

Abstract
Phosphatidylinositol 3-kinase (PIK3)/ PtdIns dependent protein kinase-1(PDPK1)/Akt signaling plays a critical role in activating proliferation and survival pathways within cancer cells. We report the molecular pharmacology and antitumor activity of PHT-427 a compound designed to bind to the pleckstrin homology (PH) binding domain of signaling molecules important in cancer. Although originally designed to bind the PH domain of Akt, we now report that PHT-427 also binds to the PH domain of PDPK1. A series of PHT-427 analogs with variable C-4 to C-16 alkyl chain length were synthesized and tested. PHT-427 itself (C-12 chain) bound with the highest affinity to the PH domains of both PDPK1 and Akt. PHT-427 inhibited Akt and PDKP1 signaling and their downstream targets in sensitive but not resistant cells and tumor xenografts. When given orally PHT-427 inhibited the growth of human tumor xenografts in immunodeficient mice with up to 80% inhibition in the most sensitive tumors and showed greater activity than analogs with C4, C6 or C8 alkyl chains. Inhibition of PDKP1 was more closely correlated to antitumor activity than Akt inhibition. Tumors with PIK3CA mutation were the most sensitive and K-Ras mutant tumors the least sensitive. Combination studies showed that PHT-427 has greater than additive antitumor activity with paclitaxel in breast cancer, and with erlotinib in NSC lung cancer. When given over 5 days PHT-427 caused no weight loss or change in blood chemistry. Thus, we report a novel PH domain binding inhibitor of PDPK1/Akt signaling with significant in vivo antitumor activity and minimal toxicity.
Keywords: Akt, PDPK1, pleckstrin homology domain, PHT-427
Introduction
The pleckstrin homology (PH) domain is a region of 100 to 120 amino acids found in over 250 human proteins (1). Although the amino acid sequence of PH domains is not universally conserved, the tertiary structure is remarkably conserved. While PH domains bind to a variety of different targets, a unique property of about 40 PH domains is the specificity with which they bind phosphorylated phosphatidylinositide (PtdIns) lipids within the biological cell membrane. PtdIns phosphorylation and the subsequent binding of PH domain-containing proteins are vital components of signal transduction pathways that regulate cell growth and survival; and thus, are opportunistic targets for up-regulation and oncogenic determinism (2).
The PtdIns-3-kinase / PtdIns dependent protein kinase-1(PDPK1)/Akt (protein kinase B) signaling pathway is critical for the proliferation and survival of many types of cancer cells (3). Its activation is found in a variety of cancers, associated with mutation or loss of the mixed function lipid phosphatase PTEN (4), or mutations of PtdIns-3-K (5). PtdIns-3-K phosphorylates PtdIns(4,5)P2 to give PtdIns(3,4,5)P3 which binds to the PH domain of Akt allowing Akt to translocate from the cytoplasm to the inner leaflet of the plasma membrane (6) where it is phosphorylated on Thr308 by PDPK1, another PH domain containing protein (7). Subsequent phosphorylation of Akt on the Ser473 residue occurs either by integrin linked kinase (ILK), by the kinase activity of Akt itself, or by mTORC2 (8), causing Akt to translocate back to the cytoplasm and to the nucleus where it phosphorylates a variety of downstream targets. These include promoters of apoptosis such as forkhead transcription factors (FKHR) and AFX, and the Bcl-2 family member Bad (3). Akt also promotes cell survival by activating CREB (9), and promotes proliferation by activating mTOR and, indirectly, p70 ribosomal S6 kinase (10) and GSK-3β which contributes to cyclin D accumulation of cell cycle entry (11). Furthermore, Akt acts as a mediator for both VEGF production and angiogenesis by phosphorylation of mammalian target of rapamycin (mTOR) (12).
Aberrant PtdIns-3-K signaling has been found to play an important role in multiple aspects of tumorigenesis including uncontrolled proliferation, resistance to apoptosis, angiogenesis and metastatic capability. This aberrant signaling may occur through dysfunction of pathways upstream of PtdIns-3-K such as mutationally activated growth factor receptors, and Ras, or activation of the pathway itself. The first mechanism discovered by which the PI3K/Akt pathway is directly activated was the loss or inactivation of the PTEN tumor suppressor (13). PIK3CA, the gene encoding PtdIns-3-K p110α, is frequently mutated in several common cancers (5). Eighty percent of these mutations occur in one of three hot spots leading to gain of function amino-acid substitutions in the helical or kinase domain of the enzyme. There appear to be at least two molecular mechanisms for the gain of function in PtdIns-3-K p110α, with helical domain mutations abolishing the binding of the p85 inhibitory subunit, and kinase domain mutations mimicking the conformational change caused by the interaction with Ras (5). The other PIK3CA mutations are widely distributed and far less potent in activating PtdIns-3-K enzyme activity than the hot spot mutations, and are only marginally oncogenic. Other PtdIns-3-K p110 isoforms do not show cancer-specific mutations. However, they are often differentially expressed in cancer and are oncogenic when over-expressed in cell culture. There is no indication that either class II PtdIns-3-K or class III PtdIns-3-K are linked to cancer. Most recently mutations in the PH domain of Akt1 which causes electrostatic alterations leading to increased binding of the Akt PH domain with PtdIns(3,4,5)P3 and increased phosphorylation, have been found to aberrantly activate the pathway (14) . Thus far, the initial mutation found at amino acid 17 of the Akt PH domain has been identified in 8% of the breast tumors studied, 6% of colorectal tumors, and 2% of ovarian cancers. Most attempts to develop inhibitors of Akt have focused on compounds that bind to the kinase ATP-binding pocket. Due to the similarity of the ATP pocket among serine/threonine kinases, particularly AGC family kinases to which AKT belongs, achieving target specificity has been extremely difficult (15). All the reported AKT ATP-pocket inhibitors also inhibit protein kinase A (PKA) which may account for the relatively high toxicity of this type of inhibitor observed in animals and in patients.
