

Abstract
Cells in vivo are constantly exposed to reactive oxygen species (ROS) generated endogenously and exogenously. To defend against the deleterious consequences of ROS, cells contain multiple antioxidant enzymes expressed in various cellular compartments to scavenge these toxic species. Under oxidative stresses, these antioxidant enzymes are upregulated to restore redox homeostasis. Such an adaptive response results from the activation of a redox-sensitive gene regulatory network mediated by nuclear factor E2-related factor 2. To more completely understand how the redox control system is designed by nature to meet homeostatic goals, we have examined the network from a systems perspective using engineering approaches. As with man-made control devices, the redox control system can be decomposed into distinct functional modules, including transducer, controller, actuator, and plant. Cells achieve specific performance objectives by utilizing nested feedback loops, feedforward control, and ultrasensitive signaling motifs, etc. Given that endogenously generated ROS are also used as signaling molecules, our analysis suggests a novel mode of action to explain oxidative stress-induced pathological conditions and diseases. Specifically, by adaptively upregulating antioxidant enzymes, oxidative stress may inadvertently attenuate ROS signals that mediate physiological processes, resulting in aberrations of cellular functions and adverse consequences. Lastly, by simultaneously considering the two competing cellular tasks - adaptive antioxidant defense and ROS signaling - we re-examine the premise that dietary antioxidant supplements is generally beneficial to human health. Our analysis highlights some possible adverse effects of these widely consumed antioxidants.
Keywords: ROS, Keap1, Nrf2, oxidative stress, antioxidant, redox homeostasis, feedback, feedforward, ultrasensitive motif, loop gain
1. Introduction
By utilizing molecular oxygen as the final electron transfer recipient, living aerobic organisms are at a tremendous evolutionary advantage over anaerobic species for effective energy abstraction from organic nutrients. However, with the emergence of this mitochondrial respiratory process, comes a number of byproducts, mainly in the form of reactive oxygen species (ROS) (Turrens, 2003; Brand et al., 2004). Highly reactive, ROS, which include superoxide anion (O2-·), hydrogen peroxide (H2O2), and hydroxyl radical (OH·), etc., are generally deleterious to the structures and functions of the macromolecular constituents of the cell. By reacting largely in a nonspecific manner with nucleic acids, proteins, and membrane lipids, they can cause gene mutation, impairment or loss of enzyme activity, and altered cell membrane permeability (Finkel, 1998; Finkel, 2003).
Besides being produced by oxidative respiration in the mitochondrion, ROS also arise as intermediates in many other metabolic processes in the cell. These processes include β-oxidation of fatty acids in the peroxisome, cytochrome P450 enzyme-catalyzed metabolic reactions in the endoplasmic reticulum, and prostaglandin synthesis from arachidonic acid at the cell membrane, etc (Thannickal and Fanburg, 2000). ROS are also generated deliberately as part of a cellular defense mechanism against invaded pathogens (Forman and Torres, 2002). Phagocytosis of bacteria by neutrophils and macrophages is frequently accompanied by a respiratory process characterized by heightened oxygen consumption. NADPH oxidase (NOX) and myeloperoxidase are employed during this so-called respiratory burst to generate O2-· and hypochlorous acid to kill invading bacteria (Nauseef, 2007). Leakage of ROS from this innate immune response may unnecessarily subject the host cells to high concentrations of the bactericidal reactive species, leading to undesirable toxicity.
Given their ubiquitous cellular toxicity, ROS need to be tightly controlled so that they do not accumulate in the cell unchecked. Living in an oxygen-rich atmosphere for thousands of millions of years, cells have evolved to contain a suite of antioxidant molecules and detoxifying enzymes that can quickly remove or detoxify reactive species. The thiol-containing tripeptide, glutathione (GSH), is the most abundant small-molecule antioxidant that scavenges ROS and neutralizes electrophiles (Meister and Anderson, 1983). Large-molecule antioxidant and detoxifying enzymes include superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase (CAT), glutathione reductase (GR), glutamate cysteine ligase (GCL), NAD(P)H:quinone oxidoreductase 1 (NQO1), heme oxygenase1 (HO-1), and other phase II xenobiotic-metabolizing enzymes such as glutathione S-transferase (GST), UDP-glucuronyl transferase (UGT), and sulfotransferase (SULT), etc (Nguyen et al., 2003). Expressed in a number of isoforms and distributed in various organelles and subcellular compartments, these enzymes cooperatively participate in a network of interconnected metabolic reactions that eliminate reactive species at the sites of origin. At basal, non-stress conditions, these enzymes are adequately expressed, keeping ROS at low non-toxic levels within the cell. Besides controlling the absolute concentrations of ROS, the antioxidant enzymes also maintain the redox states of various cellular compartments at levels that are conducive to the operation of many redox-sensitive processes, such as protein folding in the endoplasmic reticulum (Go and Jones, 2008). A primary metric of the redox state is redox potential, which is determined by the ratios of major thiol/disulfide pairs, including GSH/GSSG, cysteine/cystine, thioredoxin-1 (SH2/SS), and NADPH/NADP+ (Jones, 2006; Kemp et al., 2008). By controlling the concentrations and thus the ratios of these redox pairs, antioxidant enzymes such as GCL, GR, GPx, and glucose-6-phosphate dehydrogenase (G6PD), etc., help maintain a generally favorable intracellular redox environment.
The favorable redox environment can be disrupted by a number of oxidative events, leading to an initial imbalance between the production of ROS and the cellular antioxidant capacity. As a result, ROS rise and the intracellular milieu is shifted to a more oxidizing condition. Oxidative events (stresses) may result endogenously from increased aerobic metabolism in the mitochondrion, respiratory burst during microbial phagocytosis, or in some cases, inhibition of antioxidant enzymes. In many circumstances, oxidative stress originates from exogenous sources in the environment. Common external ROS-inducing stressors include UV light, ionizing radiation, and various oxidative chemicals. Regardless of the origin, cells need to respond quickly to adapt to the stressful condition and restore redox homeostasis. Otherwise, prolonged elevations in ROS may cause irreparable oxidative damage and even cell death.
To achieve the homeostatic goal, cells, when confronted with oxidative stressors, must augment their antioxidant capacity quickly and sufficiently to counteract the increased ROS production. Extensive research in the past decade has gradually filled out the details of a redox homeostatic gene regulatory network that centers on a master regulating transcription factor named nuclear factor erythroid 2-related factor 2 (Nrf2) (Nguyen et al., 2003; Itoh et al., 2004; Motohashi and Yamamoto, 2004; Kensler et al., 2006; Kobayashi and Yamamoto, 2006). Under oxidative and electrophilic stresses, this Nrf2-centered network is adaptively activated, enhancing the expression of a suite of antioxidant and phase II enzymes to restore redox homeostasis.
ROS have been implicated in etiology of a number of pathological conditions and diseases, such as cancer, diabetes, atherosclerosis, aging, and neurodegeneraion (Droge, 2002). The magnitude and duration of ROS elevation are likely to be important quantitative determinants for the initiation, progression, and severity of their pathological consequences. The dynamics of the fluctuating ROS concentrations and redox potentials in various intracellular compartments are expected to be dependent on the operation of the redox homeostatic control network. Thus, understanding the quantitative behavior of this Nrf2-centered adaptive system is a necessary step for quantitative risk assessment for human exposure to environmental toxicants that alter cellular ROS. Yet, as the list of parts and interactions involved in redox regulation expands, it becomes evident that the redox control network is a complex system containing nested feedback loops, fast and slow adaptation pathways, signal amplification motifs, and feedforward controls. The network complexity makes it challenging to understand the design characteristics for fast adaptation and robust redox homeostasis. The situation is further complicated by the fact that ROS, despite their deleterious effects, also serve as normal second messengers (Finkel, 2003; Buetler et al., 2004). In this signaling context, the adaptive induction of antioxidants may not be entirely beneficial if it blunts ROS signals that mediate physiological responses (Pi et al., 2007b). The structural complexity and nonlinear interactions, characteristic of the redox control network, require a computational systems biology approach to comprehend its dynamic and steady-state behaviors (Kitano, 2002). It is increasingly discovered that biological systems appear to operate by the same principles that govern many man-made devices except that the parts that implement these principles are genes and proteins, etc., rather than resistors, capacitors, and transistors as in electronic circuits (Tyson et al., 2003; Xiong and Ferrell, 2003; El-Samad et al., 2005). The principles and theories, developed by engineers, can be borrowed, with or without modifications, to study biological systems (Åström and Murray 2008).
This review first describes the engineering-oriented modular organization of the molecular circuit underlying cellular redox homeostasis. It is followed by enumeration of the composition and function of each functional module. After specifying the performance objectives of the redox homeostatic system, we show how nested feedback loops and embedded ultrasensitive motifs help achieve these goals. With this background established, the issue of physiological ROS signaling in the context of adaptive antioxidant response is examined and placed into perspective, which leads to a novel mechanism proposed for oxidative stress-induced cell malfunction. Finally, by taking into consideration the interplay between the adaptive antioxidant response and ROS signaling, we discuss, from a systems biology perspective, the potential adverse consequences of consuming dietary antioxidant supplements, which are widely used nowadays for general health promotion and chemoprevention.
2. Architecture of the Nrf2-mediated redox homeostatic control system
Mechanical and electrical engineers have worked on man-made control devices for decades. Familiar examples of these devices are thermostat for temperature control, automatic cruising system for car speed control, and auto-piloting for airplane altitude and speed control. It becomes increasingly clear that man-made control devices and biological control systems share similar structures and perhaps operate by similar principles (Yi et al., 2000; El-Samad et al., 2005; Zhang and Andersen, 2007). From an engineering perspective, a control system using negative feedback comprises several functional modules: transducer/sensor, controller, actuator, and plant (Åström and Murray 2008). Organizing the components of a biological homeostatic system into these functional modules and visually presenting them in a layout that engineers routinely use would greatly facilitate our investigation into how the system functions to meet its objectives mandated through evolution. Fig. 1 illustrates an engineering approach to describe the Nrf2-mediated redox control network. This system has multiple variables that need to be effectively controlled, including ROS, lipid peroxides, electrophiles, as well as the redox potential as represented by the ratios of GSSG/GSH and other redox pairs.
