

Abstract
The principal objective of this study was to investigate the mechanisms regulating the activity of γ-glutamylcysteine ligase (GCL; EC 6.3.2.2), the rate limiting enzyme in glutathione biosynthesis. Two phylogenetically divergent species, mouse and the fruitfly, Drosophila melanogaster were used to test the hypothesis that reversible protein phosphorylation and pyridine dinucleotide phosphate dependent allostery regulate GCL activity. GCL was almost completely inhibited under phosphorylating conditions, involving preincubations with MgATP and endogenous protein kinases. Maximal GCL inhibitions of 94%, 77%, 85%, 87%, 83%, 95% and 89% occurred, respectively, in mouse cerebellum, hippocampus, brainstem, striatum, cortex and heart, and Drosophila. These changes in GCL activity were detected using saturating levels of substrates, suggesting that Vmax was dramatically affected, whereas Km values showed no differences. In vitro activation of GCL, presumably due to dephosphorylation, was blocked by inhibitors of protein phosphatases, suggesting that GCL exists in vivo as a mixture of phosphorylated and dephosphorylated forms. The reversibility of the dephosphorylation-dependent activation was indicated by the time-dependent inactivation of the in vitro activated Drosophila GCL, by preincubation with MgATP. NADPH increased maximal GCL activity by up to 93%, whereas several other nucleotide analogues did not, thereby demonstrating specificity. Kinetic analysis using Hanes–Woolf replots of initial velocity data suggested that the NADPH-dependent stimulation of GCL activity is brought about by a change in the maximal activity, Vmax, rather than changes in substrate affinity. Results of this study suggest that mechanisms of modulation of eukaryotic GCL enzymes may include specific binding of ligands such as pyridine dinucleotide phosphates and reversible protein phosphorylation.
Keywords: γ-glutamylcysteine ligase, Signal transduction, Phosphorylation, Glutathione, Oxidative stress
1. Introduction
The tripeptide, l-γ-glutamyl-l-cysteinylglycine, i.e., glutathione (GSH), is the most abundant cellular nonprotein thiol and has multiple physiological roles in vivo, acting as a cellular redox buffer, a potent nucleophile, an antioxidant against reactive oxygen species and as a major source of the amino acid l-cysteine [1–3]. Recent evidence also suggests that intracellular GSH is a key regulator of stress-activated signal transduction pathways [4].
GSH is synthesized by two ATP-dependent sequential reactions: ligation of l-glutamate and l-cysteine via a γ-carboxyl group by glutamylcysteine ligase (GCL; EC 6.3.2.2), which is the primary rate-limiting reaction in the pathway, and the addition of glycine to γ-glutamylcysteine by glutathione synthetase (GSS; EC 6.3.2.3). In humans and many other eukaryotes, GCL is heterodimeric [5]: the catalytic subunit, GCLc (a zinc metalloprotein [6]) and the modulatory subunit, GCLm, comprise the holoenzyme. De novo GSH synthesis is thought to be induced primarily by transcriptional regulation [5,7,8], via the yAP-1 binding sites in yeast [9,10] and a cascade of signaling events leading to Nrf2 binding to promoter regions of antioxidant/electrophile response elements (ARE/EpRE) in humans [8,11–13]. GCLc and GCLm do not necessarily exist in equimolar amounts within the cell [5,6] and their relative ratios can be independently altered using a variety of oxidative stressors [5]. In humans, rats, mice and Drosophila, GCLm modifies the catalytic properties of GCLc by decreasing the Km for glutamate [14–17]. The GCLc monomer is very sensitive to feedback inhibition by GSH (Ki ∼1.8 mM) and has an extremely high Km (∼18.2 mM) for l-glutamate, suggesting that GCLc may possess exceedingly low activity in vivo unless complexed with GCLm [14,17,18].
The presently known in vitro biochemical characteristics of GCL subunits do not seem to adequately explain the mechanistic basis of the multi-fold induction of GCL catalytic activity. For instance, the induction of the GCLc subunit alone may only result in relatively small increases in GCL activity in vivo, given its very high Km for l-glutamate compared to the holoenzyme [19]. Although transcriptional induction of GCL subunits is undoubtedly a key mechanism for GSH synthesis in response to oxidative stress and for GSH homeostasis [12,20], it seems likely that additional mechanisms may also exist to regulate de novo GSH synthesis as a rapid “primary biochemical response”.
Depletion of cell GSH content by signal transduction agonists has been reported in rat, with the authors postulating that the effect may be due to GCL phosphorylation [21]. However, attempts to significantly phosphorylate GCL resulted in only marginal success, unless large amounts of commercially available protein kinase A, protein kinase C or Ca2+-calmodulin-dependent kinase II (CMK) were added [22], suggesting that phosphorylation of GCL has little physiological relevance. In essence, it is presently unknown whether endogenous kinase/phosphatases are able to modulate GCL activity in a reversible manner or at magnitudes that would enable protein phosphorylation to be considered as a potential mechanism for a de novo GSH synthetic response. Furthermore, it is not known whether such a putative mechanism would be a general phenomenon, manifest in divergent phylogenetic groups.
GCL gene expression can be induced by several stimuli, including oxidative stressors [5] and chemotherapeutic agents [23]. However, there is little information about the possible modulation of GCL activity at the protein level by signal transduction related metabolites. For example, nicotinic coenzymes are known to play a crucial function in cytosolic and mitochondrial redox balance, signaling and the cellular defense against oxidative damage [4,24]. NADPH is essential for the functioning of glutathione-dependent antioxidant defense systems [20] and has been implicated in a diverse array of functions, including direct scavenging of free radicals [25], as well as the generation of reactive oxygen species by microsomes (e.g., see [26,27] and references therein). Although substrate feedback-inhibition of GCL by l-cysteine [28,29] and ATP/ADP [30] has been inferred, the possible effect of pyridine dinucleotide phosphates has not been investigated.
Thus, the purpose of this study was to investigate the potential involvement of mechanisms such as reversible phosphorylation and pyridine dinucleotide allostery in GCL activity. Studies were conducted on mouse brain, liver and heart and Drosophila, whose tissues are primarily composed of postmitotic cells. Results of this study demonstrate the dynamic nature of the effects of reversible phosphorylating conditions and pyridine dinucleotide phosphates on the enzymatic activity of GCL.
2. Materials and methods
2.1. Chemicals and reagents
HPLC calibration standards (γ-GC, GSH, GSSG, and l-cysteine were obtained from Sigma Chemical Company (St. Louis, MO). o-Phosphoric acid was purchased from EMD Science (Gibbstown, NJ). Rottlerin and quercetin were purchased from Calbiochem-Novabiochem corporation (La Jolla, CA). Milli-Q grade water was prepared by reverse-osmosis on a Millipore® water-purification system. All chemicals were either analytical grade or of the highest purity commercially available.
2.2. Animals
Male C57BL/6 mice (13 months of age) and Fischer 344 rats (4 months of age) used in this study, were obtained from the National Institute on Aging, National Institutes of Health, and maintained under standard conditions and fed ad libitum. Male Drosophila melanogaster, of a reference strain containing the recessive markers y (yellow body) and w (white eye) were housed in groups of 25 under constant light at ∼25 ± 1 °C and maintained approximately 12 days post-eclosion, prior to experimentation.
2.3. Preparation of mouse and Drosophila tissue and protein samples
Mice and rats were killed by cervical dislocation and decapitation, respectively. Brain regions and hearts were quickly removed and frozen at −80 °C. Flies were prepared as described previously [29]. All subsequent procedures were carried out at 4 °C unless otherwise stated. Tissue homogenization was carried out in Kontes glass homogenizers (Vineland, NJ) using 10 vol of extraction buffer (320 mM sucrose, 1 mM PMSF, 1 mM ε-amino-n-caproic acid, 10 mM Tris, pH 7.4). Complete protease inhibitor cocktail tablets (Roche, Indianapolis, IN), at a concentration of 1 tablet per 10 ml of extraction buffer, were used to inhibit endogenous proteases. Crude-homogenates were centrifuged at 3000×g for 10 min at 4 °C to pellet debris. Small molecular weight compounds were removed from the supernatants by centrifugation through centrifugal filters (Pall Corp, Ann Arbor, MI) with a 10-kDa membrane cut-off (14,000×g for 15 min at 4 °C). Briefly, clarified supernatants were passed though 0.45 μm PTFE Acrodisc® syringe filters (Gelman Laboratory, Ann Arbor, MI) directly into Pall centrifugal devices. Following centrifugation, the protein samples in the centrifugal devices were washed with 100 μl of wash buffer (200 mM sucrose, 1 mM PMSF, 1 mM ε-amino-n-caproic acid, 10 mM Tris, pH 7.4) and made up to a known volume with the wash buffer. Aliquots of this preparation were either used in preincubations or used immediately for GCL assays, as described below [29,31]. Following the GCL assay, samples for HPLC analysis were either directly injected onto the HPLC column or stored at −80 °C for no longer than 24 h before analysis. A similar procedure was used for the preparation of fly tissues.