PDPK1 phosphorylates and activates many members of the AGC subfamily of Ser/Thr kinases through phosphorylation of their auto inhibitory activation loops (16). Most PDPK1 substrates do not have a PH domain and are phosphorylated by PDPK1 in the cytosol (17). Akt is the only known PH domain-containing PDPK1 target and PDPK1 has to be localized to the plasma membrane to activate Akt through binding of its PH domain to PtdIns(3,4,5)P3 (18). Substrates phosphorylated by PDPK1 in addition to Akt, include p70 ribosomal S6 kinase (19), p90 ribosomal S6 kinase (20), PKC isoforms (21), and serum/glucocorticoid regulated kinases (SGKs) (22). PDPK1 copy numbers and levels are increased in human breast cancer leading to the increased ability of upstream lesions in the PtdIns-3-K pathway to signal to Akt (23). Tumors with PIK3CA mutations appear to preferentially signal through PDPKI and its downstream target SGK3 (24). Thus, PDPK1 is also an attractive target for cancer drug discovery. However, as with Akt it has proven difficult to achieve catalytic ATP pocket inhibitor selectivity for PDPK1 compared to other serine/threonine kinases such as PKA (15). Thus the PH domain of PDPK1 presents an alternate target for the development of inhibitors with the potential for antitumor activity.
The approach we have adopted to inhibit Akt and PDPK1 uses structure-based design of small molecules that bind to the PH domains of the proteins, thus, inhibiting their activity. We have identified a novel chemical scaffold in several compounds that binds selectively to the PH domain of Akt, inducing a decrease in Akt activation, and causing apoptosis at low micromolar concentrations (25). One compound PHT-427 (4-dodecyl-N-(5-(5-(methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino)pentyl-1,3,4-thiadiazol-2-yl)benzenesulfonamide) inhibited Akt and its downstream targets in cells and showed in vivo antitumor activity when administered by the intraperitoneal route in a pancreatic cancer xenograft (26). We now report the cellular and in vivo pharmacology of PHT-427 and some analogs with different alkyl chain lengths and show that PHT-427 binds to the PH domains of both Akt and PDPK1 and inhibits the activity of both proteins in cells and in tumor xenografts. We have developed an oral formulation for PHT-427 and show its antitumor activity against a number of human tumor xenografts, particularly those with PIK3CA mutations.
Materials and Methods
Cells and reagents
BxPC-3, Panc-1 and MiaPaCa-2 pancreatic cancer cells PC-3 prostate cancer cells, SKOV-3 ovarian cancer cells, MCF-7 breast cancer cells, and A-549 and NCI-H441 non-small cell lung cancer (NSCLC) cells were obtained from the American Tissue Type Collection (Manassas, VA). All cell lines were characterized by short tandem repeat profiling before use by the MD Anderson Cell Characterization Service. The cells were grown in humidified 95% air, 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). All cell lines were tested to be mycoplasma free using a PCR ELISA kit (Roche Diagnostics Inc, Indianapolis, IN). PHT-427 (Figure 1A) and its C2 to C14 carbon chain length analogs (Figure 1B) were synthesized as previously described (26,27). The Akt inhibitors triciribine (NSC 154020), perifosine and edelfosine were purchased from Cayman Chemical (Ann Arbor, MI) and DPIEL (D1-3,4-dideoxyphosphatidylinositol ether lipid) and MK-2206 were synthesized by the MD Anderson Translational Chemistry Service. PHT-427 was formulated for oral administration suspended at 40 to 50 mg/ml in sesame seed oil. Expression and purification of recombinant human Akt2 and PDPK1 PH domains was performed as previously described (26).
Figure 1. Relative binding of PHT-427 analogs with different carbon chain lengths to the expressed PH domains of Akt and PDK.

A, structure of PHT-427; B Structure of analogs where R is a C-4 to C-14 carbon chain. C, Surface plasmon resonance spectroscopy (SPR) was used to measure the binding affinity (Ki) for the expressed PH domain of Akt2 and of D, PDPK1 by competitive binding of the compounds for the natural ligand phosphatidylinositol-(3,4,5)-P3. E, Kis for reported Akt PH domain inhibitors, and PHT-427 and its C-4- C-14 carbon chain analogs. Ki, were measured as the concentration of compound that displace 50% of the PH domain bound to lipid vesicles enriched in PdtIns(3,4,5)P3 in the absence of drug. Values are the mean of 4 determinations and bars are SE. ND is not determined
Surface plasmon resonance (SPR) spectroscopy binding assays
All interaction analyses were performed with a Biacore 2000, using SA chips, Biacore 2000 Control Software v3.2 and the BIAevaluation v4.1 analysis software (GE Healthcare) as previously described (25). Competitive binding assays and Ki determination used PtdIns(3,4,5)P3 phosphate-biotin labeled liposomes (Echelon Biosciences, Salt Lake City, UT) and increasing concentrations of the compounds being tested.
Signaling pathway inhibition
Reverse phase protein array analysis was carried out as previously described (28). Inhibition of the phosphorylation of Akt, PDPK1 and of their downstream targets was measured by Western blotting using rabbit polyclonal antibodies to phospho-Ser473-Akt, phospho-Thr308-Akt, total-Akt, phospho-Ser241-PDPK1, phospho-Ser9-GSK3β̣ phospho-Ser21-GSK3β,,phospho-Ser657-protein kinase C (PKC) and phospho-Ser240-p70S6-kinase (Cell Signaling Technology Inc., Beverly, MA.) as described previously (26). PDK1 specific inhibition was measured by phospho-Ser221-ribosomal protein S6 kinase (RSK) (R & D Systems. Minneapolis, MN). β-Actin (Santa Cruz, Santa Cruz, CA) was used as a loading control and blots quantified using an ImageQuant image analyzer (Molecular Dynamics, Sunnyvale,CA).
Cell imaging
Panc-1 cells stably transfected with green fluorescent protein (GFP) tagged Akt or PDKP1 PH domains were serum starved in phenol red free growth medium on glass-bottom 96-well imaging plates (Matrical, Spokane, WA) for 16 hours. They were then treated with PHT-427 at 1, 5, and 10µM or PI-103 (Caymen Chemical, Ann Arbor, MI) for 4 hr, and stimulated with 50ng/mL IGF-1 (R & D Systems. Minneapolis, MN) for 10 min. Images were taken before and after IGF-1 treatment using an IN Cell Analyzer 1000 (GE Healthcare Life Sciences, Piscataway, NJ) instrument with a Nikon Plan Fluor ELWD 20X/0.45 objective loaded and using a 300msec exposure time.