Figure 1.
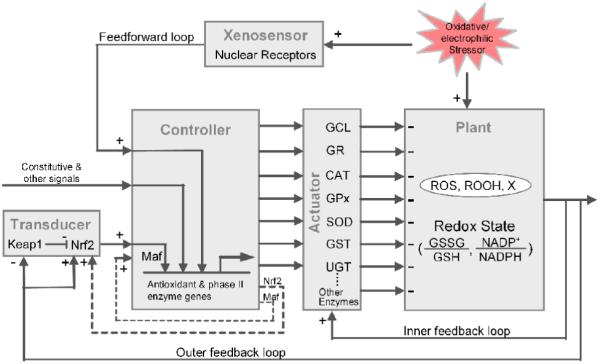
An engineering view of the Nrf2-mediated redox homeostatic control system. The system comprises several functional modules routinely found in man-made control devices - transducer/ xenosensor, controller, actuator, and plant - which are linked in tandem through feedback and feedforward loops. Refer to the main text for a detailed description of the components and functions of individual modules. The inner feedback loop contains two pathways which are not separately illustrated here: a fast loop through increasing the activities of redox-sensitive antioxidant enzymes, and a slow loop through increasing the stabilization of antioxidant mRNAs. The two dashed loops represent positive auto-regulation of Nrf2 and Maf genes. Constitutive and other signals represent inputs to the controller other than Nrf2, such as those mediated by AP-1, NF-kB, and other redox-sensitive signals, etc. Nuclear receptors acting as xenosensors include AhR, PXR, and CAR. ROS: reactive oxygen species, ROOH; lipid peroxides, X: reactive electrophiles.
2.1. Transducer
When an oxidative stressor is encountered by cells, ROS and redox potentials will initially deviate from their basal levels. The deviation, especially in ROS levels, is detected by the transducer module comprising two key proteins - Kelch-like ECH-associated protein 1 (Keap1) and Nrf2 - where Keap1 functions as the actual sensor molecule and Nrf2 relays the redox signal gathered by Keap1 to the controller module. Keap1 is a cysteine-rich protein that is anchored to the cytoskeletal actin in the cytosol (Dinkova-Kostova et al., 2002). By physically interacting with Nrf2 and acting as an adaptor protein for a Cul3-dependent E3 ubiquitin ligase complex, Keap1 promotes ubiquitination of Nrf2 and its eventual degradation by the proteasome (McMahon et al., 2003; Stewart et al., 2003; Hong et al., 2005; Kobayashi et al., 2006). Under non-stress conditions Nrf2 is rapidly degraded via Keap1, exhibiting a short half-life of 13~21 minutes (Hong et al., 2005; Kobayashi and Yamamoto, 2006). This rapid turnover keeps Nrf2 at a low basal level. The multiple cysteine residues contained in the amino acid sequence of Keap1 enable it to function as a molecular sensor dectecting changes in the cellular redox state. A rise in the intracellular levels of ROS or electrophiles results in an increase in the oxidation or conjugation of key cysteine residues in the Keap1 molecule (Zhang and Hannink, 2003; Levonen et al., 2004). Although the exact cysteine residues that become oxidized and the nature of oxidation appear to be chemical agent-specific (Kobayashi et al., 2009), modifications of the Keap1 molecule at multiple cysteine residues generally weaken its activity as an E3 ligase adaptor. As a result, Keap1 becomes less capable of promoting Nrf2 degradation, leading to stabilization of Nrf2 - its half-life can be extended to 100~200 minutes under high-level oxidative stresses (Hong et al., 2005; Kobayashi et al., 2006). With slower degradation, cytosolic Nrf2 increases from de novo protein synthesis before moving into the nucleus. It was previously thought that oxidative stress activates Nrf2 by promoting its dissociation from Keap1 sequestration, yet subsequent studies indicated this is not the case as the binding affinity between Keap1 and Nrf2 was not affected by exposure to oxidative stressors (Eggler et al., 2005; He et al., 2006; Kobayashi et al., 2006; Tong et al., 2006).
Although Keap1 is the main sensor molecule in the Keap1/Nrf2 transducer, Nrf2 itself also appears to be redox-sensitive (Li et al., 2006). The transactivation domain of Nrf2 contains a nuclear export signal (NES), which is responsible, at least partially, for shuffling Nrf2 out of the nucleus (Li and Kong, 2009). The nuclear-exporting activity of this NES sequence, which contains a cysteine residue at position 138, is regulated by the redox state of the cell. Under oxidative stress, the cysteine residue is modified, which weakens the NES activity (Li et al., 2006). As a result, the trans-nuclear equilibrium shifts away from Nrf2 exporting, leading to increased retention of Nrf2 in the nucleus. Therefore, oxidative stress-induced nuclear Nrf2 accumulation is a consequence of two independent processes - increased cellular abundance due to stabilization and increased nuclear retention due to weakening of NES activity. In the redox control system, nuclear Nrf2 is the output signal of the transducer module, which is fed into the controller to transcriptionally regulate antioxidant and phase II enzymes.
2.2. Controller
In a man-made control system, controller is the functional unit that receives the error signal, i.e., the difference between the current value of the controlled variable fed back through the transducer and the set-point (e.g., the desired room temperature or car speed). The controller then processes the signal according to a preset mathematical algorithm and at the same time integrates other regulatory inputs. In most engineering applications, the processing algorithm generates an output that is either a proportional (P), integral (I), or differential (D) function of the error signal (PID control in engineering terminology), representing the present, past, and projected future state of the controlled variable, respectively (Åström and Murray 2008). The output of the controller is then used to drive the actuator to counteract perturbations. By this analogy, the controller in the redox homeostatic system contains a set of transcriptional processes in the nucleus which receive inputs from Nrf2 and other control signals. The controller integrates all these inputs to regulate the transcription of a battery of antioxidant and phase II genes. The redox controller contains a number of transcription activators and repressors. Among them the small Maf protein is an obligatory dimeric partner of Nrf2 (Katsuoka et al., 2005b). The Nrf2-Maf dimer is a transcription activator possessing high specificity and affinity to the antioxidant response element (ARE) located in the promoters of most antioxidant and phase II genes. Bach1 is a transcriptional repressor that binds to small Maf, making it unavailable for Nrf2 (Shan et al., 2004; Dhakshinamoorthy et al., 2005). The promoters of many antioxidant and phase II genes also receive regulatory input from other transcription factors, such as AP-1 and NF-kB (Harada et al., 2008; Liu et al., 2008; Kimura et al., 2009), suggesting that the antioxidant response is coordinately regulated with other cellular processes such as the inflammatory response (Woods et al., 2009). Implicitly the controller contains the general transcription apparatus and cofactors involved in the formation of the transcription initiation complex.
2.3. Actuator
In man-made control devices, actuator is the “heavy-duty machine” that receives control signals from the controller and executes the corrective process to minimize perturbations of the system by external disturbance. In the redox homeostatic control system described here, the set of antioxidant and phase II enzymes are the molecular actuators that directly catalyze a series of reactions neutralizing and detoxifying ROS and electrophiles. The antioxidant capacity of the actuator is regulated primarily through transcriptional control of the amounts of antioxidant and phase II enzymes located in various cellular compartments. As described in details later, the antioxidant capacity is additionally regulated through redox-sensitive alteration in the activities of certain antioxidant enzymes and stabilization of mRNA molecules.
2.4. Plant
The biochemical plant is all the cellular space where ROS, redox pairs, and antioxidant and phase II enzymes co-exist. It includes the cytosol, nucleus, mitochondrion, peroxisome, and other ROS-producing organelles. In these spaces, metabolic reactions occur continuously, producing and removing ROS and other reactive species. Antioxidant enzymes, which act as molecular actuators, catalyze a number of reactions that produce small-molecule antioxidants and neutralize ROS into nontoxic or less reactive species. Examples of these reactions include the de novo synthesis of GSH by GCL and GS, recycling of GSSG back to GSH by GR, production of NADPH by G6PD, conversion of O2-· to H2O2 by SOD, and reduction of H2O2 into water by GPx and CAT.
2.5. Nested feedback loops
As illustrated in Fig. 1, the sequential connection of the functional modules - transducer → controller → actuator → plant → transducer - forms a standard negative feedback control system. Although Keap1 is the high-profile redox sensor and the Nrf2-mediated transcriptional feedback loop is the mainframe architecture of the control system, the Keap1/Nrf2 transducer is not the sole redox sensor mediating the antioxidant response. Certain antioxidant enzymes themselves are redox sensors - changes in the cellular redox state can directly modify the structures and activities of the enzyme molecules (Ochi, 1995; Ochi, 1996). Just like Keap1, the redox-sensitive antioxidants and related enzymes all contain cysteine residues in their active sites that can be reversibly modified by ROS (Schuppe-Koistinen et al., 1994; Tu and Anders, 1998).