2.4. Preincubation of protein samples prior to γ-glutamylcysteine ligase assay
Protein samples (∼1 to 3 mg ml−1; total volume of ∼200 μl), prepared as described above, were preincubated for 0 to 30 min at room temperature with a mixture of protease inhibitors (PMSF, (1 mM); ε-amino-n-caproic acid, (1 mM); benzamidine, (1 mM) or Complete protease inhibitor cocktail tablets (Roche Molecular Biochemicals, Palo Alto, CA), added immediately prior to homogenization. Wherever appropriate, the effects of classical phosphatase inhibitors (NaF (50 mM); sodium pyrophosphate (10 mM) and ammonium molybdate (0.1 mM)) and protein kinase inhibitors (rottlerin (20 μM) and quercetin (20 μM)) were investigated by their inclusion in preincubation mixtures and GCL assay buffers. Control reactions were carried out with and without phosphatase and kinase inhibitors and with 0 to 1 mM buthionine sulfoximine (BSO), a specific GCL inhibitor. A range of salt concentrations (e.g., up to 200 mM NaCl and KCl) were used to eliminate the possibility of general “salt effects” during the preincubations and assays [32]. The possibility of proteolysis, following preincubations, was checked using western analysis, as described below, with antibodies directed against the GCLc subunits of mouse and Drosophila.
2.5. HPLC based γ-glutamylcysteine ligase enzyme assay
Protein samples (∼30 to 50 μg) in aliquots of 5 to 20 μl were used for the GCL assay, either following preincubation or directly. The assay buffer was always prepared fresh and was composed of 100 mM Tris–HCl, 20 mM MgCl2, (pH 8.2), 10 mM ATP, 5 mM l-cysteine, 50 mM l-glutamate, and 500 μM acivicin in a total volume of 250 μl. Assays were usually carried out for 15 min at 25 °C. Reaction linearity of various substrate concentrations, with time and with enzyme protein content was rigorously tested in preliminary experiments. A specific and potent inhibitor of GCL, l-buthionine-SR-sulfoximine (l-BSO; [33]) was used to determine the specificity of the assay and to determine Ki values for the inhibitor. Briefly, extracts were incubated at room temperature for 10 min with up to 1 mM l-BSO and 5 mM ATP in incubation buffer (0.1 M Tris–HCl, 20 mM MgCl2, pH 8.2) and then subjected to the GCL assay. When assays were performed in the presence of signaling-related metabolites, the compounds were included in the assay mixtures at final concentrations ranging from 0 to 8 mM (more details are given in the appropriate figure legends). At the end of the assay, reactions were terminated with an equivalent volume of 15 mM o-phosphoric acid. Precipitated protein was removed by centrifugation at 14,000×g for 10 min at 4 °C, the supernatant was refiltered through 0.45 μm PTFE Acrodisc® syringe filters and injected onto the HPLC either immediately or within 24 h.
2.6. Quantitation of GCL activity
GCL activity was calculated by γ-GC quantitation in the assays. Standards (20 μl of 0 to 5 μM γ-GC) were automatically injected on to the HPLC-column. GCL activity was determined by measuring the amount of γ-GC synthesized during a preset time-period and correlated to the protein content of the sample. Assays were frequently tested to ensure linearity for protein (5 to 50 μg) and time (0 to 60 min). The specific GCL inhibitor l-buthionine sulfoximine (BSO; 1 mM) was used to test the specificity of the assay. Ki values for the inhibitor were obtained by incubation with a range of BSO concentrations (up to 1 mM). No γ-GC peak was observed following incubation of samples without any one of the three substrates (i.e., ATP, l-cysteine or l-glutamate) or in the presence of 1 mM BSO.
In extensive preliminary experiments, sample spiking with γ-GC (as an “internal standard”) indicated peak coelution and was also used to test factors such as relative sample recovery, to exclude the possibility of metabolism by the protein preparations and also to determine between run variation during HPLC analysis. Invariably, no metabolism of the γ-GC spike by the various protein preparations was observed and the variability in sample recovery and between HPLC runs was negligible. A more detailed description of the procedure has been reported elsewhere [29,31].
2.7. HPLC resolution and coulometric detection of mouse and Drosophila aminothiols
HPLC resolution and detection of γ-GC and other aminothiols was conducted as described below, and in recently published reports [31,34,35]. In brief, the mobile phase was delivered via a Waters 515 solvent pump system. Compounds were resolved on a reverse-phase C18 Luna column (particle size 5 μm; 250 × 4.6 mm; Phenomenex, Torrance, CA) using isocratic elution with 15 mM o-phosphoric acid (pH 2.0) as the mobile phase at a flow rate of 1.0 ml min−1 [31,35]. A series of calibration standards, which included γ-GC, cys-gly, GSH, GSSG and cysteine of different concentrations were prepared in 15 mM o-phosphoric acid, and were injected onto the HPLC at regular intervals to ensure uniform standardization. Each standard or experimental sample was analyzed by HPLC in duplicate and mean peak areas were calculated. Following HPLC resolution, compound detection was with a model 5011 CoulArray electrochemical detector (ESA Inc., Chelmsford, MA), equipped with a two-channel analytical cell. Potentials of +100 and +600 mV were applied on channels 1 and 2, respectively.
2.8. Western analysis of Drosophila and mouse protein preparations
Mouse and Drosophila protein samples (10 and 5 μg, respectively) were resolved by 10% SDS-PAGE. The proteins were electrotransferred to Immobilon-P PVDF membranes (Millipore, Bedford MA). The mouse protein blots were incubated with a commercially available antibody against the mouse GCLc subunit (Lab Vision, Freemont, CA). Polyclonal antibody against purified recombinant Drosophila GCLc protein was prepared in rabbits (Covance Research Products, PA). Anti-GCLc primary antibodies were diluted in TBS-T (20 mM Tris–HCl, pH 7.6, 8 g 1−1 NaCl and 0.1% Tween-20; 1:500 dilution for mouse and 1:20000 for Drosophila) containing 5% (w/v) non-fat dry milk and were incubated at 4 °C overnight. The membranes were washed four times with TBS-T before addition of horseradish peroxidase conjugated secondary antibody (diluted 1:20000 in TBS-T containing 5% (w/v) nonfat dry milk) for 1 h at room temperature. Bands were visualized with Amersham ECL Plus Western blotting detection kit (Piscataway, NJ) following the manufacturer's instructions.
2.9. Sequence alignment and phylogenetic analysis
Published cDNA or amino acid sequences for GCLc subunits were obtained using the public sequence databases and analysis services at NCBI. The mouse and Drosophila deduced amino acid sequences were used for BLAST searches (http://www.ncbi.nlm.nih.gov/BLAST). Construction of phylogenetic trees from deduced GCLc amino acid sequences was performed using public software, freely accessible on the world wide web (TreeTop-Phylogenetic tree prediction; http://www.genebee.msu.su/genebee.html). Bootstrap values are indicated above the nodes of the tree. Conserved Ser/Thr/Tyr residues were aligned using the above software in addition to visual inspection and verification.
2.10. Protein concentration determinations
The protein content of the extracts was determined in duplicate using the sodium bicinchoninate (BCA) protein assay (Pierce, Rockford, IL), according to the manufacturer's instructions. Calibration curves were constructed using BSA standards made up to between 0.2 and 50 μg ml−1 of protein [36].
2.11. Statistical analysis
Results are presented as means±S.E.M. Experiments were performed at least three to four times and typically included 10 to 12 replicates per treatment. Treatments were compared statistically using Student's t-tests on Microsoft Excel® 2002 software. P values of <0.05 were considered statistically significant.
3. Results
3.1. GCLc enzymes can be phylogenetically divided into distinct groups—conservation of putative consensus phosphorylation sites between mouse, rat and Drosophila
A BLAST analysis revealed a high degree of similarity between the rat, mouse, human and Drosophila GCLc primary protein structure. A phylogenetic tree (Fig. 1), constructed from the multiple sequence alignments (not shown), suggested that GCLc proteins can be divided into a number of distinct groups (Fig. 1; for convenience, designated as groups Ia, Ib, II and III). Members of group Ia included human, rat, mouse and Drosophila GCLc. Several Ser and Thr residues are nested in putative consensus phosphorylation sites and are strictly conserved within members of group I. For example, the seryl residues Ser658, Ser448, Ser397, Ser325, Ser321 in the Drosophila GCLc sequence are surrounded by strictly conserved kinase recognition motifs [37]. In addition, Ser658 is located in the extreme C-terminus, a region that is often surface exposed and therefore potentially accessible to kinase/phosphatases. The putative phosphorylation sites are thus conserved in eukaryotes in groups Ia and Ib, ranging from mammals to insects, nematodes and fungi. We hypothesized that, since phosphorylation of rat GCL has already been demonstrated [22], the strict conservation of phosphorylatable residues in additional species may have functional regulatory implications. This hypothesis was tested using homogenates of Drosophila and different tissues of the mouse.