Antitumor Studies
Approximately 107 BxPC-3, Panc-1, MiaPaCa-2, PC-3, SKOV-3, A-549 or MCF-7 cells in log cell growth were suspended in 0.2 ml phosphate buffered saline and injected subcutaneously into the flanks of female scid mice. The MCF-7 cells were suspended in Matrigel™ (Becton Dickinson Biosciences, Palo Alto, CA) and the mice implanted 1 day previously with a 60 day 17-β-estradiol release pellet (Innovative Research of America, Sarasota, FA). The animals were weighed weekly and tumor diameters measured twice weekly at right angles (dshort and dlong) with electronic calipers and converted to volume by the formula v = (dshort)2 × (dlong) ÷ 2 (29). When the tumors reached volumes between 150 and 300 mm3, the mice were stratified into groups of 8 animals having approximately equal mean tumor volumes and administration of PHT-427 or its analogs, dissolved on 0.1 ml sesame seed oil, was begun. Control animals received vehicle alone. When the tumor volume reached ≥1,500 mm3 or became necrotic, the animals were euthanized. The growth rate of individual tumors was measured over the 10 day period of dosing and the mean growth rate for each treatment group compared to the control using Student’s t-test. NCI-H441 non-small cell lung cancers were implanted orthotopically in the left lung of nu/nu nude mice as previously described (30) and 20 days later oral PHT-427 administration started at 200 mg/kg twice daily for 10 days. Erlotinib was administered orally, also beginning at 20 days, at 50mg/kg daily until the end of the study. All the mice were killed at day 63 when the control animals became moribund and tumor volumes measured as above.
Pharmacokinetic Studies
Female C57Bl/6 mice were administered PHT-427 as a single oral dose of 200 mg/kg. The mice were killed at different times (3 mice at each time point), blood collected into heparinized tubes and plasma prepared and stored frozen at −80 °C. For assay 0.2 ml plasma was mixed with 0.2 ml of 0.1 M sodium phosphate buffer, pH 4.0, and extracted for 1 hr by inversion with 1 ml ethyl acetate. After centrifugation 0.8 ml of the organic layer was removed, evaporated under N2 and redissolved in 0.2 ml ethanol and 10 µl injected onto a Waters Quattro Ultima tandem mass spectrometer (Waters, Milford, MA) using a Phenomenex Luna 3.0 µm, 2.0 × 50 mm C8 analytical column (Phenomenex, Torrence, CA) with detection and quantification by multiple reaction monitoring with the mass spectrometer operating in electrospray positive ionization mode. The mass transition m/z 410.2 >91.3 was used to detect and quantify PHT-427 with quantification by external standardization with a peak retention time for PHT-427 of 6.1 minutes. The lowest level of detection for plasma PHT-427 was < 1 ng/m.
Pharmacodynamic studies
1×107 BxPC-3 or MiaPaCa-2 pancreatic cancer cells were injected s.c. into the flanks of female scid mice and allowed to grow to approximately 300 mm3. Mice received a single oral dose of PHT-427 of 200 mg/kg in 0.1 ml vehicle. Mice were killed after various times, the tumors removed and immediately frozen in liquid N2. The tumors were homogenized in 50 mM HEPES buffer, pH 7.5, 50 mM NaCl, 1% Nonidet® P40 and 0.25 % sodium deoxycholate. Western blotting was performed as described above.
Toxicity studies
PHT-427 in vehicle or vehicle alone was administered orally twice a day for five days to female C57/Bl6 mice, five per group. The mice were killed 16 hr after the last dose and changes in body weight from the start of the study, spleen weight, white blood cells, red blood cells, hemoglobin, platelet or differential count blood, and serum glucose, aspartate amino transferase (AST), amino alanine transferase (ALT), blood urea nitrogen (BUN) and creatinine were measured.
Results
PH domain binding of PHT-427 analogs
The binding affinities of PHT-427 and its derivatives (Figures 1A and 1B) to the PH domains of Akt (Figure 1C and PDPK1 (Figure 1D) were measured by an SPR displacement assay using PtdIns(3,4,5)P3-rich liposomes with increasing concentrations of the compounds under investigation. Maximum displacement of the PH domain of both Akt and PDPK1 bound to PtdIns(3,4,5)P3 phosphate rich liposomes was obtained with the compound bearing a 12-carbon alkyl groupC-12 (PHT-427, hereafter) with Ki values of 2.7 ± 0.4 and 5.2 ± 0.4 µM, respectively (Table 1). Of note is that while compounds C-14,C-16 and C-18 showed lower, although still appreciable, binding to the PH domain of Akt, only C-12 and C-14 showed binding to the PH domain of PDPK1. We have not investigated the binding of PH-427 to other the PH domains of other proteins except for Tiam1 to which it does not bind (not shown). DPIEL was used as a reference compound and bound with high affinity to the PH domain of Akt but with lower affinity to the PH domain of PDPK1, as previously reported (31). We also studied other compounds that have been suggested to bind to the PH domain of Akt and to inhibit its activity including the alky-lysophospholipid compounds perifosine (32) and edelfosine (33), and triciribine (34). None of these compounds bound to the PH domain of Akt or PDKP1 at concentrations up to 50 µM. MK-2206 is a recently reported allosteric inhibitor of Akt that binds to a site outside the PH domain (35). We found that MK-2206 bound very weakly to the PH domain of Akt and not at all to the PH domain of PDPK1.
Table 1. Antitumor activity of PHT-427.
There were 8 female mice per group. Control mice received vehicle only (0.1 ml sesame seed oil). The growth rate of individual tumors was measured over the period of dosing and the mean for each treatment group compared to control using Student’s t-test to obtain p values. Values are the mean ± SE. T is the mean growth rate of test (drug treated) and C is the mean growth rate of control tumor expressed as a percent. NS is not significantly different (p > 0.05).