The most studied redox-sensitive antioxidant is GCL, the enzyme that catalyzes the first step of the de novo synthesis of GSH. The fully active GCL holoenzyme is a heterodimer comprising two monomeric protein molecules: the GCL catalytic subunit (GCLC) and GCL modulatory subunit (GCLM) (Yang et al., 2001a; Yang et al., 2001b). The catalytic activity of the GCL holoenzyme can be directly augmented under oxidative stress in the absence of any increases in the expression of the two protein subunits (Ochi, 1995; Ochi, 1996). Additionally, the reversible heterodimerization between GCLC and GCLM that forms the holoenzyme GCL is also a redox-regulated process. An oxidizing environment favors association and a reducing environment favors dissociation between the two subunits (Seelig et al., 1984; Huang et al., 1993; Tu and Anders, 1998; Fraser et al., 2002). The redox-sensitive regulation of GCL’s enzymatic activity and subunit heterodimerization appears to be mediated via cysteine residues present in both the GCLC and GCLM subunits, since the formation of intra- and inter-molecular disulfyl bonds between these cysteine residues is likely regulated by redox conditions (Tu and Anders, 1998; Fraser et al., 2003). The catalytic activity of GCL is also subject to inhibitory regulation by GSH. GSH competes with glutamate, one of GCL’s substrates, for its binding site on GCLC to inhibit the enzyme’s activity (Chen et al., 2005). It should be noted that although GSH and its oxidized form GSSG are the dominant redox couple that influences the cellular redox potentials, the competitive inhibition of GCL by GSH is not a redox-regulated process. The inhibition is sensitive only to the absolute concentration of GSH. Regardless, the redox-sensitive regulation of GCL activity and subunit heterodimerization, and competitive inhibition of GCL by GSH jointly form a short feedback loop from the plant to the actuator to regulate de novo GSH synthesis. Under oxidative stress, the intracellular redox environment becomes more oxidizing, and concomitantly the level of GSH often drops, at least initially, as it is consumed by reactions to neutralize ROS and electrophiles (Pi et al., 2008; Woods et al., 2009). These changes in the plant are fed back to the actuator through this short feedback pathway to synergistically enhance the total GCL activity and consequently the GSH synthesis rate.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a redox-sensitive metabolic enzyme involved in the glycolysis pathway. It catalyzes the reversible phosphorylation of glyeraldehyde-3-phosphate to 1,3-bisphosphoglycerate. Under oxidative stress, GAPDH’s enzymatic activity is reversibly inhibited due to modification to certain thiol groups in the protein sequence (Brodie and Reed, 1990; Schuppe-Koistinen et al., 1994). This inhibition results in a reduction in the metabolic flux along the glycolytic pathway, and a re-routing to the pentose phosphate pathway (PPP). A higher carbon flux to the PPP pathway leads to increased production of NAPDH, which serves as electron donor for both glutaredoxin (Grx) and thioredoxin reductase (TrxR)-catalyzed antioxidant reactions, and is required by GR to recycle GSSG to GSH. Strictly speaking GADPH is not an antioxidant enzyme in itself, but its redox-sensitive activity change indirectly leads to increased NADPH production and subsequent enhancement of cellular antioxidant capacity (Ralser et al., 2007). Together, the redox-sensitive regulation of GCL activity and metabolic re-routing of glucose catabolism constitute a short, fast negative feedback loop. The feedback regulation is fast because unlike the Nrf2-mediated feedback loop, it does not involve gene transcription. The response time could be in the order of seconds to minutes. Since this short/fast feedback loop is nested within that mediated via the Keap1/Nrf2 transducer, we refer to it as the fast inner feedback loop (Fig. 1).
In addition to Nrf2-mediated transcriptional activation, the levels of certain antioxidant enzymes are also regulated post-transcriptionally in a redox-sensitive manner. Notably, the stability of both the GCLC and GCLM mRNAs is redox-regulated. Their half-lives can be doubled when cells are challenged with various prooxidants (Sekhar et al., 1997; Liu et al., 1998; Song et al., 2005). The mechanism for this redox-sensitive response is not well understood. Stabilization of GCLC mRNA induced in colon cancer cells seems to involve redox-sensing through mitogen-activated protein kinase (MAPK) p38 and accumulation of HuR, a mRNA-stabilizing factor (Song et al., 2005). Upregulation of protein expression as a result of mRNA stabilization is a slow process however - the time to reach a new steady state is in the order of hours or even longer, depending on the lengthened half-life values. Since redox-sensing through antioxidant mRNA stabilization bypasses the Keap1/Nrf2 transducer, structurally it is also a nested feedback loop. Due to its longer response time, we refer to it as the slow inner feedback loop (Fig. 1).
2.6. Feedforward control via xenosensors
The cellular toxicity of many xenobiotics does not reveal itself until they are metabolized into some reactive intermediates, mainly by the phase I enzymes (Isin and Guengerich, 2007). Many of these reactive species are electrophiles that can react with DNA and protein, leading to genetic mutation and cancer. About 75% of carcinogens are actually reactive metabolites of procarcinogens after bioactivation by xenobiotic-metabolizing enzymes (Nebert and Dalton, 2006). Many of these reactive metabolites can activate Nrf2 through conjugating the sensor molecule Keap1. Subsequent induction of phase II and antioxidant enzymes, such as GST, UGT, and GCL, promotes reactions that detoxify the reactive metabolites. This negative feedback-mediated cellular detoxification and homeostasis is in some cases complemented by a separate control pathway. A number of parent xenobiotics can be detected by a small set of xenosensors, which are nuclear receptors including aryl hydrocarbon receptor (AhR), constitutive androstane receptor (CAR), and pregnane X receptor (PXR). While activation of these xenosensors by the parent chemicals can induce the phase I enzymes to accelerate their bioactivation, they can also induce the phase II enzymes either directly or indirectly through transcriptionally upregulating Nrf2 (Paulson et al., 1990; Favreau and Pickett, 1991; Emi et al., 1996; Miao et al., 2005; Sugatani et al., 2005; Chen et al., 2007). In terms of controlling the intracellular levels of electrophilic metabolites, the induction of the phase II metabolizing enzymes by the xenosensors constitutes a form of feedforward control (Zhang et al., 2009). Activation of the controller and actuator through this feedforward control pathway (Fig. 1) is independent of the controlled variable, which, in this case, is the intracellular electrophilic species derived from bioactivation of xenobiotics.
3. Performance objectives, nature’s design principles and implementations
3.1. Performance objectives
To have a better understanding of how the redox control system, as described above, performs robustly against oxidative stresses, it is necessary to be explicit about the performance objectives of the system. The ultimate goal of antioxidant defense is to limit and protect cells from overt oxidative damages and adverse consequences. The cellular redox control system fulfills this goal by minimizing the magnitude and duration of the excursions that the intracellular ROS and associated redox state deviate from their basal steady-state levels. The generalized dynamic change in ROS levels in response to continuous sublethal oxidative stress is illustrated in Fig. 2A. At the onset of the oxidative stress, ROS rise quickly to a high level that is likely proportional to the dose of the oxidative stressor. Then the cell starts to adapt by upregulating the antioxidant enzymes, which gradually lower the elevated ROS. We refer to this initial rise-and-fall in ROS as the adapting state/period. Eventually ROS settle to a new steady state that is above the basal level but can still be tolerated by the cell chronically (herein referred to as the adapted steady state). Given this ROS dynamics, there are evidently two primary performance objectives expected from the cellular redox control system. (1) The transient rise in ROS levels during the adapting period should be controlled so that it is not too high and does not last too long. In other words, a fast adaptation is necessary. Otherwise, even if the redox control system is able to return ROS to a low adapted steady state at last, a large and prolonged upward excursion of ROS at the beginning may not be comfortably tolerated by the cell without adverse consequences. (2) When the oxidative stressor persists, it is desirable that the adapted steady state, although somewhat elevated, stays close to the basal steady-state level. If the steady-state deviation is too large, the cell, which may have survived the initial transient ROS rise, could still be chronically affected with impaired fitness and functional aberrations. Therefore, the second objective is equivalent to having an adapted steady-state ROS level that is relatively insensitive to the dose of the oxidative stressor (Fig. 2B). Robust redox homeostasis is concerned with meeting both objectives. The next section discusses the design principles for achieving these objectives and elaborates on how nature implements these principles.
Figure 2. Adaptive changes in intracellular ROS levels under oxidative stress.

(A) Typical dynamic fluctuation in the ROS level in response to persistent oxidative stress. In the initial adapting state/period, ROS first rise sharply at the onset of the stress then gradually decline towards the basal steady-state level. ROS then settle to a fully adapted steady state that is close to but still above the basal level. To reduce short and long term oxidative damage, the performance objectives of the redox control system are two-fold: (1) minimize the magnitude and duration of the initial rise in ROS level during the adapting period; (2) keep ROS levels in the adapted steady state as close to the basal level as possible. (B) The desirable dose response relationship between the adapted steady-state ROS level and dose of oxidative stressor. Dashed line represents the basal steady-state ROS level.
3.2. Ultrasensitive motifs and feedback loop gain
The negative feedback structure of the cellular redox control system allows the antioxidant capacity to increase in response to elevation in ROS levels. Intuitively, keeping the adapted steady-state ROS level low (i.e., the second objective) requires a high steady-state induction of the antioxidant capacity. In quantitative terms, this means that when the ROS level increases by a fixed percentage, that change needs to be amplified somehow to drive a much greater percentage increase in the overall antioxidant activity in the cell. This sensitivity amplification requires a type of nonlinear response that is referred to as ultrasensitivity in quantitative computational biology (Ferrell, 1996; Legewie et al., 2005). A biochemical process that can amplify signals in such a way is referred to as an ultrasensitive motif. The degree of ultrasensitivity is quantitatively evaluated by the ratio of the percentage change in the output signal to that in the input signal. By convention, the ratio is referred to as response coefficient or logarithmic gain (or gain for simplicity) given that the ratio of percentage changes is mathematically equivalent to the ratio of logarithmic changes (Savageau, 1976; Fell, 1992). When multiple ultrasensitive motifs are linked in tandem, the overall gain is the mathematic product of individual gains of all motifs. In the case where ultrasensitive motifs are linked sequentially to form a loop, the product of the motif gains is usually called loop gain.