Fig. 1.

Phylogenetic tree, based on amino acid sequence alignments, showing the relationship between GCLc subunits from a variety of divergent species. Multiple alignment of sequences (not shown) used in the phylogenetic analysis could be separated into 3 broad groups, designated as Ia, Ib, II and III. The cloned GCLc subunits included in the analysis reflect the specific comparative aims of the present study and are not exhaustive.
3.2. Preincubation of protein preparations under phosphorylating conditions followed by HPLC-based direct assay of γ-glutamylcysteine ligase activity
The HPLC based direct GCL assay used in this study is similar to that reported recently [29,31]. HPLC calibration curves of γ-GC, cys-gly, GSH, GSSG, and l-cysteine standards were invariably linear (R2 ≥ 0.995; data not presented).
The spectrophotometric assay for GCL, which employs pyruvate kinase and lactate dehydrogenase, linking NADH oxidation to ADP production by GCL [38], has thwarted previous attempts to investigate GCL dephosphorylation due to interference of effectors with exogenously added coupling enzymes (e.g., see [22]). Recent technological advances in HPLC, and appropriately designed preincubation strategies, have enabled us to directly (i.e., by direct determination of γ-GC) investigate the effect of dephosphorylation/phosphorylation cycles on the activity of GCL [31,39]. The results shown in Fig. 2 suggest that the GCL from mouse can be almost totally “switched off” by preincubation under phosphorylating conditions.
Fig. 2.
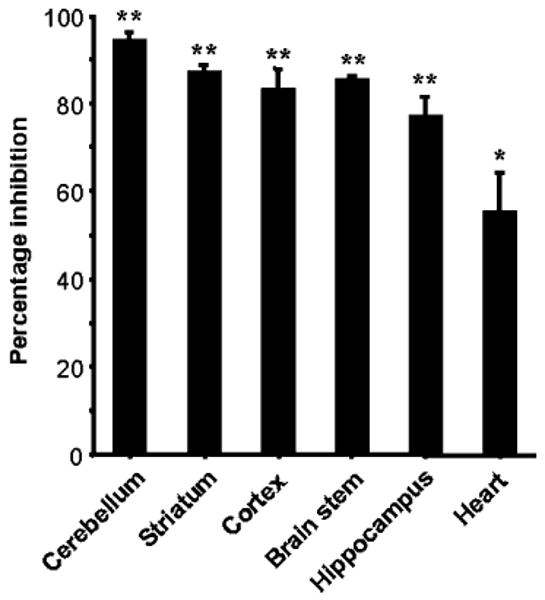
Percentage inactivation of mouse GCL under phosphorylating conditions. Protein samples (200 μl of ∼2 mg ml−1) from homogenates of mouse brain regions (cerebellum, striatum, cortex, brain stem and hippocampus) and heart were prepared as described in Materials and methods and immediately used for experimentation. Preincubations were for 20 min at ambient temperature with a mixture of protease and phosphatase inhibitors (see Materials and methods for details). The possibility of proteolytic degradation was eliminated using Western analysis with antibodies directed against the GCLc subunits of mouse and Drosophila. Experiments were repeated three times with similar results. Bars represent means ± S.E.M. of 10 to 12 replicates and P values from Student's t-test were invariably <0.001 when compared to preincubations in the absence of 5 mM MgATP. Each sample was analyzed by HPLC in duplicate. Percentage inhibitions refer to the percentage decrease in activity following treatment under the phosphorylating conditions described in Materials and methods. Maximal GCL activities for the various samples, calculated as nmoles of γ-GC produced min−1 mg−1 protein as described in Materials and methods, were as follows: 3.8, 4.2, 3.7, 2.9, 3.1, and 1.6 nmol min−1 mg−1 for cerebellum, striatum, cortex, brain stem, hippocampus and heart, respectively.
3.3. GCL inactivation under phosphorylating conditions is not due to proteolysis
A mixture of protease inhibitors was used in the current study to avoid possible confounding effects that can be brought about by protease degradation. For example, the possibility of ATP-dependent proteolysis [40], subsequent to the preincubation and the assaying period, was investigated by western analysis, as well as by monitoring GCL activity (Figs. 3 and 4). Following the periods of the preincubation and assay, tissue extracts were routinely separated by SDS-PAGE and analyzed by western blotting using antibodies against the GCL catalytic subunit from corresponding species. Invariably, there was no evidence suggestive of proteolytic degradation of GCLc catalytic subunits, following preincubations of up to 2 h or during the assay (Figs. 3 and 4). Protein incubations in the presence of protease inhibitors alone showed no significant effects on GCL activity (data not presented). It was concluded that the precautions employed in this study precluded any proteolysis.
Fig. 3.
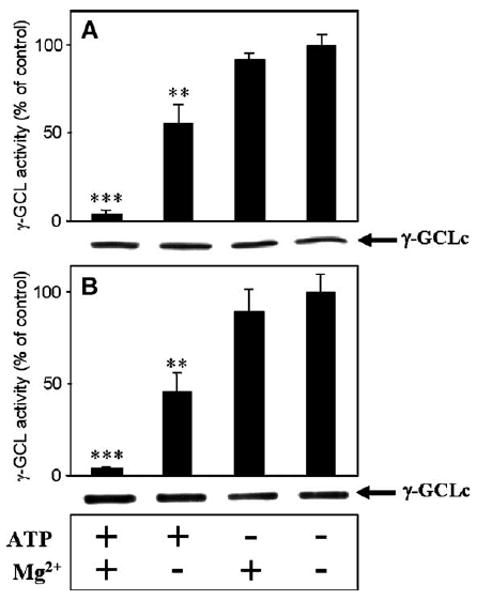
Preincubation of mouse heart and brain cortex GCL under phosphorylating conditions decreases GCL enzyme activity. Homogenates of mouse brain cortex (A) and heart (B) were preincubated according to the procedures described in Materials and methods. GCL activity assays were usually for 15 min. The experiment was repeated three times and similar results were obtained. ***P <0.000001; **P <0.001. n =12 for each treatment, and HPLC analysis was conducted in duplicate. Control values for GCL activities were taken as the maximal GCL activity obtained without the addition of Mg2+ and ATP and were similar to the activities presented in the legend to Fig. 1.
Fig. 4.
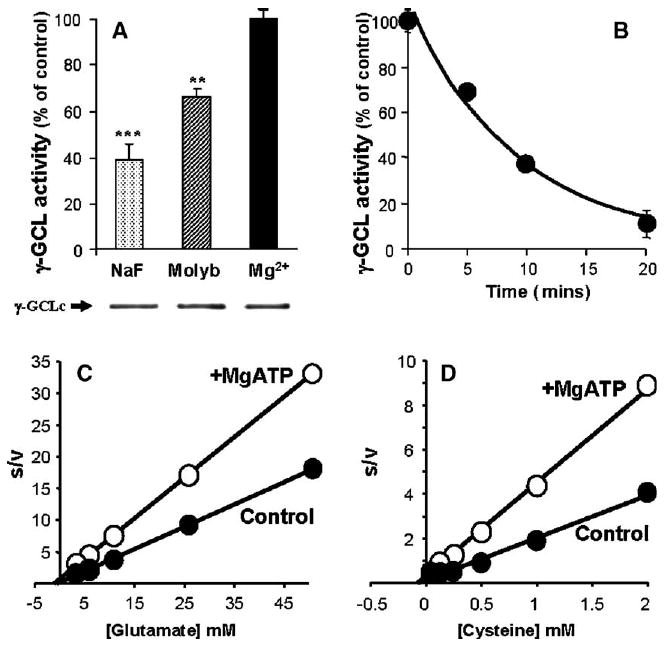
Inhibition of spontaneous activation of GCL activity with phosphatase inhibitors (A); time-dependent reversibility of in vitro activated GCL with subsequent MgATP preincubations (B); determination of the apparent Km for Drosophila GCL substrates l-glutamate (C) and l-cysteine (D) using Hanes–Woolf plots. Various protein phosphatase inhibitors, NaF (50 mM), ammonium molybdate (0.1 mM), EDTA (0.1 mM; not shown), and sodium pyrophosphate (1 mM; not shown) were preincubated with Drosophila protein preparations for 0, 5 10 and 20 min and subsequently assayed for GCL activity. Time-dependent inactivation was performed using preincubations with MgATP and 25 mM NaF. Hanes–Woolf analysis (e.g., [glutamate]/v vs. [glutamate]) of the Michaelis–Menten plots from in vitro assays of Drosophila GCL are presented. “Control” samples in panel A are defined as incubated for 30 min at room temperature with 5 mM Mg2+ (solid bar), the phosphatase inhibitor treatments (hashed and shaded bars) are expressed as a percentage of the maximal GCL activity obtained using the (maximally activated) GCL controls. Mg2+ incubated samples were then further used for Mg2+ inactivation experiments shown in panel B to demonstrate reversibility. The protein preparation used in the representative experiment presented above had a maximal GCL activity of 4.2 nmol min−1 mg−1. Experiments were carried out at least 3 to 4 times, with essentially identical results. Each sample was analyzed in duplicate and representative data are presented. Data represent means ± S.E.M. for 12 replicates. ***P <0.001; **P <0.01.