|
Tumor |
volume at start mm3 |
dose mg/kg |
schedule |
tumor growth rate mm3/10 days |
T/C % |
p value |
|---|---|---|---|---|---|---|
| BxPC-3 | 156 | control | BID × 5D | 228 ± 46 | ||
| pancreatic | 125 | BID × 5D | 67 ± 35 | 29.4 | 0.030 | |
| 250 | BID × 5D | 46 ± 53 | 20.1 | 0.027 | ||
| 97 | control | BID × 10D | 279 ± 37 | |||
| 100 | BID × 10D | 181 ± 52 | 64.8 | NS | ||
| 200 | BID × 10D | 77 ± 44 | 27.6 | 0.004 | ||
| Panc-1 | 200 | control | BID × 10D | 318 ± 53 | ||
| pancreatic | 200 | BID × 10D | 308 ± 59 | 97.1 | NS | |
| MiaPaCa-2 | 200 | control | BID × 10D | 318 ± 53 | ||
| pancreatic | 200 | BID × 10D | 308 ± 59 | 97.1 | NS | |
| PC3 | 229 | control | BID × 5D | 780 ± 161 | ||
| prostate | 125 | BID × 5D | 470 ± 121 | 60.3 | NS | |
| SKOV-3 | 192 | control | BID × 10D | 432 ± 59 | ||
| ovarian | 250 | BID × 10D | 122 ± 16 | 28.3 | 0.001 | |
| A549 | 157 | control | BID × 10D | 413 ± 37 | ||
| nsc lung | 200 | BID × 10D | 182 ± 47 | 44.1 | 0.016 | |
| MCF-7 | 142 | control | BID × 10D | 410 ± 101 | ||
| breast | 100 | BID × 10D | 383 ± 139 | 93.4 | NS | |
| 200 | BID × 10D | 156 ± 30 | 38.0 | 0.042 | ||
RPPA Studies
The effects of PHT-427 on cell signaling were investigated by RPPA using a panel of 86 antibodies to phospho- and non-phosphorylated signaling protein related to PtdIns-3-K/PDPK1/Akt signaling in PC-3 prostate cells where PtdIns-3-K/PDPK1/Akt signaling is activated because of homozygous PTEN mutation. Wortmannin a PtdIns-3-K inhibitor was used as a positive control. A heat map of the results is shown in Figure 2A and quantitations of relevant proteins are illustrated in Figure 2B. After 16 hours, a reduction was observed in phospho-Ser241-PDPK1 phospho-Thr308-Akt by both 10 µM PH-427 and 0.1 µM wortmannin. In contrast, the PDPK1-independent phospho-Ser473-Akt was slightly increased by PHT-427, but completely inhibited in by wortmannin. Finally, phospho-Ser657-protein kinase C (PKC) and total SGK1, both previously shown to be dependent on the activation of PDPK1 (36,37) were decreased by treatment with both PHT-427 and wortmannin. These results suggest that at 10 µM PHT-427 inhibits both Akt and PDKP1.
Figure 2. Reverse phase protein array of PHT-427 effects in PC-3 prostate cancer cells.

A, Heat map of PC-3 cells were exposed to serum free medium for 8 hr and wortmannin or PHT-427 at 0.1, 0.5, 1, 5,10 µM, (shown by increasing arrow), or DMSO vehicle for 16 hr, protein isolated and subjected to quantitative RPPA with a panel of 74 validated antibodies related to PtdIns-3-K/Akt.PDKP1 signaling. Values are expressed as mean of 3 determinations of expression relative to DMSO control, red is high, green is inhibition, black is no change. B, histograms of phosph Ser241-PDK1, phospho-Akt-Thr308, phospho-Akt-Ser473, phosphor-PKC-Ser657 and total SGK1 levels with DMSO vehicle control, 10 µM PHT-427 and 0.1 µM wortmannin (wort).
Western Blotting
The BxPC-3 and MiaPaCa-2 pancreatic cancer cell lines were probed by Western blotting following up to 24 hr exposure to 10 µM PHT-427, which is below the IC50 for cell growth inhibition of around 30 µM (26), to determine the effects of PHT-427 on of the PtdIns-3-K/PDPK1/Akt signaling pathway components. We have previously reported that the BxPC-3 cells which give xenografts that are sensitive to the antitumor activity of PHT-427 (see below) showed a decrease of both phospho-Ser473-Akt and phospho-Thr308-Akt at 12 and 16 hours (Figure 3A), while both were increased at these time points in MiaPaCa-2 cells which give resistant xenografts (Figure 3B). Total Akt was decreased at 16 hr in both cell lines. Phospho-Ser241-PDPK1 was inhibited in both lines at 12–16 hr while phospho-Ser240 S6 ribosomal kinase was decreased in the sensitive BxPC-3 cells but not the resistant MiaPaCa-2 line.
Figure 3. Effects of PHT-427 in cells.
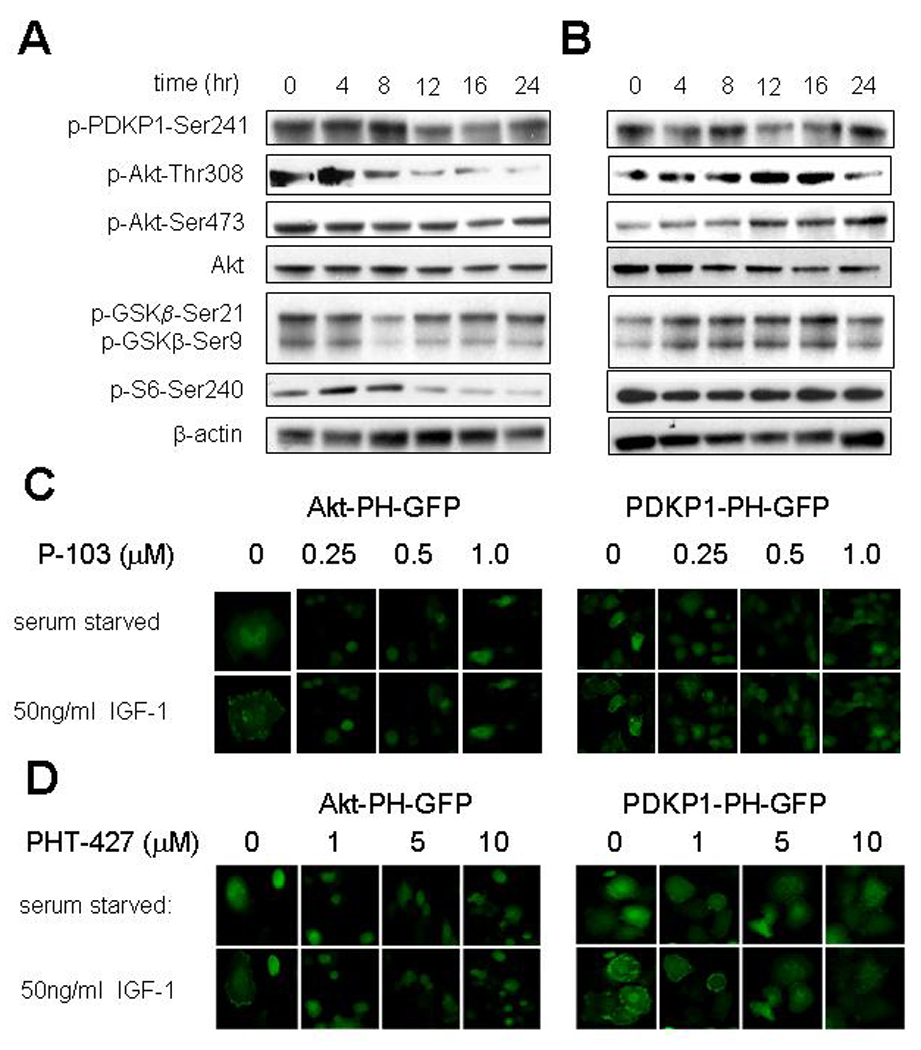
Western blots of Akt activity measured by phospho-Ser473-Akt and PDPK1 activity by phospho-Ser241-PDPK1 and activation of selected downstream targets. β-Actin was used as a loading control. A, BxPC-3 pancreatic cancer cells exposed to 10 µM PHT-427 in media with 10% fetal bovine serum (FBS) for various times; B, MiaPaCa-2 pancreatic cancer cells exposed to 10 µM PHT-427 in media with 10% FBS for various times; C, Panc-1 pancreatic cancer cells stably transfected with Akt-PH domain-GFP or PDKP-1-PH domain-GFP were cultured in serum free medium for 16 hr, exposed to the PtdIns-3-K inhibitor P-103 at different concentrations for 4 hr, and then stimulated with IGF-1 50 ng/ml for 20 min or no stimulation. Cellular fluorescence was measured with an IN Cell Analyzer 1000; D, similar studies with Akt-PH domain-GFP or PDKP-1-PH domain-GFP Panc-1 cells exposed to PHT-427 at different concentrations.