To demonstrate the importance of ultrasensitive motifs in redox homeostasis, it is helpful to conceptually simplify the negative feedback-mediated redox control (Fig. 3A). According to our previous study on anti-stress cellular systems (Zhang and Andersen, 2007), the steady-state ROS level in relation to the dose of the oxidative stressor (S) can be approximately captured by the systems-level response coefficient RSROS (Fig. 3B, Equation 1 and 2). Rloop is the overall loop gain which is the sum of both the inner and outer feedback loop gains, and the latter comprises primarily the product of individual gains of the transducer, controller, actuator, and plant as they are linked sequentially. Since the value of RSROS is usually less than 1, a fixed percentage increase in the dose of the oxidative stressor results in a smaller percentage increase in the steady-state ROS level. This loop performance gives rise to a nonlinear dose response that has a downward-concave appearance (Fig. 3C), which is highly desirable for homeostatic purpose. Obviously, for ROS to be minimal, a high Rloop is desired, which would in turn require that the functional modules along the feedback loop process signals in a highly ultrasensitive manner. Within limits, the higher the gain conferred by each module, the more resistant the system is to oxidative perturbations. The mammalian redox control system seems to have evolved along this line of design principle by using various response motifs that can transfer signal ultrasensitively. The following section enumerates these ultrasensitive motifs.
Figure 3. Loop gain analysis and associated steady-state dose response curve for redox homeostatic control.
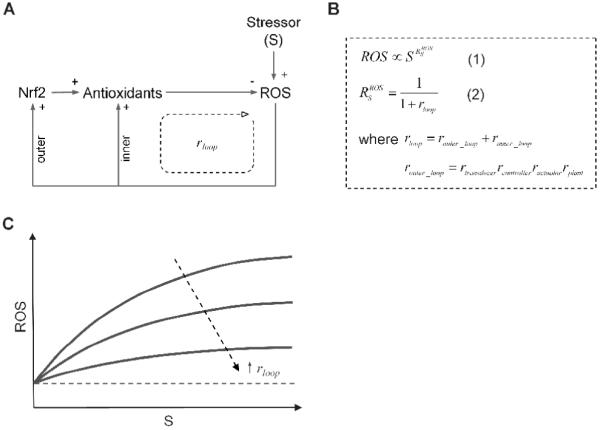
(A) Conceptually simplified redox control system. (B) Equation 1 indicates that the adapted steady-state ROS level is a nonlinear function of the stressor dose S - proportional to S raised to the power of RSROS. The latter is the systems-level response coefficient ROS in response to S, as expressed by Equation 2. rloop is the overall loop gain which is the sum of the outer (router_loop) and inner (rinner_loop) loop gains. The outer loop gain is the product of individual gains of the transducer, controller, actuator, and plant. (C) Since RSROS is usually less than 1, the dose response curve between the adapted steady-state ROS level and the dose of the stressor S has a curvature that is concave downward. The higher the overall loop gain, the less the ROS level increases with S. Dashed line represents the basal steady-state ROS level.
3.2.1. Nrf2 activation via stabilization
One mechanism of Nrf2 activation is through protein stabilization, as Keap1 loses its ability under oxidative stress to promote ubiquitination and degradation of Nrf2 (Hong et al., 2005; Kobayashi et al., 2006). Depending on the binding affinity between Nrf2 and Keap1 and their respective intracellular concentrations, the Keap1-mediated Nrf2 degradation may operate close to saturation, making the degradation essentially a near zero-order process. Under this circumstance, a small percentage decline in the concentration of the reduced form of Keap1 would result in a larger percentage increase in the steady-state concentration of Nrf2. Thus, the Keap1/Nrf2 transducer itself may be designed as an ultrasensitive module that not only detects but also amplifies cellular redox fluctuations. This potential ultrasensitive Nrf2 response is mechanistically analogous to zero-order ultrasensitivity proposed for reversible covalent modification of proteins, such as phosphorylation and dephosphorylation of signaling proteins by kinases and phosphatases (Goldbeter and Koshland, 1981). The exception is that Nrf2 degradation is an irreversible process without being counterbalanced. The mode of Nrf2 activation is very similar to that of hypoxia inducible factor 1α (HIF1α), which is stabilized via inhibition of proline hydroxylase in response to hypoxia. A highly ultrasensitive increase in HIF1α abundance via zero-order degradation has been proposed to explain the observed switch-like response to molecular oxygen (Jiang et al., 1996; Kohn et al., 2004).
3.2.2. Auto-regulation of Nrf2 and small Maf genes
In response to oxidative and electrophilic stresses, Nrf2 accumulates in the nucleus, where it associates with small Maf proteins to form a heterodimer in order to bind to AREs in the target genes (Katsuoka et al., 2005b). Among the target genes are Nrf2 and small Maf themselves. Both genes have been demonstrated to contain functional AREs in the promoter regions (Kwak et al., 2002; Katsuoka et al., 2005a), and in response to stresses, the mRNAs of both genes are upregulated (Kwak et al., 2002; Pi et al., 2003; Katsuoka et al., 2005b). Although the increases in the mRNA levels (2- to 3-fold) are not as robust as observed with antioxidant and phase II genes, the two positive feedback loops mediated by Nrf2 and small Maf gene auto-regulation are likely to serve as ultrasensitive response motifs to increase the transducer and control’s gains (Kholodenko, 2000; Legewie et al., 2005). Removal of the Nrf2 auto-regulation was shown to impair the steady-state homeostatic performance (Zhang and Andersen, 2007).
3.2.3. MAPK cascade: a conserved ultrasensitive motif
Extensive studies have demonstrated that protein phosphorylation is a potential mechanism for Nrf2 activation. A variety of cytosolic kinases, including protein kinase C, phosphatidylinositol 3-kinase, casein kinase 2, and MAPK, can modify Nrf2 and its activity posttranslationally (Huang et al., 2002; Kang et al., 2002; Xu et al., 2006; Pi et al., 2007a). It is particularly interesting to note that oxidative stresses can activate MAPK such as p38 and JNK (Yu et al., 2000; Zipper and Mulcahy, 2000; Xu et al., 2006; Yuan et al., 2006). The role of the MAPK cascade as an ultrasensitive motif has been well established in a variety of cell types (Huang and Ferrell, 1996; Bagowski et al., 2003). By phosphorylating Nrf2 and other regulatory proteins, MAPK may mediate ultrasensitive activation of transcription factors involved in cellular antioxidant defense (Iles et al., 2002; Xu et al., 2006; Harada et al., 2008). The ultrasensitive role played by the MAPK cascade has been demonstrated in numerous signaling processes, such as Xenopus oocyte maturation and sustained MAPK activation in response to growth factors in fibroblasts (Ferrell and Machleder, 1998; Bhalla et al., 2002).
Ultrasensitivity in the MAPK cascade originates from three independent sources: (1) multi-step signaling, (2) zero-order ultrasensitivity, and (3) cascading arrangement (Huang and Ferrell, 1996). It was recognized that the MAPK kinase (MKK) and MAPK each contain two of either serine, threonine, or tyrosine residues that can be phosphorylated by the upstream kinases. Phosphorylation of the two sites involves two separate collisions between the enzyme and substrate molecules (Ferrell and Bhatt, 1997). This non-processive mechanism of dual phosphorylation for MKK and MAPK activation provides a basis for multi-step signaling and thus some degree of ultrasensitivity. Secondly, phosphorylation and dephosphorylation of MKK and MAPK by respective kinases and phosphatases is a typical covalent modification cycle. Some of these enzymes in the cascade may work under conditions that are close to saturation, providing the possibility for zero-order ultrasensitivity (Goldbeter and Koshland, 1981). The third source of ultrasensitivity lies in the fact that the MAPK cascade has a layered signaling structure that is conserved throughout evolution. This structure allows the output of one layer to be fed as an input into the next layer; therefore the gain of each layer can be multiplied as the signal propagates down the cascade. As a result of these ultrasensitive mechanisms, the overall stimulus-response property of the MAPK cascade is highly sigmoid and may actually be a switch (Bagowski et al., 2003). It is likely that the activation of the MAPK cascade under oxidative stress may contribute to an ultrasensitive activation of Nrf2, which in turn is responsible for ultrasensitive inductions of antioxidant and phase II enzymes.
3.2.4. Homo-multimerization of antioxidant enzymes
In cell signaling networks, an important ultrasensitive response motif is protein homo-multimerization (or oligomerization), which includes homo-dimerization, homo-trimerization, and homo-tetramerization, and so on. Protein homo-multimerization is usually a reversible process involving association of monomers into multimers and dissociation of multimers into monomers. According to the kinetic law of mass action, the rate of monomer association is a nonlinear function, proportional to the concentration of the monomer raised to the power of the multimeric order (i.e., the power is 2 for dimer, 3 for trimer, and so on). Then the steady-state concentration of the multimer will increase in proportion to the concentration of the monomer raised to the multimeric order, constituting a classical ultrasensitive response. Ultrasensitivity via protein homo-multimerization is widely utilized in cellular systems for signal amplification. For example, in response to heat stress, three monomers of heat shock factor 1 (HSF1) form a homo-trimer, which then induces robust expression of chaperone proteins (Liu and Thiele, 1999). In the cellular redox control system, the fully active forms of most antioxidant and phase II enzymes are homo-multimers (Table 1). For instance, GR, Grx, Prx, and GST are homo-dimers (Carlberg and Mannervik, 1975; Evrard et al., 2004; Vargo et al., 2004; Lillig et al., 2005). It is particularly interesting to note that GPx and CAT, the two key enzymes responsible for H2O2 detoxification, are homo-tetramers (Kirkman and Gaetani, 1984; Asayama et al., 1994). G6PD, the enzyme involved in NADPH generation through the PPP pathway, is also a homo-tetramer (Lee et al., 1979). Some isoforms of bacterial CAT even exist as homo-hexamers (Loewen and Switala, 1986; Loewen and Switala, 1987). The functional implications of the homo-multimerization process for metabolic enzymes have long escaped satisfying explanations. It has been argued that multimerization might be required for enzymes to achieve full activity, or to become more stable. Here in the context of cellular redox control, we propose that homo-multimerization of antioxidant enzymes is likely to act as crucially important ultrasensitive motifs. With such a motif, a fractional increase in the transcription rate of an antioxidant gene would result in a much greater fractional increase in the abundance of the fully-active, multimeric antioxidant enzyme. Thus homo-multimerization is one means by which the cellular antioxidant capacity is amplified in response to oxidative stress, contributing to robust redox homeostasis (Zhang and Andersen, 2007).
