129:57–68. doi: 10.1016/j.cell.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 32.Somesh BP, Reid J, Liu WF, Søgaard TM, Erdjument-Bromage H, Tempst P, Svejstrup JQ. Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell. 2005;121:913–923. doi: 10.1016/j.cell.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Sarasin A, Stary A. New insights for understanding the transcription-coupled repair pathway. DNA Repair. 2007;6:265–269. doi: 10.1016/j.dnarep.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Lainé JP, Egly JM. When transcription and repair meet: a complex system. Trends Genet. 2006;22:430–436. doi: 10.1016/j.tig.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Mellon I. Transcription-coupled repair: a complex affair. Mutat Res. 2005;577:155–161. doi: 10.1016/j.mrfmmm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 36.Tornaletti S. Transcription arrest at DNA damage sites. Mutat Res. 2005;577:131–145. doi: 10.1016/j.mrfmmm.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 37.Svejstrup JQ. Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell Biol. 2002;3:21–29. doi: 10.1038/nrm703. [DOI] [PubMed] [Google Scholar]
- 38.Svejstrup JQ. Rescue of arrested RNA polymerase II complexes. J. Cell Sci. 2003;116:447–451. doi: 10.1242/jcs.00271. [DOI] [PubMed] [Google Scholar]
- 39.Hanawalt PC. Transcription-coupled repair and human disease. Science. 1994;266:1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- 40.Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18:73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- 41.Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc. Natl Acad. Sci. USA. 1996;93:11586–11590. doi: 10.1073/pnas.93.21.11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalogeraki VS, Tornaletti S, Hanawalt PC. Transcription arrest at a lesion in the transcribed DNA strand in vitro is not affected by a nearby lesion in the opposite strand. J. Biol. Chem. 2003;278:19558–19564. doi: 10.1074/jbc.M301060200. [DOI] [PubMed] [Google Scholar]
- 43.Lee KB, Wang D, Lippard SJ, Sharp PA. Transcription-coupled and DNA damage dependent ubiquitination of RNA polymerase II in vitro. Proc. Natl Acad. Sci. USA. 2002;99:4239–4244. doi: 10.1073/pnas.072068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-Bromage H, Tempst P, Svejstrup JQ. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature. 2002;415:929–933. doi: 10.1038/415929a. [DOI] [PubMed] [Google Scholar]
- 45.Yu SL, Lee SK, Johnson RE, Prakash L, Prakash S. The stalling of transcription at abasic sites is highly mutagenic. Mol. Cell. Biol. 2003;23:382–388. doi: 10.1128/MCB.23.1.382-388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tornaletti S, Hanawalt PC. Effect of DNA lesions on transcription elongation. Biochimie. 1999;81:139–146. doi: 10.1016/s0300-9084(99)80046-7. [DOI] [PubMed] [Google Scholar]
- 47.Malik S, Bagla S, Chaurasia P, Duan Z, Bhaumik SR. Elongating RNA polymerase II is disassembled through specific degradation of its largest but not other subunits in response to DNA damage in vivo. J. Biol. Chem. 2008;283:6897–6905. doi: 10.1074/jbc.M707649200. [DOI] [PubMed] [Google Scholar]
- 48.Christians FC, Hanawalt PC. Inhibition of transcription and strand-specific DNA repair by α-amanitin in Chinese hamster ovary cells. Mutat. Res. 1992;274:93–101. doi: 10.1016/0921-8777(92)90056-9. [DOI] [PubMed] [Google Scholar]