Subcellular translocation
GFP tagged PH domains were used to follow the subcellular localization of Akt and PDK1 upon IGF-1 stimulation in Panc-1 cells. There was diffuse cytoplasmic fluorescence in unstimulated cells with movement of the fluorescence to the plasma membrane upon stimulation with IGF-1 (Figure 3C and D). The plasma membrane translocation of both the Akt and PDKP1 PH domains was inhibited by PHT-427 at between 5 and 10 µM. This inhibition was comparable to that obtained when cells are treated with the PtdIns-3-K inhibitor P-103.
Antitumor Activity
Effect of the carbon chain length
Mice with BxPC-3 pancreatic, MCF-7 breast or A-549 NSCL cancer xenografts were administered PHT-427, or its analogs with a C-4, C-6 or C-8 alkyl chain by oral gavage twice a day for 10 days. The results show that PHT-427 had the greatest antitumor activity with the C-8 chain analog having less activity, and analogs with a C-4 or C-6 chain very little activity (Figure 4A). All further antitumor studies were conducted using compound PHT-427.
Figure 4. Antitumor activity of PHT-427 analogs.

Panel A, Effect of carbon chain length. Mice with BxPC-3 pancreatic, MCF-7 and A549 nscl lung cancer sc xenografts were treated with vehicle alone (C) or PHT-427 and its analogs with different C chain lengths at 200 mg/kg twice a day for 10 days and tumor growth rate measured. n = 8 mice per group, bars are SE of mean. * p < 0.05 and ** p < 0.01 compared to control. Panel B. Tumor growth inhibition by PHT-427 in different tumors. Doses and schedules: PC-3 prostate cancer 125 mg/kg twice a day (BID) × 5 days; A549 nsc lung cancer 200 mg/kg BID × 10 days; MCF-7 breast cancer 200 mg/kg BID × 10 days; SKOV-3 ovarian cancer 250 mg/kg BID × 10 days; BxPC-3 pancreatic cancer 250 mg/kg BID × 5 days. Results are expressed as the growth rate of the PHT-427 treated tumors relative to the control tumors. n = 8 mice per group, bars are SE of mean. * p < 0.05 and ** p < 0.01 compared to control. C, Combination of PHT-427 with paclitaxel in MCF-7 human breast cancer xenografts. Female scid mice with a sc implanted 60 day estradiol release pellet were injected sc with 107 MCF-7 human breast cancer cell. When the tumors reached ~180 mm3 dosing was started on day 13 shown by ↑. ( ) vehicle control po twice a day for 10 days; (◇) PHT-427 200 mg/kg po twice a day for 10 days; (□) paclitaxel 10 mg/kg ip every other day for 5 doses;(△) PHT-427 200 mg/kg po twice a day for 10 days and paclitaxel 10 mg/kg ip every other day for 5 doses. D, Mice were implanted orthotopically with NCI-H441 non-small cell lung cancer cells into their lungs and twenty days later treatment was begun with daily oral erlotinib 50mg/kg until the end of the experiment or daily orally of 200mg/kg PHT-427 for 10 days. The mice were killed and autopsied at day 63 when control animals became moribund and primary left lung tumor volume was measured. Values are the mean of 10 animals per group and bars are SE, * = p < 0.05
) vehicle control po twice a day for 10 days; (◇) PHT-427 200 mg/kg po twice a day for 10 days; (□) paclitaxel 10 mg/kg ip every other day for 5 doses;(△) PHT-427 200 mg/kg po twice a day for 10 days and paclitaxel 10 mg/kg ip every other day for 5 doses. D, Mice were implanted orthotopically with NCI-H441 non-small cell lung cancer cells into their lungs and twenty days later treatment was begun with daily oral erlotinib 50mg/kg until the end of the experiment or daily orally of 200mg/kg PHT-427 for 10 days. The mice were killed and autopsied at day 63 when control animals became moribund and primary left lung tumor volume was measured. Values are the mean of 10 animals per group and bars are SE, * = p < 0.05
Activity of PHT-427 in different tumors
The antitumor activity of PHT-427 at doses of 125 to 250 mg/kg in different tumors is shown in Table 1 and graphically in Figure 4B. PHT-427 gave up to an 80% inhibition of tumor growth in the most sensitive tumors. Tumors with PIK3CA mutation were among the most sensitive. Tumors with a K-Ras mutation were less sensitive irrespective of the dose of PHT-427. The pattern of inhibition in different tumors is similar to that we have seen using the PtdIns-3-K inhibitor PX-866 (38) suggesting that the antitumor activity of PHT-427 is due to inhibition of the PtdIns-3K/PDPK1/Akt signaling pathway.
Antitumor activity in combination with chemotherapy
PHT-427 administered orally to mice with subcutaneous MCF-7 human breast cancer xenografts showed antitumor that was additive with that of paclitaxel (Figure 4C). PHT-427 administered orally for 10 days to mice with orthotopic NCI-H441 NSC lung cancer xenografts increased the antitumor activity of continuous daily erlotinib which by itself had no antitumor activity in this model (Figure 4D). NCI-H441 has a mutant K-Ras which is known to be a negative predictor of erlotinib activity in NSCL cancer.