3.4. Glutamylcysteine ligase is dependent on MgATP for enzymatic activity and can also be inactivated by MgATP preincubations
Protein preparations from mouse heart and brain, and Drosophila, were assayed in the presence or absence of each of the following subtrates: l-cysteine, l-glutamate and ATP. All GCL enzyme preparations showed an absolute requirement for each substrate, as well as ATP and Mg2+ for activity (data not presented). Preliminary experiments showed that γ-GC formation was essentially abolished if tissue extracts were preincubated with 500 μM l-BSO and 5 mM ATP for 5 min before the initiation of the assay [33].
Although enzyme preparations showed a strict requirement for MgATP for γ-GC synthesis, preincubation of the homogenates under phosphorylating conditions with MgATP also caused a maximal GCL inhibition of 94%, 77%, 85%, 87%, 83%, 95% and 89% in mouse cerebellum, hippocampus, brainstem, striatum, cortex, heart and Drosophila, respectively (Figs. 2–4). The phosphorylation effect on GCL was routinely detected at saturating concentrations of substrates, suggesting that maximal activity (Vmax) was one of the kinetic parameters affected.
3.5. Activation of Drosophila and mouse γ-glutamylcysteine ligase
Preincubation of protein preparations from mouse brain and heart, in the absence of ATP, resulted in a noticeable increase in the activity of GCL (Figs. 3A, B and 4). It was hypothesized, based on the inhibitory effect of preincubations with MgATP, “spontaneous activation” of GCL may also occur in Drosophila protein preparations, demonstrating the reversibility of GCL inactivation. A series of broad range phosphatase inhibitors were used to determine if protein phosphatases were involved in the GCL activation phenomenon. In vitro preincubations were performed with NaF (50 mM), molybdate (0.1 mM) and Mg2+ (5 mM as control). These phosphatase inhibitors significantly inhibited the activation of GCL (Fig. 4A), suggesting that dephosphorylation is at least partially responsible for the spontaneous increase in in vitro GCL activity. In preliminary studies, a range of other salts at various concentrations were also tested in preincubations (data not presented) with mouse brain, heart and Drosophila GCL. GCL activity was relatively insensitive to high ionic strength (data not presented) with NaCl and KCl concentrations of ≥200 mM causing maximal reductions of <6%. The findings from protein phosphatase inhibitor treatments and the activation of GCL in the absence of such inhibitors suggest that mouse and Drosophila GCL exists in vivo as a mixture of phospho/dephospho-GCL species.
3.6. Reversibility of GCL activation: in vitro γ-glutamylcysteine ligase activation can be reversed with MgATP in a time-dependent manner
Since the data presented as Figs. 2–4A suggest that protein dephosphorylation activates GCL, it was reasoned that reversibility of activation by dephosphorylation could be achieved by subsequent incubation of the activated (dephosphorylated) enzyme under phosphorylating conditions. Time-course experiments were performed to test whether activated GCL (aliquots of preparations that yielded activity data similar to that represented by the 3rd bar in Fig. 4A) could be subsequently inactivated, thereby demonstrating the reversibility of this modulatory process. Taken together, the results, presented in Fig. 4A and B, show, for the first time, that GCL could be activated under dephosphorylating conditions (Fig. 4A), and subsequently be inactivated (Fig. 4B), in a time-dependent manner, under phosphorylating conditions, thereby indicating reversibility of the process.
It is noteworthy that all GCL assays conducted in the present study were based on the saturating substrate assay described recently by Toroser and Sohal [29]. From the Hanes–Woolf analysis of kinetic data presented as Fig. 4C and D, it can be hypothesized that a significant decrease in Vmax of GCL is caused by phosphorylation, and that a change in the Km for these substrates is not apparent under the described conditions. In brief, the analysis shown in Fig. 4C and D suggests that the Vmax is the kinetic parameter affected by reversible phosphorylation of GCL in both mouse and Drosophila.
3.7. Effect of protein kinase inhibitors on GCL inactivation
Several protein kinase inhibitors exhibit relatively high degrees of specificity, and may be useful for identifying the physiological substrates and cellular functions of these enzymes [41]. The small hydrophobic poison “rottlerin” (also known as mallotoxin) and the ubiquitous flavenoid “quercetin” (3,3′,4′,5,7-pentahydroxyflavone) are potent protein kinase inhibitors [42–44]. Quercetin can inhibit PKA [42] and rottlerin has often been reported to inhibit PKC and CaM-kinase III isoforms, with reports of IC50 values ranging from 3 to 100 μM [43].
Liver was chosen as a model system as it contains relatively high amounts of GSH and GCL activity [20]. To test the effect of protein kinase inhibitors on the possible regulation of GCL by phosphorylation, GCL preparations were made from rat and mouse liver and also Drosophila as described in “Materials and Methods”. Inclusion of rottlerin in preincubations, at a concentration of 20 μM, resulted in 36 (P <0.000005), 113 (P <0.000005) and 17% (not significant at the P <0.05 level) increase for rat, mouse and Drosophila GCL, respectively. Inclusion of quercetin resulted, respectively, in a 21 (P <0.000005), 203 (P <0.000005) and 50% (P <0.000005) increase in GCL activity compared to controls devoid of kinase inhibitors (Fig. 5).
Fig. 5.
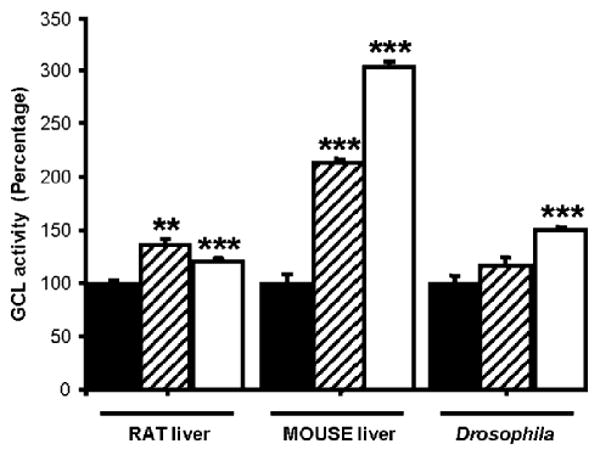
Inhibition of GCL-kinase by rottlerin (hashed bars) and quercetin (open bars). GCL-activities are compared to the MgATP incubated control, which is set as 100% (black bars; incubations were as described in Materials and methods, except that they were carried out for 10 min) devoid of protein kinase inhibitors. The inhibitors were each included, at a concentration of 20 μM, in preincubations containing rat, mouse and Drosophila protein preparations and 5 mM ATP. Each bar represents the mean of three replicates. The vertical bars represent standard error of the mean. ***P <0.000005; **P <0.00002. Data is representative of two independent experiments.
Since in vitro inhibition of GCL activity of ≥80% was routinely observed in the absence of PKC cofactors such as phosphatidylserine and 1,2-dioleoyl-sn-glycerol (Figs. 2–4; [45]), these results suggest that PKC activity may not be an absolute requirement for regulatory GCL phosphorylation. It is noteworthy that recent studies suggest that rottlerin may also inactivate p-38 regulated/activated kinase (PRAK) and MAPK-activated protein kinase-2 (MAPKAP-K2) [42]. The results obtained here, with rottlerin and quercetin, suggest that kinase mediated protein phosphorylation is involved in rat, mouse and Drosophila GCL inactivation. Furthermore, the differences in the protein kinase inhibitor sensitivity between the rat, mouse and Drosophila samples (Fig. 5) imply that specific kinase activities involved in the regulatory process may not have equivalent significance in GCL phosphorylation in each of the tested species. Surprisingly, the percentages of GCL activity controlled by phosphorylation, as assessed by the effect of rottlerin and quercetin, were more alike between the rat and Drosophila versus the mouse GCL enzyme (Fig. 5). Given that significant tissue-specific differences in phosphorylation-dependent GCL regulation were also observed within the same species, e.g., heart versus the brain in mouse (Fig. 2), it is tempting to speculate that the basis for these differences may be due to factors other than the GCL enzyme itself. Interconverting kinase/phosphatase activities and their effector requirements within a given tissue and species may vary. Further work is required to isolate and identify these modulatory GCL-kinase/phosphatase activities.