- 49.Sweder KS, Hanawalt PC. Preferential repair of cyclobutane pyrimidine dimers in the transcribed strand of a gene in yeast chromosomes and plasmids is dependent on transcription. Proc. Natl Acad. Sci. USA. 1992;89:10696–10700. doi: 10.1073/pnas.89.22.10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pingle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 51.Bhaumik SR, Green MR. Differential requirement of SAGA components for recruitment of TATA-box-binding protein to promoters in vivo. Mol. Cell. Biol. 2002;22:7365–7371. doi: 10.1128/MCB.22.21.7365-7371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhaumik SR, Green MR. Interaction of Gal4p with components of transcription machinery in vivo. Methods Enzymol. 2003;370:445–454. doi: 10.1016/S0076-6879(03)70038-X. [DOI] [PubMed] [Google Scholar]
- 53.Shukla A, Stanojevic N, Duan Z, Sen P, Bhaumik SR. Ubp8p, a histone deubiquitinase whose association with SAGA is mediated by Sgf11p, differentially regulates lysine 4 methylation of histone H3 in vivo. Mol. Cell. Biol. 2006;26:3339–3352. doi: 10.1128/MCB.26.9.3339-3352.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hall DB, Struhl K. The VP16 activation domain interacts with multiple transcriptional components as determined by protein-protein cross-linking in vivo. J. Biol. Chem. 2002;277:46043–46050. doi: 10.1074/jbc.M208911200. [DOI] [PubMed] [Google Scholar]
- 55.Bhaumik SR, Raha T, Aiello DP, Green MR. In vivo target of a transcriptional activator revealed by fluorescence resonance energy transfer. Genes Dev. 2004;18:333–343. doi: 10.1101/gad.1148404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leuther KK, Johnston SA. Nondissociation of GAL4 and GAL80 in vivo after galactose induction. Science. 1992;256:1333–1335. doi: 10.1126/science.1598579. [DOI] [PubMed] [Google Scholar]
- 57.Sil K, Alam S, Xin P, Ma L, Morgan M, Lebo CM, Woods MP, Hopper JE. The Gal3p-Gal80p-Gal4p transcription switch of yeast: Gal3p destabilizes the Gal80p Gal4p complex in response to galactose and ATP. Mol. Cell Biol. 1999;19:7828–7840. doi: 10.1128/mcb.19.11.7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhaumik SR, Green MR. SAGA is an essential in vivo target of the yeast acidic activator Gal4p. Genes Dev. 2001;15:1935–1945. doi: 10.1101/gad.911401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larschan E, Winston F. The S. cerevisiae SAGA complex functions in vivo as a coactivator for transcriptional activation by Gal4. Genes Dev. 2001;15:1946–1956. doi: 10.1101/gad.911501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SK, Yu SM, Prakash L, Prakash S. Requirement for yeast RAD26, a homologue of the human CSB gene, in elongation by RNA polymerase II. Mol. Cell. Biol. 2001;21:8651–8656. doi: 10.1128/MCB.21.24.8651-8656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee SK, Yu SL, Prakash L, Prakash S. Yeast RAD26, a homolog of the human CSB gene, functions independently of nucleotide excision repair and base excision repair in promoting transcription through damaged bases. Mol. Cell Biol. 2002;22:4383–4389. doi: 10.1128/MCB.22.12.4383-4389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caspari T, Dahlen M, Kanter-Smoler G, Lindsay HD, Hofmann K, Papadimitriou K, Sunnerhagen P, Carr AM. Characterization of Schizosaccharomyces pombe Hus1: a PCNA-related protein that associates with Rad1 and Rad9. Mol. Cell Biol. 2000;20:1254–1262. doi: 10.1128/mcb.20.4.1254-1262.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998;12:382–395. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harris S, Kemplen C, Caspari T, Chan C, Lindsay HD, Poitelea M, Carr AM, Price C. Delineating the position of rad4+/cut5+ within the DNA-structure checkpoint pathways in Schizosaccharomyces pombe. J. Cell Sci. 2003;116:3519–3529. doi: 10.1242/jcs.00677. [DOI] [PubMed] [Google Scholar]