Table 1. Homo-multimeric Antioxidant and Phase II enzymes.
| Enzyme | Multimeric order | References |
|---|---|---|
| CAT | Tetramer | (Kirkman and Gaetani, 1984) |
| G6PD | Tetramer | (Lee et al., 1979) |
| GPx | Tetramer | (Asayama et al., 1994) |
| GR | Dimer | (Carlberg and Mannervik, 1975) |
| Grx | Dimer | (Lillig et al., 2005) |
| GS | Dimer | (Huang et al., 1995) |
| GST | Dimer | (Vargo et al., 2004) |
| NQO1 | Dimer | (Faig et al., 2000) |
| Prx | Dimer | (Evrard et al., 2004) |
| SOD | Dimer (SOD1) | (Lindberg et al., 2004) |
| Tetramer (SOD2) | (Matsuda et al., 1990) | |
| Tetramer (SOD3) | (Antonyuk et al., 2009) | |
| SULT | Dimer | (Pedersen et al., 2000) |
| Trx | Dimer | (Weichsel et al., 1996) |
| TrxR | Dimer | (Oblong et al., 1993) |
| UGT | Dimer | (Meech and Mackenzie, 1997) |
3.2.5. Ultrasensitivity via multi-step signaling
An important way to achieve ultrasensitive response is via multi-step signaling, in which a signaling molecule controls a target molecule in more than one way, and the controls are synergistic rather than additive. For instance, if a signal X not only increases the transcription of protein Y, but also decreases the degradation of Y, then a fractional increase in X would lead to a fractional increase in Y that is twice as much, corresponding to a gain of 2. In the redox homeostatic network, multi-step signaling is utilized extensively at various levels to achieve a high loop gain.
As noted above, oxidative stress not only increases the total abundance of Nrf2 via stabilization, but also increases its nuclear retention by inhibiting one of the NES domains (Li et al., 2006). As a result of this coherent dual-regulation, the amount of Nrf2 that eventually accumulates in the nucleus is expected to be a highly nonlinear function of the intracellular ROS level. Another example of multi-step signaling is the induction of the holoenzyme GCL. Both of its two subunits, GCLC and GCLM, can be upregulated simultaneously by Nrf2, which then heterodimerize to form the fully active holoenzyme (Dickinson et al., 2004). Convergence of the two separate induction events at GCL confers ultrasensitivity to the induction of the holoenzyme by Nrf2. At a higher architectural level of the redox control network, the inner feedback loop itself is also an ultrasensitive pathway containing multi-step signaling. It comprises at least two converging paths: redox-sensitive regulation of antioxidant enzyme activities (fast loop) and redox-sensitive regulation of antioxidant mRNA stability (slow loop). More globally, the inner and outer feedback loops also work synergistically to generate ultrasensitivity, as they separately regulate the transcription, degradation, and activity of the antioxidant enzymes, which are independent signaling steps that would impact the total antioxidant capacity in a multiplicative manner.
Another location in the feedback loop where ultrasensitivity may arise is the reversible binding between the Nrf2-Maf heterodimer and the promoters of antioxidant genes. Ultrasensitivity arises if a promoter contains multiple ARE copies clustered in close proximity and Nrf2-Maf heterodimers bind to these AREs cooperatively. With positive cooperative binding, the occupancy of one ARE by an Nrf2-Maf molecule facilitates the binding of neighboring AREs by other Nrf2-Maf molecules, and so on. Amplification of gene induction through cooperative transcription factor binding at gene promoters has been demonstrated in the heat shock response, wherein HSF1 transcriptionally induces heat shock proteins (Erkine et al., 1999). It is likely that certain antioxidant genes such as HO-1 and NQO1, which are often induced by hundreds and sometimes even thousands of folds, may involve cooperative Nrf2-Maf dimer binding to their promoters. Taken together, multiple ultrasensitive motifs distributed along the feedback loops contribute to a high overall loop gain. And by employing these multiple ultrasensitive motifs, cells are able to accomplish the second performance objective, i.e., keeping the adapted steady-state ROS as close as possible to the basal level.
In theory, a similar loop gain can be achieved by replacing the multiple ultrasensitive steps described above with one or two highly ultrasensitive motif(s). Concentrating the loop gain in one or two ultrasensitive motifs, however, may make the redox control system extremely vulnerable to functional failures of these few motifs, jeopardizing redox homeostasis. By using multiple ultrasensitive motifs, which appear to be “redundant” at first glance, the redox control system is better designed against internal component failures. A high loop gain can be achieved even when each motif is only fractionally ultrasensitive. In this way a failing motif in terms of its ultrasensitive function will only reduce the loop gain by a small amount, limiting its impact on the homeostatic performance of the system.
3.3. Fast inner feedback loop
Under persistent oxidative stress, intracellular ROS first rise then fall before settling to an adapted steady-state level (Fig. 2A). While keeping the adapted steady-state ROS level low is one objective of the redox control system, another important performance objective is to minimize the initial upward excursion of ROS so that it does not cause irreversible adverse consequences. To have a fast adaptation, it requires that the antioxidant capacity be upregulated also at a fast pace. However, the majority of the adaptive increase in the antioxidant capacity is driven by Nrf2-mediated transcriptional upregulation, which is a slow process. The time for antioxidant enzymes to reach new steady states is usually in the order of hours or even days, depending largely on the half-lives of antioxidant mRNAs and proteins (Glass and Doyle, 1972; Geisler et al., 1978; Knight and Sunde, 1987; Liu et al., 1998; Chen et al., 2005). This slow response indicates that the Nrf2-mediated antioxidant upregulation is unable to launch a fast adaptation to limit the initial rise in ROS. In contrast, the redox-sensitive antioxidant and related enzymes, such as GCL and GAPDH, which constitute the fast feedback loop that bypasses the Nrf2-mediated outer loop (Fig. 1), can upregulate the antioxidant capacity quickly, albeit to a limited extent. The redox-sensitive changes in GCL activity, heterodimerization, and GAPDH-mediated re-routing of glucose metabolism to the PPP do not involve transcriptional and translational events and can occur almost instantaneously within seconds to minutes. This extremely fast feedback response would limit the initial sharp rise in ROS at the onset of the oxidative stress, reducing potential cellular damage before the Nrf2-mediated transcriptional mechanism is fully activated. Moreover, the fast feedback loop also seems better positioned to cope with transient oxidative stresses. A transient oxidative stress may be too short to engage the transcriptionally-mediated antioxidant upregulation, yet it may cause a large, albeit short-lived, spike in ROS levels. The increase may be limited through the instantaneous activation of the fast feedback loop mediated by the redox-sensitive antioxidant enzymes. Moreover, the role of the fast feedback loop is not restricted to handling minute-to-minute changes in the redox state; it also contributes to the overall loop gain of the entire feedback system as part of the inner loop (Fig. 3B). In the absence of the fast feedback loop, the system would not only be less capable of dealing with quick rises in ROS, but also - as a result of lowered overall loop gain - has an impaired control over the the adapted steady-state ROS level (Fig. 4). Lastly, the differential response times of the fast and slow feedback loops may increase the stability of the redox control network by reducing the probability of oscillation, which is an issue intrinsic to negative feedback systems with high loop gains (Kholodenko, 2000).
Figure 4.
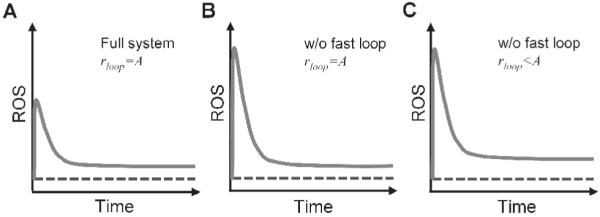
Illustration of the dual role of the fast inner feedback loop as mediated by redox-sensitive antioxidant and related enzymes. (A) The fast loop (1) limits the initial ROS rise at the onset of the oxidative stress, and (2) helps to keep the adapted steady-state ROS at low levels by contributing to the overall loop gain (rloop). (B) Disabling the fast loop and increasing the slow loop gain to keep the same overall loop gain will result in a higher initial ROS rise but same adapted steady-state ROS level as the full system. (C) Disabling the fast loop without compensating for the loss in the overall loop gain will result in a higher initial ROS rise and a higher adapted steady-state ROS level. Dashed line represents the basal steady-state ROS level.
In summary, nature has designed the redox control system to meet specific performance goals. By using a combination of fast and slow feedback loops and multiple ultrasensitive motifs, the control system is not only able to keep the adapted steady-state ROS close to the basal level, but also limit the initial sharp rise in ROS shortly after the onset of oxidative stresses.