Pharmacokinetics
Plasma levels of PHT-427 following oral administration to mice of a dose of 200 mg/kg showed rapid absorption, without a lag phase, Cmax was 8.2 µg/ml 1 hr following dosing, and the elimination half-life was 1.4 hr with a terminal PHT-427 concentration of 0.1 µg/ml 10 hr after dosing (Figure 5A). The plasma concentration of PH-427 was above the level which gave inhibition of Akt and PDPK1 signaling in cells of 10 µM (4 µg/ml) for at least 3 hr.
Figure 5. In vivo effects of PHT-427.

A, C57Bl/6 female mice were administered PHT-427 at 200 mg/kg po in 0.1 ml sesame oil and plasma concentrations of parent compound measured by HPLC-MS. Values are the mean of 3 mice per group and bars are SE. Western blots of B, BxPC-3 pancreatic cell xenografts and C, MiaPaCa-2 pancreatic cell xenografts in scid mice administered a single oral dose of PHT-427 of 200 mg/kg in 0.1 ml vehicle. Mice were killed after various times, the tumors removed and immediately frozen in liquid N2.
Pharmacodynamics
Xenografts derived from both the MiaPaCa-2 and BxPC-3 lines were excised and subjected to Western blotting for markers of PtdIns-3-K/PDPK1/Akt signaling after a single 200 mg/kg dose at times up to 12 hr (Figures 5B and 5C). Quantitation of the blots for MiaPaCa-2 and BxPC-3 xenografts showed maximal decreases at 8 hr compared to non-treated control, for phospho-Ser473-Akt of 68.7% and 10%, phospho-Thr308-Akt of 74.1% and 100%, and phospho-Ser241-PDPK1 of 69.6% and 14.8%, respectively. Total Akt itself showed a small decrease of about 25% at 8 hr. phospho-Ser221-RSK which has been shown to be phosphorylated independently of Akt activation (24) was decreased by 85.5% in MiaPaCa-2 and 37.2% in BxPC-3xenografts. phospho-Ser240 ribosomal S6-kinase levels showed a decrease at 0.5 hr and again at 4 to 6 hr in BxPC3 xenografts but not in MiaPaCa-2 xenografts. Thus, the xenograft studies confirm the previous cell line studies with inhibition of phospho-Ser241-PDPK1, phospho-Ser473-Akt and phospho-Thr308-Akt in both BxPC3 and MiaPaCa-2, while inhibition of phospho-Ser240 S6 ribosomal kinase was only seen in the sensitive BxPC-3 but not the resistant MiaPaCa-2.
Toxicity
Groups of 4 female C57/Bl6 mice were administered 0.1 ml vehicle alone or PHT-427 in 0.1 ml vehicle at 200 mg/kg twice a day for five days and blood collected 16 hr after the last dose. PHT-427 administration gave no significant change in blood chemistry (white blood cells, red blood cells, hemoglobin, platelet or differential count), serum glucose, creatinine or blood urea nitrogen, but there was a significant decrease in serum AST from 167.2 ± 29.4 to 86.4 ± 13.3 U/L (p < 0.05) and in serum ALT from 28.2 ± 2.3 to 20.8 ± 0.9 U/L (p < 0.05), possibly suggestive of mild liver dysfunction. There was no significant change in body weight or the weight of the spleens.
Discussion
Because of their roles in cellular apoptosis and survival pathways, the AGC family serine/threonine kinases Akt and PDPK1 have emerged as attractive therapeutic targets for cancer (3,15). Attempts to develop Akt and PDPK1 inhibitors targeting the ATP binding pocket have generally produced compounds that also inhibit other ACG family kinases, such as PKA, which may account for the toxicity of this type of inhibitor seen in animals and in patients (15). Recently, an allosteric Akt inhibitor MK-2206 that binds to a region outside the ATP catalytic site has been described and is in early clinical trial (35). We have developed an alternative approach to inhibiting Akt and other signaling proteins through compounds that bind to the PH domain (25,26,31,39). The PH domain is essential for the binding of a number of cytosolic signaling proteins to plasma membrane PtdIns(3,4,5)P3 formed by the activity of PtdIns-3-K, thus, causing allosteric activation, or bringing the proteins into proximity with their effectors and substrates, leading to the activation of signaling cascades (1).
Although PHT-247 was originally developed as an inhibitor of the PH domain of Akt (26), new results suggest that it is also an inhibitor of PDPK1. We first showed by SPR studies that PHT-427 binds to the expressed PH domain of PDPK1 with an affinity similar to that of binding to the PH domain of Akt (Ki for PDPK1 5.2 µM and for Akt 2.7 µM). We also investigated the effect of varying the length of the alkyl chain attached to the benzene ring, which our modeling studies (not shown) suggested fits into a shallow channel in the PH domain. For both Akt and PDPK1 the optimum chain length giving maximum binding was C-12 to C-14. While the C-14, C-16 and C-18 analogs also showed some binding to the Akt PH domain only the C-12 and C-14 analogs showed appreciable binding to the PH domain of PDPK1. Other compounds that have been suggested to bind to the PH domain of Akt include perifosine (32) and triciribine (34), and both compounds are currently in clinical trial as Akt inhibitors. However, in our hands neither of them bound to the PH domains of Akt or PDPK1. MK-2206 is an allosteric inhibitor of Akt that was reported to bind to a region outside the ATP site of Akt (35). Our SPR studies confirmed that it does not bind significantly to the PH domain of Akt or PDK1. Thus, PHT-427 distinguishes itself from other inhibitors by showing high affinity binding for the PH domains of Akt and PDPK1.