- 65.Wolkow TD, Enoch T. Fission yeast Rad26 is a regulatory subunit of the Rad3 checkpoint kinase. Mol. Biol. Cell. 2002;13:480–492. doi: 10.1091/mbc.01-03-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jansen LE, Belo AI, Hulsker R, Brouwer J. Transcription elongation factor Spt4 mediates loss of phosphorylated RNA polymerase II transcription in response to DNA damage. Nucleic Acids Res. 2002;30:3532–3539. doi: 10.1093/nar/gkf475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bucheli M, Sweder K. In UV-irradiated Saccharomyces cerevisiae, overexpression of Swi2/Snf2 family member Rad26 increases transcription-coupled repair and repair of the non-transcribed strand. Mol. Microbiol. 2004;52:1653–1663. doi: 10.1111/j.1365-2958.2004.04081.x. [DOI] [PubMed] [Google Scholar]
- 68.Hahn S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 2004;11:394–403. doi: 10.1038/nsmb763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koleske AJ, Young RA. The RNA polymerase II holoenzyme and its implications for gene regulation. Trends Biochem. Sci. 1995;20:113–116. doi: 10.1016/s0968-0004(00)88977-x. [DOI] [PubMed] [Google Scholar]
- 70.Myer VE, Young RA. RNA polymerase II holoenzymes and subcomplexes. J. Biol. Chem. 1998;273:27757–27760. doi: 10.1074/jbc.273.43.27757. [DOI] [PubMed] [Google Scholar]
- 71.Woychik NA, Young R. A. RNA polymerase II: subunit structure and function. Trends Biochem. Sci. 1990;15:347–351. doi: 10.1016/0968-0004(90)90074-l. [DOI] [PubMed] [Google Scholar]
- 72.Young RA. RNA polymerase II. Annu Rev. Biochem. 1991;60:689–715. doi: 10.1146/annurev.bi.60.070191.003353. [DOI] [PubMed] [Google Scholar]
- 73.Li S, Smerdon MJ. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J. 2002;21:5921–5929. doi: 10.1093/emboj/cdf589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhatia PK, Verhage RA, Brouwer J, Friedberg EC. Molecular cloning and characterization of Saccharomyces cerevisiae RAD28, the yeast homolog of the human Cockayne syndrome A (CSA) gene. J. Bacteriol. 1996;178:5977–5988. doi: 10.1128/jb.178.20.5977-5988.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jansen LE, den Dulk H, Brouns RM, de Ruijter M, Brandsma JA, Brouwer J. Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J. 2000;19:6498–6507. doi: 10.1093/emboj/19.23.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Teng Y, Waters R. Excision repair at the level of the nucleotide in the upstream control region, the coding sequence and in the region where transcription terminates of the Saccharomyces cerevisiae MFA2 gene and the role of RAD26. Nucleic Acids Res. 2000;28:1114–1119. doi: 10.1093/nar/28.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Gool AJ, Verhage R, Swagemakers SM, van de Putte P, Brouwer J, Troelstra C, Bootsma D, Hoeijmakers JH. RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J. 1994;13:5361–5369. doi: 10.1002/j.1460-2075.1994.tb06871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Verhage RA, van Gool AJ, de Groot N, Hoeijmakers JH, van de Putte P, Brouwer J. Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol. Cell. Biol. 1996;16:496–502. doi: 10.1128/mcb.16.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edwards RJ, Bentley NJ, Carr AM. A Rad3-Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nat. Cell Biol. 1999;1:393–398. doi: 10.1038/15623. [DOI] [PubMed] [Google Scholar]