3.4. Feedforward control and hormesis
As noted earlier, as far as controlling the intracellular levels of electrophilic metabolites is concerned, the induction of the phase II metabolizing enzymes by the xenosensors constitutes a form of feedforward control (Zhang et al., 2009). The emergence of this xenosensor-mediated feedforward control mechanism provides additional evolutionary advantage for cellular fitness and survival. If a biological organism frequently encounters a xenobiotic compound that, after phase I bioactivation, becomes a harmful reactive species, it would be highly beneficial when the parent chemical can directly engage a detoxification mechanism to reduce the level of its toxic metabolite. With feedforward control, a nearly perfect adaptation may be achieved if the feedforward gain, which is contributed to by ultrasensitive motifs in the xenosensor, controller and actuator, matches closely the bioactivation gain. In this way this control motif may generate a threshold response (Zhang et al., 2009). Moreover, if the ultrasensitive motifs located in the feedforward loop are able to produce a greater feedforward gain than the bioactivation gain, a J-shaped change in the level of the electrophilic metabolite may result with respect to the dose of the parent chemical (Zhang et al., 2009). Such a hormetic change in the level of the intracellular electrophilic species may underpin a similar downstream endpoint response such as mutagenesis and carcinogenesis as seen with many carcinogenic chemicals (Camurri et al., 1983; Kitano et al., 1998; Masuda et al., 2001; Puatanachokchai et al., 2006).
4. ROS signaling and redox homeostasis
Despite their well-known deleterious effects on biomacromolecules, ROS, particularly H2O2, also function as physiological signaling molecules (Finkel, 2003; Buetler et al., 2004). Like nitric oxide, the fast turnover and high diffusibility of H2O2 make it a good candidate as a second messenger (Rhee et al., 2003; Rhee, 2006). The signaling role of ROS has been demonstrated in a variety of physiological processes, including glucose-stimulated insulin secretion in β-cells, glucose sensing in the hypothalamus, and growth factor-induced differentiation, etc (Denu and Tanner, 1998; Suzukawa et al., 2000; Goldstein et al., 2005; Leloup et al., 2006; Pi et al., 2007b; Leloup et al., 2009).
Employing ROS as signaling molecules implies that the magnitude of the ROS signal is likely to be inversely correlated to the antioxidant capacity in the cell. In this regard, it is interesting to note that β-cells, which are believed to use glucose-induced ROS to trigger insulin secretion (Pi et al., 2007b; Leloup et al., 2009), have very low levels of antioxidant enzymes compared with other cell types (Tiedge et al., 1997). This low antioxidant capacity may provide an intracellular redox environment that is conducive to robust ROS signaling. Conversely if the intracellular antioxidant capacity becomes elevated, signaling ROS are expected to be attenuated. Such attenuation effects occur with epidermal growth factor (EGF)-induced AKT activation, a process mediated by H2O2 generated following EGF binding to its membrane receptors. When cells were overexpressed with GPx1 and CAT, the two primary H2O2-degrading antioxidants, activation of AKT is significantly reduced (Handy et al., 2009). The generally inverse relationship between ROS signaling and antioxidant capacity has important implications for the mechanism by which oxidative stresses impairs cellular functions. When chronically under oxidative stress, cells may have their antioxidant enzymes upregulated through the Nrf2-mediated transcriptional induction. The adaptive augmentation of antioxidant capacity is usually widespread, involving various isoforms of antioxidant enzymes located throughout the cytosol and major ROS-producing organelles. Indiscriminately, these high-level antioxidants would not only work to degrade the ROS molecules generated as metabolic byproducts, but as a side effect, may also dampen the nearby ROS generated as physiological signals.
The pathological consequences of oxidative and electrophilic stresses have long been proposed to result from direct impairment of the molecular components of the cell by toxic reactive species. Except for certain occupational exposure scenarios, oxidative stresses imposed on the general population by environmental factors are more likely to be minimal to mild. After compensated by the redox homeostatic control network, oxidative stresses at low or even moderate levels may not produce significant oxidative damages that have pathological consequences. Instead, we propose the following mode of action to explain some pathological conditions rendered by environmentally-derived oxidative stresses. When chronically under mild to moderate oxidative stress, most cells are able to adequately adapt by engaging the redox homeostatic control system. Robust upregulation of the antioxidant enzymes maintains ROS at low adapted levels, keeping oxidative damage-associated cell malfunctioning at a minimum. However, persistently high antioxidant content in the cell would attenuate the signaling ROS generated for a variety of physiological responses (compare Fig. 5A and 5B). Thus by tampering with ROS signaling, mild to moderate oxidative stresses may disrupt a number of ROS-mediated cellular functions. This unconventional mode of action appears to underlie the suppression by oxidative stress of glucose-stimulated insulin secretion in β-cells, contributing to the progression of type 2 diabetes (Pi et al., 2007b). For a review on this topic, see the article by Pi, et al. in the same issue.
Figure 5.
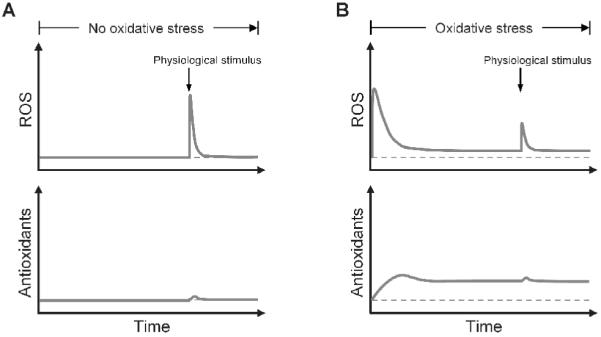
Illustration that chronic oxidative stress attenuates ROS signaling by adaptively upregulating antioxidant enzymes. (A) When there is no oxidative stress, signaling ROS generated by physiological stimulus is transient and high, which do not significantly upregulate the antioxidant enzymes. (B) Under chronic oxidative stress, antioxidant enzymes are upregulated to high levels, which attenuate signaling ROS generated by physiological stimulus. Note that the oxidative stress can also generate ROS as signals at the onset, but the rise in ROS is temporary as adaptation occurs after antioxidant enzymes are upregulated. Dashed lines represent the basal steady-state ROS level and antioxidant level.
For this unconventional mode of action of oxidative stress to gain acceptance, a couple of confounding issues need to be clarified. One question is whether endogenously generated signaling ROS would reach a magnitude and spatial extent that constitute an oxidative stress to the cell such that the Nrf2-mediated antioxidant response is triggered. The other question is whether the ROS generated by oxidative stressors can also serve as signals to trigger physiological responses normally mediated by endogenously generated ROS. Answers to these questions are likely to hinge on several important aspects of ROS signaling that have not been well understood, including concentration, duration, and subcellular location of physiological ROS signals. The predominant source of signaling ROS identified so far seems to come from NOX located on the plasma membrane (Fisher, 2008; Ushio-Fukai, 2008). NOX can be activated by membrane events such as growth factor binding to cognate receptors and glucose uptake by transporters (Sturrock et al., 2006; Morgan et al., 2009). Activated NOX first generates O2-· on the extracellular side of the plasma membrane. O2-· is then converted to H2O2 which diffuses through aqueous channels into the cell to continue the signal transduction process. Because the cytosol is filled with potent H2O2-degrading enzymes such as GPx and Prx, H2O2 is not expected to diffuse very far to the interior of the cell before its concentration drops precipitously (Forman, 2007). Therefore a steep H2O2 concentration gradient may exist in the cytosolic space near the plasma membrane where signaling H2O2 first appears (Forman, 2007; Fisher, 2008). It is perhaps within this highly compartmentalized space which contains highly concentrated H2O2 that downstream ROS signaling occurs. A primary target of signaling H2O2 is protein tyrosine phosphatase (PTP) (Denu and Tanner, 1998). By inhibiting the phosphatase activity of PTP, H2O2 increases the phosphorylation level of target proteins further downstream. The averaged physiological concentration of H2O2 in the cell has been estimated to fluctuate in the low nanomolar to sub-micromolar range, whereas activation/inactivation of target effector proteins such as PTP requires H2O2 at micromolar concentrations (Stone, 2004). This concentration mismatch suggests that NOX-generated H2O2 may have to reach micromolar concentrations in the subcellular space close to the plasma membrane to effectively inactivate PTP molecules. This requirement is consistent with a number of experimental observations showing that micromolar exogenous H2O2 is usually needed to generate significant cellular signaling responses (Meng et al., 2002; Salmeen et al., 2003; Pi et al., 2007b). With respect to the duration of ROS signaling, experimental evidence indicates that the increase in ROS generated endogenously as messenger molecules are usually short-lived (Bae et al., 1997; Bae et al., 2000; Suzukawa et al., 2000). In summary, the endogenously generated ROS functioning as second messengers, such as those produced by NOX, are likely to be highly localized, concentrated, and transient.
If the second messenger ROS is indeed highly localized and transient, they are not expected to activate the Nrf2-mediated redox control system. To induce antioxidant enzymes, the redox sensor Keap1 has to be in contact with elevated ROS to become modified at cysteine residues. Since Keap1 molecules are largely immobilized to the cytoskeletal actin filaments in the cytosol, and NOX-generated signaling ROS is highly compartmentalized to the immediate vicinity of the plasma membrane, physical contacts between Keap1 and the signaling ROS is likely to be rather limited. Moreover, transiently elevated signaling ROS would not provide enough time to allow Nrf2-mediated transcriptional induction of antioxidant enzymes to reach significant levels. Based on these considerations, it is unlikely that signaling ROS constitute an oxidative stress that can trigger the Nrf2-mediated antioxidant upregulation in a tangible manner, which may attenuate subsequent ROS-mediated signaling.
Upon exposure to exogenous oxidative stressors, the intracellular ROS first rise then fall. Depending on the dose of the stressor, the initial rise in ROS levels may be high enough to effectively activate/inactive redox-sensitive protein molecules that are normally targeted by physiological signaling ROS. Such effects, mimicking physiological responses, have been clearly demonstrated in cell cultures to which oxidative agents such as H2O2 were added (Maechler et al., 1999; Pi et al., 2007b). However, if the oxidative stressor is present continuously, the adaptive upregulation of antioxidant enzymes through the Keap1/Nrf2 transducer will likely cut the stress-induced ROS short and keep them down at low adapted levels (Fig. 5B). Therefore, depending on the dose of the stressor, persistent oxidative stress may generate transient ROS to mimic signaling effects.