RPPA studies in PC-3 prostate cancer cells showed that PHT-427 causes a reduction in phospho-Thr308-Akt, and also in phospho-Ser241-PDPK1 and its downstream targets, phospho-Ser657-PKC and total SGK1 (36,37). Western blotting using BxPC-3 pancreatic cancer cells showed that PHT-427 causes a decrease at both phospho-Thr308-Akt and phospho- Ser473-Akt, and also in phospho-Ser241-PDPK1 and phospho-Ser9-GSK3β. In contrast MiaPaCa-2 pancreatic cancer cells showed an increase in phospho-Thr308-Akt and phospho- Ser473-Akt with no decrease in phospho-Ser9-GSK3β upon treatment with PHT-427. In vivo studies in mice with BxPC-3 and MiaPaCa-2 xenografts treated with a 200 mg/kg dose of PHT-427 showed a decrease in phospho-Ser473-AKT, phospho-Thr308-Akt, phospho-Ser241-PDPK1 and in the PDKP1-specific downstream target phospho-Ser221-RSK (24). Of note was that phospho-Ser240 ribosomal S6-kinase was decreased by PHT-427 in BxPC-3 cells and xenografts but not in MiaPaCa-2 cells or xenografts that were resistant to the antitumor effects of PH-427. Phospho-Ser240 ribosomal S6-kinase has been previously reported to be a biomarker for Akt pathway inhibition in sensitive tumors (40). Thus, our studies show that PHT-427 inhibits both Akt and PDPK1 signaling in sensitive cancer cells, but that resistant cancer cells have a rebound increase in Akt activity. Consequently PDPK1 inhibition may be more important for the antitumor activity of PHT-427 than inhibition of Akt.
Two mechanisms have been suggested for the inhibition of PH domain proteins by small molecule inhibitors. A study of the binding of 2-hydroxymethyl-carbonyletherlipid, a compound related to DPIEL identified by our group as a PH domain inhibitor of Akt (31), suggested that the binding to the PtdIns pocket of the PH domain causes an open interdomain conformation where the N-terminal PH domain and C-terminal regulatory domains move away from the kinase domain preventing translocation of Akt to the plasma membrane, thus blocking its activation (41). Another study using an allosteric inhibitor of Akt which did not bind directly the PtdIns pocket of the PH domain of Akt but instead interacted with Trp80 outside of the pocket, found that the PH domain was folded back on the kinase domain, thus blocking its activity (42). We have found that PHT-427 displaces PtdIns(3,4,5)P3 from the Akt PH domain in a manner similar to DPIEL, while the allosteric inhibitor MK-2206 did not displace PtdIns(3,4,5)P3, suggesting that the binding of PHT-427 causes an open conformation preventing the translocation of Akt to the plasma membrane. This is consistent with our observation of the inhibition of the cellular translocation of both an Akt and a PDKP1 PH domain GFP construct by PHT-427. The inhibition of translocation occurs even if there is no antitumor response to PHT-427 because of the presence of a K-Ras mutation.
Activating mutations in the p110 alpha catalytic subunit (PIK3CA) are present in a number of human tumors (5) with the most common sites clustering around the amino acid 1047 site in the kinase domain and amino acid 545 in the helical domain (43). Recent data suggests that cells with PIK3CA mutation may be divided into two classes. The first are cells that show dependency on Akt signaling, an effect that is augmented by additional activation of the pathway, for example, and loss of the tumor suppressor PTEN. The second class of cells have decreased Akt signaling and an increased dependence on PDPK1 signaling and the effects it has on targets independent of Akt signaling, most notably the AGC kinase SGK (22). It has recently been shown that in PIK3CA mutant breast cancer cells, PDPK1 can signal independently of Akt by phosphorylating and activating SGK3 due to altered PtdIns(3,4,5)P3 levels caused by mutant PIK3CA (22). Mutant PIK3CA cells are therefore dependent on PDPK1 and SGK3 for viability. We observed good antitumor activity of PHT-427 in one tumor xenograft with a PIK3CA mutation, SKOV-3 ovarian cancer, which was better than with a PC-3 prostate cancer xenograft which has loss of PTEN, although this was with a lower dose of PHT-427. Thus, although not conclusive the weight of evidence suggests that it is the PDPK1 inhibitory activity of PHT-427, a consequence of its binding to the PH domain of PDPK1 that could be primarily responsible for the antitumor activity of PHT-427. Inhibition of Akt through binding to the PH domain of Akt cannot however be discounted as a contributing to PHT-427’s antitumor activity, and may vary depending upon the cell type.
We found that the presence of mutant K-Ras predicts for resistance. This is similar to the pattern of sensitivity to the antitumor activity of a PtdIns-3-K inhibitor, with K-Ras bypassing Akt signaling through activation of the MEK pathway (38). However, the by-pass is not complete because A-549 non small cell lung cancer which has mutant K-Ras shows some sensitivity to the antitumor activity of PHT-427, as it does to PtdIns-3-K inhibition (38). Orthotopic H441 non small cell lung cancer, which also has mutant K-Ras, was resistant to the antitumor activity of PHT-427 alone, but showed synergy when PHT-427 was combined with the EGFR inhibitor erlotinib.
In summary, a series of PHT-427 analogs with C-4 to C-16 alkyl chain length were synthesized and tested. PHT-427 (C-12 chain) and the C-14 analog bound with the highest affinity to the PH domains of PDPK1 and Akt. PHT-427 gives a transient inhibition of Akt signaling in cells and tumor xenografts, but a longer-lasting inhibition of PDPK1 signaling that correlated with decreased activation of down stream signaling targets for both proteins. PHT-427 given orally on a twice daily schedule inhibited the growth rate of human tumor xenografts in immunodeficient mice as long as it was given, with up to an 80% inhibition in the most sensitive tumors. The pharmacokinetics of PHT-427 showed a relatively short plasma half life of 1.4 hr and levels that corresponded to the time course of the inhibition of signaling pathways in tumor xenografts. There was no weight loss or change in blood chemistry associated with the administration of PHT-427. Tumors with a K-Ras mutation were less sensitive to PHT-427 as has been observed for other inhibitors of the PtdIns-3-K signaling pathway. Combination antitumor studies showed that PHT-427 has greater than additive activity with paclitaxel in breast cancer and with erlotinib in non small cell lung cancer.
Acknowledgments
This work was supported by grants RO1 CA 061015 and P30 CA 23074 from the National Cancer Institute to GP. SM was supported in part by the MGE@MSA Fellowship and a minority supplement for CA 061015, and JJ by the Lung Cancer Research Foundation.