Millimolar levels of ATP were used in the present study to test the effects of the protein kinase inhibitors. Subphysiological (μM) levels of ATP, which are often used in the literature, can lead to higher rates of inhibition by these inhibitors since most protein kinase inhibitors are “ATP-competitive” [46].
3.8. The effect of signal transduction related compounds on GCL activity
A wide search was conducted to determine whether a variety of signal transduction and redox related low molecular weight compounds may have a modulatory effect on in vitro GCL activity when included directly in GCL assays (Fig. 6A). ATP, ADP, GMP, NAD+, NADH, NADP+, NADPH, glucose (glu), phosphocreatine (pCr), fructose-1,6 bisphosphate (fru-1,6BP), phosphoenol pyruvate (PEP) (at 2 mM final assay concentrations) and resveratrol (trans-3,5,4′-trihydroxystilbene; 1 μM) were added directly to the GCL assay mixture. Majority of the tested compounds either had little effect on GCL activity or caused inhibition (Fig. 6A), except for NADPH, which caused a significant increase in GCL activity. The order of decreasing inhibition was: NADP+>PEP>fru-1,6BP > resrevatrol > ADP > glu > NADH > NAD > GMP > p-Cr>ATP. NADPH increased the activity of GCL by 93% (P <0.002; Student's t-test) compared to its respective control. The effect of NADPH (most potent activator; Fig. 6A) and NADP+ (most potent inhibitor; Fig. 6A) on GCL activity was tested using effector concentrations between 0 and 8 mM (Fig. 6B). The results presented in Fig. 6B and subsequent kinetic experiments, which are described below, were performed at 500 μM and suggest that reduced pyridine dinucleotide phosphates are effective at regulating GCL activity at physiological concentrations.
Fig. 6.
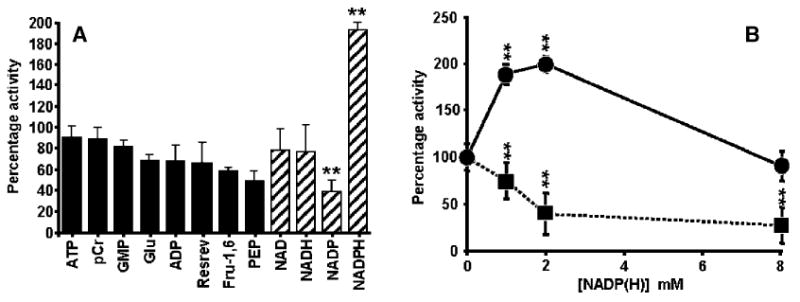
Effect of pyridine dinucleotides and signal transduction related metabolites on the activity of Drosophila GCL. Activation of GCL specifically by NADPH but not by other nucleotide analogues or a panel of other signal transduction related metabolites (A). The effect of NADPH (●) and NADP (■) concentration on in vitro Drosophila GCL activity (B). For the experiments presented in panels A and B, freshly prepared effectors were included directly in the GCL assays at 2 mM final concentration, except for resveratrol which was included at 1 μM. Assay details were as described in Materials and methods. **P <0.002; each data point represents the mean of at least six replicates; each sample was analyzed twice; vertical bars represent ± S.E.M. Each experiment was performed five times using independent protein preparations.
3.9. Kinetics of γ-GC formation with reduced pyridine dinucleotide phosphates
To date, GCL enzymes remain unexamined in terms of their kinetics in the presence of nucleotide diphosphates. In the present study, substrate affinity data were obtained for Drosophila GCL for the three substrates in the presence and absence of near physiological concentrations of NADPH (filled and open symbols, respectively; Fig. 7) [47]. Initial velocity data were replotted using Hanes–Woolf analysis (Fig. 7A–C).
Fig. 7.
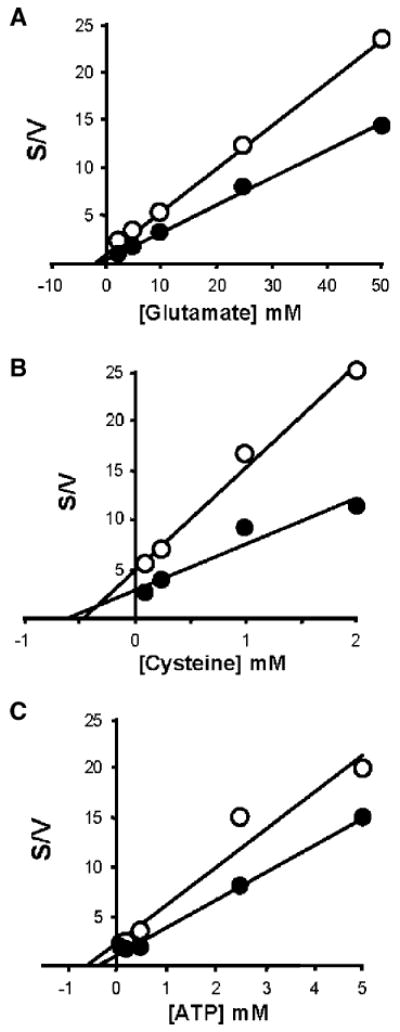
Determination of apparent kinetic constants for Drosophila GCL in the presence of reduced pyridine dinucleotide phosphate effectors. GCL assays were carried out in the presence (●) and absence (○) of NADPH. Various concentrations of l-glutamate (A), l-cysteine (B) and ATP (C) were used to obtain Hanes–Woolf (s/v vs. [substrate]) replots from the initial velocity data. Assay details for the determination of initial velocities were as described under Materials and methods. Substrate concentrations were as follows: 0 to 50 mM glutamate; 0 to 2 mM l-cysteine; 0 to 5 mM ATP. Sample analysis by HPLC was in duplicate. The maximal GCL activity of the “control” (○; devoid of any effectors) was 2.9 nmol min−1 mg−1. Each data point represents the mean of at least six replicates.
Due to its proposed role as a hydride (hydrogen ion donor; [25]), it was of particular interest to determine whether a possible “mode of action”, by which NADPH may effect GCL activity involves disruption of disulfide bonds, a mechanism usually attributed to GSH [18] and known to bring about a diagnostic increase in the Km of GCL for its l-glutamate substrate. Diagnostic GCL assays were performed in the presence and absence of NADPH (500 μM) at various concentrations of l-glutamate (0 to 50 mM). No differences in l-glutamate affinity (as determined by Hanes–Woolf plots; linear regression extrapolated to the x axis) were detected between the NADPH activated (Fig. 7A; closed symbols) and control (Fig. 7A; open symbols) treatments. In brief, from currently accepted models of GCL regulation, the absence of a change in the affinity of GCL towards glutamate ( = ∼1.4 mM; Fig. 7A) is consistent with the notion that NADPH does not bring about a change in the disulfide linkages between GCLc and GCLm that are thought to govern the affinity towards glutamate (Fig. 7A; [48]). The Km for l-glutamate determined in the present study is well below the in vivo concentrations in many tissues, of ∼3 mM [14], with the possible exception of red blood cells, where lower concentrations of glutamate have been reported [49]. The (∼1.4 mM; Fig. 6A) is also consistent with biochemical reports which imply that the predominant molecular species that contributes to GCL activity in vivo is the GCL holoenzyme [17], an area of considerable debate in recent literature [50]. Interestingly, the GCLc subunit has recently been estimated to comprise ∼2 to 8% of the total GCL activity in vivo [17]. The effect of reduced pyridine dinucleotide phosphates may represent an additional mechanism which acts in addition to ATP utilization and cysteine supply that have been demonstrated to influence GCL activity in recent reports [17,50,51].
As stated above, a major regulator of GSH production by GCL is the availability of cysteine, the major limiting substrate [20,51]. The determined in the present study was 0.59 and 0.46 mM in the presence and absence of NADPH, respectively (Fig. 7B). Kinetic analysis suggested that the differences between the enzymes was due to an increase in Vmax (Fig. 7B). It is noteworthy that the Km for cysteine is thought to be independent of the GCLc:GCLm interaction [20]. Recent data suggests that the for GCL can be up to 5.7-fold higher in the absence of the GCLm subunit [17]. In the present study the was ∼0.38 and 0.59 mM in the presence and absence of NADPH, respectively.