5. Antioxidant supplements revisited
Decades of research emphasizing the detrimental aspect of ROS in biological organisms has led to the premise that by eliminating reactive species, generated either endogenously or exogenously by environmental pollutants, antioxidant supplements are universally beneficial to human health. This consensus has led to a widespread consumption of antioxidant supplements as part of people’s daily diet. Most of these supplements are originally derived from vegetables, fruits, and other food sources. Among them, vitamin C, E, and β-carotene have been popular choices for their effectiveness to directly scavenge ROS, terminating the chain reactions that lead to oxidative damage. In contrast to these direct antioxidants, a number of plant-derived antioxidant compounds are actually weak prooxidants, which include resveratrol from grape seeds, isothiocyanate from cruciferous vegetables, catechins from green tea, and curcumin from turmeric (Keum et al., 2004; Surh et al., 2005; Son et al., 2008). The antioxidant effect of these phytochemicals is indirect. Acting as weak prooxidants, they have limited ability to cause oxidative damage, yet they are potent enough to activate the Nrf2-mediated adaptive response (Surh, 2003; Surh et al., 2005; Kohle and Bock, 2006). The enhanced expression of endogenous ROS-scavenging and electrophile-detoxifying enzymes is believed to protect cells from potential DNA and protein damage caused by oxidative and electrophilic stresses. For this reason these phytochemicals have been widely taken as chemopreventive agents to retard carcinogenesis initiated from environmentally-induced genotoxicity. Numerous population-based studies show consumption of fruits and vegetables containing these phytochemicals can reduce the risk of developing cancer by up to 50% (Surh, 2003).
Despite the widely self-prescribed ingestion of antioxidant supplements in the general human population, a number of well-controlled clinical studies aiming to evaluate the health effects of these antioxidant compounds have so far produced inconclusive results; in some cases, they even increased incidence for certain pathological conditions, such as cancer and diabetes, or all-cause death (Wiernsperger, 2003; Bjelakovic et al., 2007; Ristow et al., 2009). In retrospect, the biological effects of dietary antioxidants may need to be evaluated more comprehensively from a systems perspective. Although intuitively sound, the idea to artificially raise our antioxidant capacity to ward off potential oxidative damages may be naïve when the concept is re-examined in the context of Nrf2-mediated redox homeostasis. The feedback nature of the redox control network has to be taken into consideration. An oversupply of direct antioxidants, such as vitamin C and E, can indeed lower ROS and free radical levels in cells and extracellular fluids. However, this would result in a more reducing intracellular environment, which keeps more Keap1 molecules in the reduced configuration. With less oxidized Keap1 molecules around, ubiquitination and degradation of Nrf2 increases, leading to a lower basal steady-state Nrf2 level. A decline in Nrf2 would result in lower basal levels of endogenous antioxidant and phase II enzymes. In addition, a more reducing intracellular environment will also cause a decrease in the activities of redox-sensitive antioxidant enzymes such as GCL. Therefore, daily ingestion of direct antioxidants may lead to a chronically diminished endogenous antioxidant capacity and phase II enzyme expression. With less GSH and phase II detoxification enzymes, detoxification of electrophilic metabolites of certain environmental procarcinogens will be compromised, with attendant increases in genotoxicity and carcinogenesis. This may explain why many randomized human studies with vitamin C, E, and β-carotene did not observe unambiguous improvement in cancer reduction by these direct antioxidants, and in some cases there were increased cancer incidence and associated mortality (Bjelakovic et al., 2004; Bairati et al., 2006; Hercberg et al., 2007; Omenn, 2007; Plummer et al., 2007; Lin et al., 2009). This potential side effect of direct antioxidants is consistent with the concept of hormesis, which would argue that conversely, taking low-dose prooxidants should be beneficial (Calabrese, 2001). Low-dose prooxidants such as isothiocyanate and resveratrol may activate the Nrf2 antioxidant system, providing protection against existing or future high-level stresses.
Clearly, the overall cellular antioxidant capacity can be generally enhanced by exogenous supply of either direct antioxidants such as vitamin C and E, or prooxidant type of antioxidants which, by activating Nrf2, augment the endogenous antioxidant content. While the enhanced antioxidant capacity is generally good for preventing and limiting oxidative damages and related pathological consequences, it could become a damper to physiological responses mediated by signaling ROS, such as glucose-stimulated insulin secretion (Pi et al., 2007b). Therefore, whether antioxidant supplements are beneficial or detrimental may well depend on the health endpoint evaluated. It is likely that the basal intracellular redox environment and antioxidant capacity is already at a tradeoff optimally tuned through evolution, which on one hand provides adequate protection against the damaging effect of ROS, and on the other hand is also conducive to cell type-specific ROS signaling. From time to time cells may have to juggle between these two competing tasks and in some circumstances favor one over the other. Artificial interventions aiming to alter the redox balance one way or the other seem unlikely to be precise enough to produce overall beneficial consequences. Thus, antioxidant supplements are unlikely to be a one-size-fits-all panacea as we hoped that would categorically prevent, eradicate, or moderate many oxidative stress-related pathological conditions without adverse consequences. In the absence of more detailed understanding of redox homeostasis, ROS signaling, and the relationship between them, it may be unwise for healthy people to tamper with their antioxidant system through excessive intake of dietary antioxidants. To protect populations from oxidative stresses exerted by environmental factors, shifting our efforts from indiscriminate antioxidant consumption to identifying the oxidative sources and avoiding future exposures may be a better option for now. More personalized and targeted therapeutic approaches may be available in the future as we gain a more complete quantitative understanding of the biochemical networks underlying redox homeostasis and ROS signaling. Choosing the therapeutic parameters, such as antioxidant/prooxidant type, dose, timing, and frequency, may one day require computer simulations of the underlying biological system in individual patients and for specific endpoints.
6. Final remarks
The details of the molecular circuitry underlying redox homeostatic control demonstrate a robust, well-designed feedback and feedforward structure. To understand the quantitative behaviors of such a system, intuitive reasoning is inadequate and more quantitative tools are necessary. Ideas borrowed from engineers who design and make man-made control devices play an increasingly important role in reverse-engineering and understanding complicated biochemical networks. Using an engineering-oriented approach, we have re-examined the redox homeostatic control system from a systems biology perspective. This approach elucidates nature’s design principles for robust redox homeostasis and how these principles are implemented at the molecular level. The systems biology of redox homeostasis has also prompted us to rethink the pathological modes of action for oxidative stressors and to re-evaluate the popular practice of including antioxidant supplements in our diet and its promised benefits. The health effects of antioxidants need to be evaluated more comprehensively without focusing solely on a subset of problems. As we gain further understanding of the cellular redox control system and its interactions with ROS signaling, the systems biology approach begun here will allow us to move closer to multi-target, multi-stage therapeutic interventions that may reverse many pathological conditions stemming from cellular redox disruptions.
Acknowledgments
This work was supported by funds from NIEHS-ONES-R01ES016005, NIEHS-SBRP-P42ES04911, and the Long-Range Research Initiative of the American Chemistry Council.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors have declared that there are no conflicts of interest.