- 80.Hermand D, Nurse P. Cdc18 enforces long-term maintenance of the S phase checkpoint by anchoring the Rad3-Rad26 complex to chromatin. Mol. Cell. 2007;26:553–563. doi: 10.1016/j.molcel.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 81.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 82.Sung P, Prakash L, Matson SW, Prakash S. RAD3 protein of Saccharomyces cerevisiae is a DNA helicase. Proc. Natl Acad. Sci. USA. 1987;84:8951–8955. doi: 10.1073/pnas.84.24.8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Floer M, Bryant GO, Ptashne M. HSP90/70 chaperones are required for rapid nucleosome removal upon induction of the GAL genes of yeast. Proc. Natl Acad. Sci. USA. 2008;105:2975–2980. doi: 10.1073/pnas.0800053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 85.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 86.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. The Tudor domain ‘Royal Family': Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 88.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H, Haw RA, Pootoolal J, Tong A, Canadien V, et al. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol. Cell. 2003;12:1565–1576. doi: 10.1016/s1097-2765(03)00497-0. [DOI] [PubMed] [Google Scholar]
- 90.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 91.Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 92.Li B, Gogol M, Carey M, Pattenden SG, Seidel C, Workman JL. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007;21:1422–1430. doi: 10.1101/gad.1539307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li B, Jackson J, Simon MD, Fleharty B, Gogol M, Seidel C, Workman JL, Shilatifard A. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 2009;284:7970–7976. doi: 10.1074/jbc.M808220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li B, Gogol M, Carey M, Lee D, Seidel C, Workman JL. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007;316:1050–1054. doi: 10.1126/science.1139004. [DOI] [PubMed] [Google Scholar]
- 95.Tantin D, Kansal A, Carey M. Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol. Cell. Biol. 1997;17:6803–6814. doi: 10.1128/mcb.17.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 97.Escargueil AE, Poindessous V, Soares DG, Sarasin A, Cook PR, Larsen A. Influence of irofulven, a transcription-coupled repair-specific antitumor agent, on RNA polymerase activity, stability and dynamics in living mammalian cells. J. Cell Sci. 2008;121:1275–1283. doi: 10.1242/jcs.023259. [DOI] [PubMed] [Google Scholar]
- 98.Brueckner F, Cramer P. DNA photodamage recognition by RNA polymerase II. FEBS Lett. 2007;581:2757–2760. doi: 10.1016/j.febslet.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 99.Damsma GE, Alt A, Brueckner F, Carell T, Cramer P. Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat. Struct. Mol. Biol. 2007;14:1127–1133. doi: 10.1038/nsmb1314. [DOI] [PubMed] [Google Scholar]
- 100.Guzder SN, Habraken Y, Sung P, Prakash L, Prakash S. RAD26, the yeast homolog of human Cockayne’s syndrome group B gene, encodes a DNA dependent ATPase. J. Biol. Chem. 1996;271:18314–18317. doi: 10.1074/jbc.271.31.18314. [DOI] [PubMed] [Google Scholar]
- 101.Selby CP, Sancar A. Human transcription repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 1997;272:1885–1890. doi: 10.1074/jbc.272.3.1885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
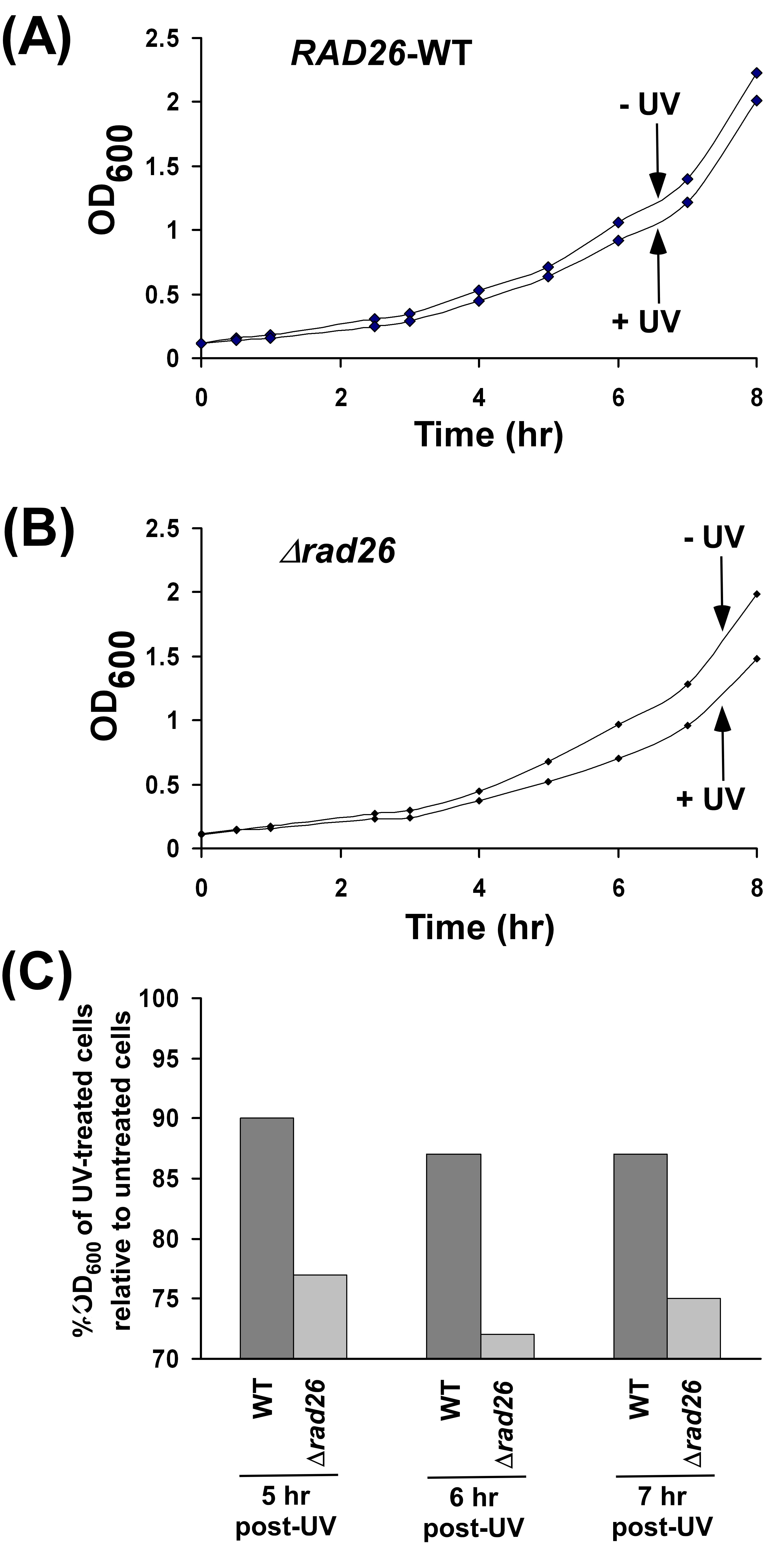{kind=link}
{kind=link}