The absence of a change in substrate affinity as shown by Hanes–Woolf analysis in the presence of both NADPH (effector) and all three of the GCL substrates, and the lack of a significant stimulatory effect by other adenylates and pyridine dinucleotides is consistent with the hypothesis that mechanisms such as substrate-stabilization or non-specific stabilization are not involved in the NADPH activation of GCL activity [52]. Additionally, these results suggest that, in the presence of up to 2 mM NADPH, substrate binding to the GCL active site is not occluded.
In summary, Hanes–Woolf analysis of initial velocity data for GCL showed that the kinetic differences observed with reduced pyridine dinucleotide phosphates suggest no differences in substrate affinity, rather, a change in Vmax is responsible for the observed differences.
3.10. Inhibition of γ-GCL by buthionine sulfoximine
l-Buthionine sulfoximine (BSO) is a potent inhibitor of GCL, and is useful for probing specific aspects of the reaction catalyzed by GCL. The GCL enzyme acts by the initial formation of a γ-glutamylphosphate intermediate, followed by nucleophilic substitution by cysteine, the crucial steps which imparts the detoxification capacity to glutathione (by way of the chemical reactivity of the SH group). However, the extent of inhibition by BSO has been reported to differ from one enzyme to another, with the differences seemingly correlated with the substrate specificity of each enzyme [53]. In the present study, a range of BSO concentrations were used to determine whether any differences could be detected for the inhibition of GCL by BSO in the presence of reduced pyridine dinucleotide phosphates, no significant differences were observed in the Ki values in the presence or absence of NADPH. BSO inactivated the rat, mouse and Drosophila GCL enzymes with Ki values at ∼110 − 150 μM (data not presented). Omission of ATP or MgCl2 lead to a lack of GCL inhibition by BSO, suggesting that phosphorylation of BSO is a prerequisite to the inactivation. The inactivation was not reversible for rat, mouse or Drosophila, suggesting the inhibition potency of BSO in the presence or absence of the NADPH effector is indistinguishable in these species (data not presented).
3.11. Determination of pH optimum in the presence of NADPH
GCL activity was determined at various pH values, ranging between 5.5 and 10. The pH optimum for Drosophila GCL was very close to the generally accepted pH maximum of ∼8.2 (Fig. 8, closed symbols). In the presence of NADPH, however, there was a shift in the pH dependence curve, with an optimum around 7.5 (Fig. 8, open symbols).
Fig. 8.
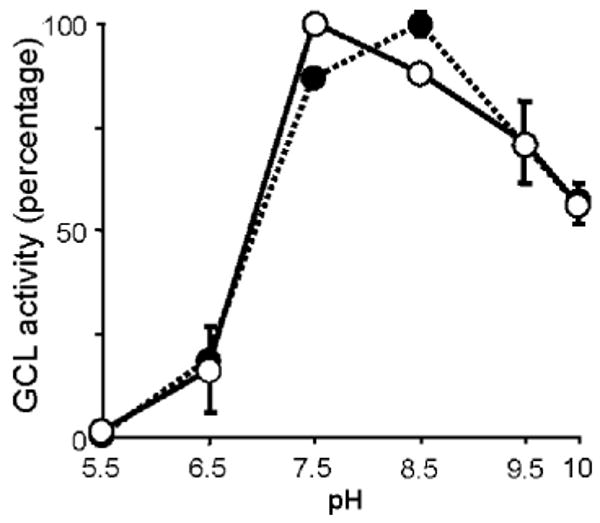
Effect of pH on GCL activity in the presence of NADPH. Drosophila GCL was assayed at pH values of 5.5 to 10.0, in the following buffers: MES 50 mM (2-(N-morpholino) ethanesulphonic acid; pH 5.5–6.5); MOPS 50 mM (3-(N-morpholino) propanesulphonic acid; pH 6.5–7.5); Tricine (N-[tris(hydroxymethyl) methyl] glycine; pH 7.5–8.5) and CHES (2-(cyclohexylamino)ethanesulphonic acid; pH 8.5–10). pH dependence curves are shown for NADPH (○), which was included in the GCL assays at a concentration of 1 mM and control assays (●) which were devoid of NADPH. Values have been normalized and represent means of five replicates. Vertical bars are ± S.E. of the mean.
4. Discussion
Results of the present study demonstrate for the first time that the MgATP dependent activation/inactivation of GCL is reversible, suggesting a potentially pivotal “primary response” regulatory mechanism, which may be operating during de novo GSH synthesis. Another novel finding was that the activation/inactivation cycles of GCL can essentially be regarded as an “on/off switch”. Previous reports [22] with rat hepatocyte GCL suggested only marginal modulatory effects (a reduction in activity of ∼10–15%). Additionally, due to inherent shortcomings of the spectrophotometric GCL assay, it had not previously been possible to determine whether GCL could undergo activation/inactivation cycles. This study demonstrates, for the first time, the reversibility of the process. For instance, an up to 2.5 fold increase (Fig. 4A) in endogenous GCL activity can be caused by preincubations under dephosphorylating conditions and this increase can be reversed by ∼89% (Fig. 4B) following a 20-min preincubation under phosphorylating conditions.
Replenishment of depleted GSH levels during oxidative stress, by de novo synthesis, is of immense physiological relevance due to the multiplicity of the functional roles of GSH (e.g., see [54] and references therein). Although the transcriptional regulation of GCL has been the subject of numerous studies (see [55] for review), little information is currently available about the possible regulation of the enzyme at the protein level via reversible protein phosphorylation. The results presented in this study suggest that phosphorylation related mechanisms may be a potential first line response to a severe oxidative insult.
GCL as well as GCL-kinase(s) are dependent on ATP and Mg2+ for enzymatic activity. It may seem paradoxical that MgATP had to be included for both the γ-GC synthesis as well as inactivation of GCL. A plausible explanation is that the “preincubation treatments” provided requisite substrates (e.g., GCL protein and MgATP) for the endogenous protein kinases; whereas in the subsequent GCL activity assay, the local substrate concentrations for GCL-kinase (i.e., GCL itself) were diluted, evidently below critical concentrations required for phosphorylation, thereby separating the GCL-kinase activity from the GCL catalytic activity in a convenient 2 stage procedure. In the preincubations, the degree of phosphorylation was optimized by maintaining relatively high protein concentrations (1 to 3 mg ml−1) and exclusion of GCL substrates (l-cysteine and l-glutamate). In the subsequent GCL activity assay, phosphorylation was minimized by a 25- to 30-fold dilution of the GCL protein mixture and addition of the full complement GCL substrates (ATP, l-cysteine and l-glutamate). It is noteworthy that in contrast to the highly purified protein preparations and recombinant proteins used in previous studies [14–16,19], the preparations used here were not exhaustively purified, a factor that may have helped to maintain proper folding, thereby facilitating possible GCL-kinase and -phosphatase docking, and minimizing the loss of protein cofactors which may be necessary.
It is possible that when GCL is engaged in enzymatic activity (e.g., when its substrates l-glutamate, l-cysteine and ATP are available in optimal levels, as they are in the Vmax assay), the enzyme may be a poor substrate for GCL kinase. Such a regulatory mechanism has previously been described for HMG-CoA reductase [56], which is also not efficiently phosphorylated when carrying out its enzymatic function, but phosphorylation leads to its inactivation in the absence of substrates. It seems possible that GCL modulation by reversible protein phosphorylation may be a key determinant of the activity of this enzyme as a primary response to oxidative stress.
The time- and MgATP-dependence of GCL inactivation (Figs. 2–4), and the effect of protein kinase inhibitors (Fig. 5) found in the present study, implicate the action of endogenous GCL-kinase(s) in preincubation mixtures. The specific requirement for MgATP during the preincubation treatments suggests that the γ-GCL inactivation is not merely a non-specific effect, as evidenced by the relatively minor inhibitory effects observed by inclusion of other adenylates, in lieu of ATP (data not presented). Furthermore, the requirement, during the preincubations, for the presence of relatively high protein concentrations, comparable to the intracellular milieu (1 to 3 mg ml−1), implies that the MgATP-dependent GCL inactivation (Figs. 2–4) and the up to 2.5-fold activation (Fig. 4) depends on the presence of relatively high concentrations of GCL protein, as well as the putative GCL-kinase and -phosphatases, whose isolation and characterization has not yet been accomplished.