References
- 1.Rebecchi MJ, Scarlata S. Pleckstrin homology domains: a common fold with diverse functions. Ann Rev Biophys Biomol Struct. 1998;27:503–528. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- 2.Workman P, Clarke PA, Guillard S, Raynaud FI. Drugging the PI3 kinome. Nature Biotechnol. 2006;24:794–796. doi: 10.1038/nbt0706-794. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 4.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular ransformation Oncogene. 2008;27:5486–5496. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 7.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 8.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 9.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 10.Chung J, Grammer TC, Lemon KP, Kazlauskas A, Blenis J. PDGF- and insulin-dependent pp70S6k activation mediated by phosphatidylinositol-3-OH kinase. Nature. 1994;370:71–75. doi: 10.1038/370071a0. [DOI] [PubMed] [Google Scholar]
- 11.van Weeren PC, de Bruyn KM, de Vries-Smits AM, van Lint J, Burgering BM. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J Biol Chem. 1998;273:13150–13156. doi: 10.1074/jbc.273.21.13150. [DOI] [PubMed] [Google Scholar]
- 12.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 13.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 14.Carptener JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 15.Li Q. Recent progress in the discovery of Akt inhibitors as anticancer agents. Expert Opin Ther Patents. 2007;17:1077–1130. [Google Scholar]
- 16.Alessi D. Discovery of PDK1, one of the missing links in insulin signal transduction. Biochem Soc Trans. 29:1–14. doi: 10.1042/0300-5127:0290001. 200. [DOI] [PubMed] [Google Scholar]
- 17.Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDPK1 is essential for activation of S6K and SGK, but not PKB. EMBO. 2001;20:4380–4390. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casamayor A, Morrice NA, Alessi DR. Phosphorylation of ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem J. 1999;342:287–292. [PMC free article] [PubMed] [Google Scholar]
- 19.Avruch J, Belham C, Weng Q, Hara K, Yonezawa K. The p70 S6 kinase integrates nutrient and growth signals to control translational capacity. Prog Mol Subcell Biol. 2001;26:115–154. doi: 10.1007/978-3-642-56688-2_5. [DOI] [PubMed] [Google Scholar]
- 20.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 21.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1(PDK-1) Curr Biol. 1998;8:1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide-3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDPK1) and PDK2. Biochem J. 1999;339:319–328. [PMC free article] [PubMed] [Google Scholar]
- 23.Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidyinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009;69:6299–6306. doi: 10.1158/0008-5472.CAN-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, et al. PDK-1-SGK3 signaling in the absence of AKT activation in PIK3CA-mutant cancers. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahadevan D, Powis G, Mash EA, et al. Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol Cancer Ther. 2008;7:2621–2632. doi: 10.1158/1535-7163.MCT-07-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moses SA, Ali A, Zuohe S, et al. In vitro and in vivo activity of novel small molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/AKT. Cancer Res. 2009;69:5073–5081. doi: 10.1158/0008-5472.CAN-08-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du-Cuny L, Song Z, Moses S, et al. Computational modeling of novel inhibitors targeting the Akt pleckstrin homology domain. Bioorg Med Chem. 2009;1(17):6983–6992. doi: 10.1016/j.bmc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, He X, Baggerly KA, Coombes KR, Hennessy BT, Mills GB. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- 29.Paine GD, Taylor CW, Curtis RA, et al. Human tumor models in the severe combined immune deficient scid mouse. Cancer Chemo. Pharmacol. 1997;40:209–214. doi: 10.1007/s002800050648. [DOI] [PubMed] [Google Scholar]
- 30.Onn A, Isobe T, Itasaka S, Wu W, O'Reilly MS. Development of an orthotopic model to study the biology and therapy of primary human lung cancer in nude mice. Clin Can Res. 2003;9:5532–5539. [PubMed] [Google Scholar]
- 31.Meuillet EJ, Mahadevan D, Vankayalapati H. Specific Inhibition of the Akt1 pleckstrin homology domain by D-3-deoxy-phosphatidyl-myo-inositol analogues. Cancer Res. 2003;2:389–399. [PubMed] [Google Scholar]
- 32.Poradosu E, Lemmon M, Keleti D. Perifosine selectively inhibits binding of Akt PH domain to PtdIns(3,4)P2. Amer Assoc Cancer Res Proc Ann Meeting. 2007 Abstract #645, [Google Scholar]
- 33.Ruiter GA, Zerp SF, Bartelink H, et al. Anti-cancer alkyl-lysophospholipids inhibit the phosphatidylinositol 3-kinase-Akt/PKB survival pathway. Anticancer Drug Des. 2003;14:167–173. doi: 10.1097/00001813-200302000-00011. [DOI] [PubMed] [Google Scholar]
- 34.Kim D, Cheng GZ, Lindsley CW, Yang H, Cheng JQ. Targeting the phosphatidylinositol-3 kinase/Akt pathway for the treatment of cancer. Curr Opin Inv Drugs. 2009;6:1250–1258. [PubMed] [Google Scholar]
- 35.Lu W, Defeo-Jones D, Davis LJ, et al. In vitro and in vivo antitumor activities of MK-2206, a new allosteric Akt inhibitor. Amer Assoc Cancer Res, Proc Annual Meeting. 2009 Abstract #3714. [Google Scholar]
- 36.Sonnenburg ED, Gao T, Newton AC. The phosphoinositide-dependent kinase, PDK-1, phosphorylates conventional protein kinase C isozymes by a mechanism that is independent of phosphoinositide 3-kinase. J Biol Chem. 2001;276:45289–45297. doi: 10.1074/jbc.M107416200. [DOI] [PubMed] [Google Scholar]
- 37.Carlisle J, Townley I, Dodge-Kafka K, et al. Spatial restriction of PDK1 activation cascades by anchoring to mAKAPα. Molecular Cell. 2005;20:661–672. doi: 10.1016/j.molcel.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Ihle NT, Lemos R, Wipf P. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–148. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meuillet EJ, Ihle N, Baker AF, et al. In vivo molecular pharmacology and antitumor activity of the targeted Akt inhibitor PX-316. Oncol Res. 2004;14:513–527. doi: 10.3727/0965040042380487. [DOI] [PubMed] [Google Scholar]
- 40.Hennessy B, Lu Y, Poradosu E, et al. Pharmacodynamic markers of perifosine efficacy. Clin Cancer Res. 2007;13:7421–7431. doi: 10.1158/1078-0432.CCR-07-0760. [DOI] [PubMed] [Google Scholar]
- 41.Huang BX, Kim H-Y. Probing Akt-inhibitor interaction by chemical cross-linking and mass spectrometry. J Amer Soc Mass Spectrometry. 2009;20:1504–1513. doi: 10.1016/j.jasms.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel ph-kinase domain interface in Pkb/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:189–200. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103:1289–1294. doi: 10.1073/pnas.0510772103. [DOI] [PMC free article] [PubMed] [Google Scholar]