The regulatory reversible phosphorylation sites have not yet been mapped in γ-GCL protein from any species. However, it is tempting to speculate that, based on the evidence presented in the present study and the crystal structure of E. coli GCL [57], phosphorylation of amino acid residues near the substrate binding sites may interfere with the catalytic activity of the GCLc subunit. Phosphorylation near substrate-binding domains of enzymes is often a regulatory mechanism that affects the Vmax activity of enzymes (e.g., see [58] and references therein). Although amino acid sequence alignments with the E. coli GCL enzyme and the GCLc homologues, used in the present study, show little overall sequence identity (data not presented), specific l-BSO and l-cysteine binding site residues [59] are conserved between E. coli and the group I species. Several putative phosphorylation sites near the conserved l-cysteine substrate binding pocket are also strictly conserved between species of group I (Fig. 1; amino acid alignments not presented; see also [59] references therein). Whether the conservation of phosphorylatable residues near substrate binding sites of group I species is of any functional significance remains to be experimentally determined.
The ratio of the reduced to total pyridine nucleotide pool is generally accepted as an indicator of the cellular redox status [47]. The introduction of an “NADPH generating system” can result in a decrease in sensitivity to oxidants [60]. NADPH is essential in GSH recycling and related antioxidant functions [27]. Conversely, elevated rates of superoxide production have been reported by microsomes supplied with NADPH [26]. Based on our data with GCL, which shows an up to 93% stimulation of GC production in the presence of NADPH, we propose that reduced pyridine dinucleotide phosphates may have a function in the regulation of the GSH synthetic machinery. Although putative binding motifs for pyridine dinucleotide phosphates could be proposed using the primary structure of GCL, clearly further work is required to unequivocally map the binding site(s), as has been achieved for enzymes such as glutathione reductase [61]. Consistent with the regulatory effects of NADPH/NADP on GCL activity, this redox couple is primarily represented in the cytosol [62], i.e., it is colocalized with GCL, whereas the NADH/NAD redox couple is mainly represented within the mitochondrial compartment, where GCL has not been detected [62].
An increase in intracellular H2O2 production leads to cytosolic acidification [63,64]. It is tempting to speculate that the shift in the pH optimum of GCL (Fig. 8) in the presence of NADPH may sustain an efficient production of GSH intermediates due to an increase in the catalytic efficiency of GCL during periods of stress-induced acidosis.
In summary, results of this study imply that it may be possible to modulate de novo GSH synthesis and to control GSH homeostasis using additional, presently neglected, components of the GSH biosynthetic machinery, such as GCL kinase/phosphatase activities (e.g., see [46] and references therein) as well as levels of pyridine dinucleotide phosphates.
Acknowledgments
This study was supported by the grants RO1 AG13563 and RO1 AG15122 from National Institute on Aging-National Institutes of Health.
Abbreviations
- acivicin
(αS,5S)-alpha-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid
- l-BSO
l-buthionine-SR-sulfoximine
- ECD
electrochemical detection
- γ-GC
γ-glutamylcysteine
- GSH
glutathione (reduced)
- GSSG
glutathione (oxidized)
- HPLC
high-performance liquid chromatography
References
- 1.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 2.Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 4.Buchanan BB, Balmer Y. Redox regulation: a broadening horizon. Annu Rev Plant Biol. 2004;3:3. doi: 10.1146/annurev.arplant.56.032604.144246. [DOI] [PubMed] [Google Scholar]
- 5.Krzywanski DM, Dickinson DA, Iles KE, Wigley AF, Franklin CC, Liu RM, Kavanagh TJ, Forman HJ. Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch Biochem Biophys. 2004;423:116–125. doi: 10.1016/j.abb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Soltaninassab SR, Sekhar KR, Meredith MJ, Freeman ML. Multifaceted regulation of γ-glutamylcysteine synthetase. J Cell Physiol. 2000;182:163–170. doi: 10.1002/(SICI)1097-4652(200002)182:2<163::AID-JCP4>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Kim SJ, Park EH, Lim CJ. Stress-dependent regulation of the gene encoding γ-glutamylcysteine synthetase from the fission yeast. Mol Biol Rep. 2004;31:23–30. doi: 10.1023/b:mole.0000013505.12111.5b. [DOI] [PubMed] [Google Scholar]
- 8.Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med. 2004;37:1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Grant CM, Collinson LP, Roe JH, Dawes IW. Yeast glutathione reductase is required for protection against oxidative stress and is a target gene for yAP-1 transcriptional regulation. Mol Microbiol. 1996;21:171–179. doi: 10.1046/j.1365-2958.1996.6351340.x. [DOI] [PubMed] [Google Scholar]
- 10.Wu AL, Moye-Rowley WS. GSH1, which encodes γ-glutamylcysteine synthetase, is a target gene for yAP-1 transcriptional regulation. Mol Cell Biol. 1994;14:5832–5839. doi: 10.1128/mcb.14.9.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moinova HR, Mulcahy RT. Up-regulation of the human γ-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem Biophys Res Commun. 1999;261:661–668. doi: 10.1006/bbrc.1999.1109. [DOI] [PubMed] [Google Scholar]
- 12.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33:974–987. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 13.Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–492. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]
- 14.Huang CS, Chang LS, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 15.Fraser JA, Kansagra P, Kotecki C, Saunders RD, McLellan LI. The modifier subunit of Drosophila glutamate-cysteine ligase regulates catalytic activity by covalent and noncovalent interactions and influences glutathione homeostasis in vivo. J Biol Chem. 2003;278:46369–46377. doi: 10.1074/jbc.M308035200. [DOI] [PubMed] [Google Scholar]
- 16.Fraser JA, Saunders RD, McLellan LI. Drosophila melanogaster glutamate-cysteine ligase activity is regulated by a modifier subunit with a mechanism of action similar to that of the mammalian form. J Biol Chem. 2002;277:1158–1165. doi: 10.1074/jbc.M106683200. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem. 2005;280:33766–33774. doi: 10.1074/jbc.M504604200. [DOI] [PubMed] [Google Scholar]
- 18.Huang CS, Anderson ME, Meister A. Amino acid sequence and function of the light subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem. 1993;268:20578–20583. [PubMed] [Google Scholar]
- 19.Meister A. Glutathione metabolism. Methods Enzymol. 1995;251:3–7. doi: 10.1016/0076-6879(95)51106-7. [DOI] [PubMed] [Google Scholar]
- 20.Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- 21.Lu SC, Kuhlenkamp J, Garcia-Ruiz C, Kaplowitz N. Hormone-mediated down-regulation of hepatic glutathione synthesis in the rat. J Clin Invest. 1991;88:260–269. doi: 10.1172/JCI115286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun WM, Huang ZZ, Lu SC. Regulation of γ-glutamylcysteine synthetase by protein phosphorylation. Biochem J. 1996;320:321–328. doi: 10.1042/bj3200321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sekhar KR, Crooks PA, Sonar VN, Friedman DB, Chan JY, Meredith MJ, Starnes JH, Kelton KR, Summar SR, Sasi S, Freeman ML. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–5645. [PubMed] [Google Scholar]
- 24.Berger F, Ramirez-Hernandez MH, Ziegler M. The new life of a centenarian: signalling functions of NAD(P) Trends Biochem Sci. 2004;29:111–118. doi: 10.1016/j.tibs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Kirsch M, De Groot H. NAD(P)H, a directly operating antioxidant? FASEB J. 2001;15:1569–1574. doi: 10.1096/fj.00-0823hyp. [DOI] [PubMed] [Google Scholar]
- 26.Rashba-Step J, Turro NJ, Cederbaum AI. Increased NADPH- and NADH-dependent production of superoxide and hydroxyl radical by microsomes after chronic ethanol treatment. Arch Biochem Biophys. 1993;300:401–408. doi: 10.1006/abbi.1993.1054. [DOI] [PubMed] [Google Scholar]
- 27.Rinaldi R, Aniya Y, Svensson R, Eliasson E, Swedmark S, Shimoji M, Morgenstern R. NADPH dependent activation of microsomal glutathione transferase 1. Chem-Biol Interact. 2004;147:163–172. doi: 10.1016/j.cbi.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 28.Jackson RC. Studies in the enzymology of glutathione metabolism in human erythrocytes. Biochem J. 1969;111:309–315. doi: 10.1042/bj1110309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toroser D, Sohal RS. Kinetic characteristics of native γ-glutamylcysteine ligase in the aging housefly, Musca domestica L. Biochem Biophys Res Commun. 2005;326:586–593. doi: 10.1016/j.bbrc.2004.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yanari S, Snoke JE, Bloch K. Energy sources in glutathione synthesis. J Biol Chem. 1953;201:561–571. [PubMed] [Google Scholar]
- 31.Gegg ME, Clark JB, Heales SJ. Determination of glutamate-cysteine ligase (γ-glutamylcysteine synthetase) activity by high-performance liquid chromatography and electrochemical detection. Anal Biochem. 2002;304:26–32. doi: 10.1006/abio.2001.5607. [DOI] [PubMed] [Google Scholar]
- 32.Schaffer SW, Ahmed AK, Wetlaufer DB. Salt effects in the glutathione-facilitated reactivation of reduced bovine pancreatic ribonuclease. J Biol Chem. 1975;250:8483–8486. [PubMed] [Google Scholar]
- 33.Huang CS, Moore WR, Meister A. On the active site thiol of γ-glutamylcysteine synthetase: relationships to catalysis, inhibition, and regulation. Proc Natl Acad Sci U S A. 1988;85:2464–2468. doi: 10.1073/pnas.85.8.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birago C, Marchei E, Pennino R, Valvo L. Assay of γ-glutamylcysteine synthetase activity in Plasmodium berghei by liquid chromatography with electrochemical detection. J Pharm Biomed Anal. 2001;25:759–765. doi: 10.1016/s0731-7085(01)00379-x. [DOI] [PubMed] [Google Scholar]
- 35.Rebrin I, Bayne AC, Mockett RJ, Orr WC, Sohal RS. Free aminothiols, glutathione redox state and protein mixed disulphides in aging Drosophila melanogaster. Biochem J. 2004;382:131–136. doi: 10.1042/BJ20040506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoscheck CM. Quantitation of protein. Methods Enzymol. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- 37.Gast R, Glokler J, Hoxter M, Kiess M, Frank R, Tegge W. Method for determining protein kinase substrate specificities by the phosphorylation of peptide libraries on beads, phosphate-specific staining, automated sorting, and sequencing. Anal Biochem. 1999;276:227–241. doi: 10.1006/abio.1999.4285. [DOI] [PubMed] [Google Scholar]
- 38.Seelig GF, Meister A. γ-glutamylcysteine synthetase. Interactions of an essential sulfhydryl group. J Biol Chem. 1984;259:3534–3538. [PubMed] [Google Scholar]
- 39.Volohonsky G, Tuby CN, Porat N, Wellman-Rousseau M, Visvikis A, Leroy P, Rashi S, Steinberg P, Stark AA. A spectrophotometric assay of γ-glutamylcysteine synthetase and glutathione synthetase in crude extracts from tissues and cultured mammalian cells. Chem-Biol Interact. 2002;140:49–65. doi: 10.1016/s0009-2797(02)00017-0. [DOI] [PubMed] [Google Scholar]
- 40.Goldberg AL. ATP-dependent proteases in prokaryotic and eukaryotic cells. Semin Cell Biol. 1990;1:423–432. [PubMed] [Google Scholar]
- 41.Cohen P. The development and therapeutic potential of protein kinase inhibitors. Curr Opin Chem Biol. 1999;3:459–465. doi: 10.1016/S1367-5931(99)80067-2. [DOI] [PubMed] [Google Scholar]
- 42.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava AK. Inhibition of phosphorylase kinase, and tyrosine protein kinase activities by quercetin. Biochem Biophys Res Commun. 1985;131:1–5. doi: 10.1016/0006-291x(85)91761-9. [DOI] [PubMed] [Google Scholar]
- 45.Casabona G. Intracellular signal modulation: a pivotal role for protein kinase C. Prog Neuro-Psychopharmacol Biol Psychiatry. 1997;21:407–425. doi: 10.1016/s0278-5846(97)00011-0. [DOI] [PubMed] [Google Scholar]
- 46.Cohen P. Protein kinases—The major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 47.Adams JD, Jr, Klaidman LK, Chang ML, Yang J. Brain oxidative stress—Analytical chemistry and thermodynamics of glutathione and NADPH. Curr Top Med Chem. 2001;1:473–482. doi: 10.2174/1568026013394778. [DOI] [PubMed] [Google Scholar]
- 48.Richman PG, Meister A. Regulation of γ-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J Biol Chem. 1975;250:1422–1426. [PubMed] [Google Scholar]
- 49.Divino Filho JC, Hazel SJ, Furst P, Bergstrom J, Hall K. Glutamate concentration in plasma, erythrocyte and muscle in relation to plasma levels of insulin-like growth factor (IGF)-I, IGF binding protein-1 and insulin in patients on haemodialysis. J Endocrinol. 1998;156:519–527. doi: 10.1677/joe.0.1560519. [DOI] [PubMed] [Google Scholar]
- 50.Lee JI, Kang J, Stipanuk MH. Differential regulation of glutamate-cysteine ligase subunit expression and increased holoenzyme formation in response to cysteine deprivation. Biochem J. doi: 10.1042/BJ20051111. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stipanuk MH. Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu Rev Nutr. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 52.Andersen JP, le Maire M, Moller JV. Properties of detergent-solubilized and membranous (Ca2++Mg2+)-activated ATPase from sarcoplasmic reticulum as studied by sulfhydryl reactivity and ESR spectroscopy. Effect of protein–protein interactions. Biochim Biophys Acta. 1980;603:84–100. doi: 10.1016/0005-2736(80)90393-4. [DOI] [PubMed] [Google Scholar]
- 53.Hiratake J, Irie T, Tokutake N, Oda J. Recognition of a cysteine substrate by E. coli γ-glutamylcysteine synthetase probed by sulfoximine-based transition-state analogue inhibitors. Biosci Biotechnol Biochem. 2002;66:1500–1514. doi: 10.1271/bbb.66.1500. [DOI] [PubMed] [Google Scholar]
- 54.Weijl NI, Elsendoorn TJ, Lentjes EG, Hopman GD, Wipkink-Bakker A, Zwinderman AH, Cleton FJ, Osanto S. Supplementation with antioxidant micronutrients and chemotherapy-induced toxicity in cancer patients treated with cisplatin-based chemotherapy: a randomised, double-blind, placebo-controlled study. Eur J Cancer. 2004;40:1713–1723. doi: 10.1016/j.ejca.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 55.Forman HJ, Dickinson DA, Iles KE. HNE-signaling pathways leading to its elimination. Mol Aspects Med. 2003;24:189–194. doi: 10.1016/s0098-2997(03)00013-x. [DOI] [PubMed] [Google Scholar]
- 56.Toroser D, Huber SC. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase kinase and sucrose-phosphate synthase kinase activities in cauliflower florets: Ca2+ dependence and substrate specificities. Arch Biochem Biophys. 1998;355:291–300. doi: 10.1006/abbi.1998.0740. [DOI] [PubMed] [Google Scholar]
- 57.Hibi T, Hisada H, Nakatsu T, Kato H, Oda J. Escherichia coli B γ-glutamylcysteine synthetase: modification, purification, crystallization and preliminary crystallographic analysis. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:316–318. doi: 10.1107/s0907444901019886. [DOI] [PubMed] [Google Scholar]
- 58.Toroser D, Athwal GS, Huber SC. Site-specific regulatory interaction between spinach leaf sucrose-phosphate synthase and 14-3-3 proteins. FEBS Lett. 1998;435:110–114. doi: 10.1016/s0014-5793(98)01048-5. [DOI] [PubMed] [Google Scholar]
- 59.Hibi T, Nii H, Nakatsu T, Kimura A, Kato H, Hiratake J, Oda J. Crystal structure of ?-glutamylcysteine synthetase: insights into the mechanism of catalysis by a key enzyme for glutathione homeostasis. Proc Natl Acad Sci U S A. 2004;101:15052–15057. doi: 10.1073/pnas.0403277101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scott MD, Zuo L, Lubin BH, Chiu DT. NADPH, not glutathione, status modulates oxidant sensitivity in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Blood. 1991;77:2059–2064. [PubMed] [Google Scholar]
- 61.Danielson UH, Jiang F, Hansson LO, Mannervik B. Probing the kinetic mechanism and coenzyme specificity of glutathione reductase from the cyanobacterium Anabaena PCC 7120 by redesign of the pyridine-nucleotide-binding site. Biochemistry. 1999;38:9254–9263. doi: 10.1021/bi9903300. [DOI] [PubMed] [Google Scholar]
- 62.Ceconi C, Bernocchi P, Boraso A, Cargnoni A, Pepi P, Curello S, Ferrari R. New insights on myocardial pyridine nucleotides and thiol redox state in ischemia and reperfusion damage. Cardiovasc Res. 2000;47:586–594. doi: 10.1016/s0008-6363(00)00104-8. [DOI] [PubMed] [Google Scholar]
- 63.Pervaiz S, Clement MV. A permissive apoptotic environment: function of a decrease in intracellular superoxide anion and cytosolic acidification. Biochem Biophys Res Commun. 2002;290:1145–1150. doi: 10.1006/bbrc.2001.6274. [DOI] [PubMed] [Google Scholar]
- 64.Wu ML, Tsai KL, Wang SM, Wu JC, Wang BS, Lee YT. Mechanism of hydrogen peroxide and hydroxyl free radical-induced intracellular acidification in cultured rat cardiac myoblasts. Circ Res. 1996;78:564–572. doi: 10.1161/01.res.78.4.564. [DOI] [PubMed] [Google Scholar]