References
- Antonyuk SV, Strange RW, Marklund SL, Hasnain SS. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 A resolution: insights into heparin and collagen binding. J Mol Biol. 2009;388:310–326. doi: 10.1016/j.jmb.2009.03.026. [DOI] [PubMed] [Google Scholar]
- Asayama K, Yokota S, Dobashi K, Hayashibe H, Kawaoi A, Nakazawa S. Purification and immunoelectron microscopic localization of cellular glutathione peroxidase in rat hepatocytes: quantitative analysis by postembedding method. Histochemistry. 1994;102:213–219. doi: 10.1007/BF00268898. [DOI] [PubMed] [Google Scholar]
- Åström KJ, Murray RM. Feedback Systems: An Introduction for Scientists and Engineers. Princeton University Press; Princeton, NJ: 2008. [Google Scholar]
- Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- Bae YS, Sung JY, Kim OS, Kim YJ, Hur KC, Kazlauskas A, Rhee SG. Platelet-derived growth factor-induced H(2)O(2) production requires the activation of phosphatidylinositol 3-kinase. J Biol Chem. 2000;275:10527–10531. doi: 10.1074/jbc.275.14.10527. [DOI] [PubMed] [Google Scholar]
- Bagowski CP, Besser J, Frey CR, Ferrell JE., Jr. The JNK cascade as a biochemical switch in mammalian cells: ultrasensitive and all-or-none responses. Curr Biol. 2003;13:315–320. doi: 10.1016/s0960-9822(03)00083-6. [DOI] [PubMed] [Google Scholar]
- Bairati I, Meyer F, Jobin E, Gelinas M, Fortin A, Nabid A, Brochet F, Tetu B. Antioxidant vitamins supplementation and mortality: a randomized trial in head and neck cancer patients. Int J Cancer. 2006;119:2221–2224. doi: 10.1002/ijc.22042. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science. 2002;297:1018–1023. doi: 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Jama. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: a systematic review and meta-analysis. Lancet. 2004;364:1219–1228. doi: 10.1016/S0140-6736(04)17138-9. [DOI] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- Brodie AE, Reed DJ. Cellular recovery of glyceraldehyde-3-phosphate dehydrogenase activity and thiol status after exposure to hydroperoxides. Arch Biochem Biophys. 1990;276:212–218. doi: 10.1016/0003-9861(90)90028-w. [DOI] [PubMed] [Google Scholar]
- Buetler TM, Krauskopf A, Ruegg UT. Role of superoxide as a signaling molecule. News Physiol Sci. 2004;19:120–123. doi: 10.1152/nips.01514.2003. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ. Overcompensation stimulation: a mechanism for hormetic effects. Crit Rev Toxicol. 2001;31:425–470. doi: 10.1080/20014091111749. [DOI] [PubMed] [Google Scholar]
- Camurri L, Codeluppi S, Pedroni C, Scarduelli L. Chromosomal aberrations and sister-chromatid exchanges in workers exposed to styrene. Mutat Res. 1983;119:361–369. doi: 10.1016/0165-7992(83)90186-0. [DOI] [PubMed] [Google Scholar]
- Carlberg I, Mannervik B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J Biol Chem. 1975;250:5475–5480. [PubMed] [Google Scholar]
- Chen X, Zhang J, Baker SM, Chen G. Human constitutive androstane receptor mediated methotrexate induction of human dehydroepiandrosterone sulfotransferase (hSULT2A1) Toxicology. 2007;231:224–233. doi: 10.1016/j.tox.2006.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: Dependence on ATPand modifier subunit for regulation of tissue glutathione levels. J Biol Chem. 2005 doi: 10.1074/jbc.M504604200. [DOI] [PubMed] [Google Scholar]
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med. 2004;37:1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Samad H, Kurata H, Doyle JC, Gross CA, Khammash M. Surviving heat shock: control strategies for robustness and performance. Proc Natl Acad Sci U S A. 2005;102:2736–2741. doi: 10.1073/pnas.0403510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emi Y, Ikushiro S, Iyanagi T. Xenobiotic responsive element-mediated transcriptional activation in the UDP-glucuronosyltransferase family 1 gene complex. J Biol Chem. 1996;271:3952–3958. doi: 10.1074/jbc.271.7.3952. [DOI] [PubMed] [Google Scholar]
- Erkine AM, Magrogan SF, Sekinger EA, Gross DS. Cooperative binding of heat shock factor to the yeast HSP82 promoter in vivo and in vitro. Mol Cell Biol. 1999;19:1627–1639. doi: 10.1128/mcb.19.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrard C, Capron A, Marchand C, Clippe A, Wattiez R, Soumillion P, Knoops B, Declercq JP. Crystal structure of a dimeric oxidized form of human peroxiredoxin 5. J Mol Biol. 2004;337:1079–1090. doi: 10.1016/j.jmb.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Faig M, Bianchet MA, Talalay P, Chen S, Winski S, Ross D, Amzel LM. Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: species comparison and structural changes with substrate binding and release. Proc Natl Acad Sci U S A. 2000;97:3177–3182. doi: 10.1073/pnas.050585797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favreau LV, Pickett CB. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J Biol Chem. 1991;266:4556–4561. [PubMed] [Google Scholar]
- Fell DA. Metabolic control analysis: a survey of its theoretical and experimental development. Biochem J. 1992;286(Pt 2):313–330. doi: 10.1042/bj2860313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE., Jr. Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21:460–466. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Jr., Bhatt RR. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J Biol Chem. 1997;272:19008–19016. doi: 10.1074/jbc.272.30.19008. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Jr., Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Fisher AB. Redox Signaling Across Cell Membranes. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med. 2007;42:926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- Fraser JA, Kansagra P, Kotecki C, Saunders RD, McLellan LI. The modifier subunit of Drosophila glutamate-cysteine ligase regulates catalytic activity by covalent and noncovalent interactions and influences glutathione homeostasis in vivo. J Biol Chem. 2003;278:46369–46377. doi: 10.1074/jbc.M308035200. [DOI] [PubMed] [Google Scholar]
- Fraser JA, Saunders RD, McLellan LI. Drosophila melanogaster glutamate-cysteine ligase activity is regulated by a modifier subunit with a mechanism of action similar to that of the mammalian form. J Biol Chem. 2002;277:1158–1165. doi: 10.1074/jbc.M106683200. [DOI] [PubMed] [Google Scholar]
- Geisler RW, Roggeveen AE, Hansen RJ. The effects of insulin on the turnover of glucose-6-phosphate dehydrogenase in epididymal adipose tissue of the rat. Biochim Biophys Acta. 1978;544:284–293. doi: 10.1016/0304-4165(78)90097-1. [DOI] [PubMed] [Google Scholar]
- Glass RD, Doyle D. On the measurement of protein turnover in animal cells. J Biol Chem. 1972;247:5234–5242. [PubMed] [Google Scholar]
- Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldbeter A, Koshland DE., Jr. An amplified sensitivity arising from covalent modification in biological systems. Proc Natl Acad Sci U S A. 1981;78:6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy DE, Lubos E, Yang Y, Galbraith JD, Kelly N, Zhang YY, Leopold JA, Loscalzo J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J Biol Chem. 2009;284:11913–11921. doi: 10.1074/jbc.M900392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Sugimoto R, Watanabe A, Taketani S, Okada K, Warabi E, Siow R, Itoh K, Yamamoto M, Ishii T. Differential roles for Nrf2 and AP-1 in upregulation of HO-1 expression by arsenite in murine embryonic fibroblasts. Free Radic Res. 2008;42:297–304. doi: 10.1080/10715760801975735. [DOI] [PubMed] [Google Scholar]
- He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 x Keap1 x Cul3 complex and recruiting Nrf2 x Maf to the antioxidant response element enhancer. J Biol Chem. 2006;281:23620–23631. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- Hercberg S, Ezzedine K, Guinot C, Preziosi P, Galan P, Bertrais S, Estaquio C, Briancon S, Favier A, Latreille J, Malvy D. Antioxidant supplementation increases the risk of skin cancers in women but not in men. J Nutr. 2007;137:2098–2105. doi: 10.1093/jn/137.9.2098. [DOI] [PubMed] [Google Scholar]
- Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- Huang CS, Chang LS, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- Huang CS, He W, Meister A, Anderson ME. Amino acid sequence of rat kidney glutathione synthetase. Proc Natl Acad Sci U S A. 1995;92:1232–1236. doi: 10.1073/pnas.92.4.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Ferrell JE., Jr. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1996;93:10078–10083. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- Iles KE, Dickinson DA, Watanabe N, Iwamoto T, Forman HJ. AP-1 activation through endogenous H(2)O(2) generation by alveolar macrophages. Free Radic Biol Med. 2002;32:1304–1313. doi: 10.1016/s0891-5849(02)00840-7. [DOI] [PubMed] [Google Scholar]
- Isin EM, Guengerich FP. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta. 2007;1770:314–329. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996;271:C1172–1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- Kang KW, Choi SH, Kim SG. Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide. 2002;7:244–253. doi: 10.1016/s1089-8603(02)00117-9. [DOI] [PubMed] [Google Scholar]
- Katsuoka F, Motohashi H, Engel JD, Yamamoto M. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J Biol Chem. 2005a;280:4483–4490. doi: 10.1074/jbc.M411451200. [DOI] [PubMed] [Google Scholar]
- Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol. 2005b;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp M, Go YM, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu Rev Pharmacol Toxicol. 2006 doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Keum YS, Jeong WS, Kong AN. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res. 2004;555:191–202. doi: 10.1016/j.mrfmmm.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur J Biochem. 2000;267:1583–1588. doi: 10.1046/j.1432-1327.2000.01197.x. [DOI] [PubMed] [Google Scholar]
- Kimura T, Kawasaki Y, Okumura F, Sone T, Natsuki R, Isobe M. Ethanol-induced expression of glutamate-cysteine ligase catalytic subunit gene is mediated by NF-kappaB. Toxicol Lett. 2009;185:110–115. doi: 10.1016/j.toxlet.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kirkman HN, Gaetani GF. Catalase: a tetrameric enzyme with four tightly bound molecules of NADPH. Proc Natl Acad Sci U S A. 1984;81:4343–4347. doi: 10.1073/pnas.81.14.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- Kitano M, Ichihara T, Matsuda T, Wanibuchi H, Tamano S, Hagiwara A, Imaoka S, Funae Y, Shirai T, Fukushima S. Presence of a threshold for promoting effects of phenobarbital on diethylnitrosamine-induced hepatic foci in the rat. Carcinogenesis. 1998;19:1475–1480. doi: 10.1093/carcin/19.8.1475. [DOI] [PubMed] [Google Scholar]
- Knight SA, Sunde RA. The effect of progressive selenium deficiency on anti-glutathione peroxidase antibody reactive protein in rat liver. J Nutr. 1987;117:732–738. doi: 10.1093/jn/117.4.732. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, Yamamoto M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Kohle C, Bock KW. Activation of coupled Ah receptor and Nrf2 gene batteries by dietary phytochemicals in relation to chemoprevention. Biochem Pharmacol. 2006;72:795–805. doi: 10.1016/j.bcp.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Kohn KW, Riss J, Aprelikova O, Weinstein JN, Pommier Y, Barrett JC. Properties of switch-like bioregulatory networks studied by simulation of the hypoxia response control system. Mol Biol Cell. 2004;15:3042–3052. doi: 10.1091/mbc.E03-12-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Yuan JH, Moser D, Kramer JM. Purification and characterization of mouse glucose 6-phosphate dehydrogenase. Mol Cell Biochem. 1979;24:67–73. doi: 10.1007/BF00314887. [DOI] [PubMed] [Google Scholar]
- Legewie S, Bluthgen N, Herzel H. Quantitative analysis of ultrasensitive responses. Febs J. 2005;272:4071–4079. doi: 10.1111/j.1742-4658.2005.04818.x. [DOI] [PubMed] [Google Scholar]
- Leloup C, Magnan C, Benani A, Bonnet E, Alquier T, Offer G, Carriere A, Periquet A, Fernandez Y, Ktorza A, Casteilla L, Penicaud L. Mitochondrial reactive oxygen species are required for hypothalamic glucose sensing. Diabetes. 2006;55:2084–2090. doi: 10.2337/db06-0086. [DOI] [PubMed] [Google Scholar]
- Leloup C, Tourrel-Cuzin C, Magnan C, Karaca M, Castel J, Carneiro L, Colombani AL, Ktorza A, Casteilla L, Penicaud L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes. 2009;58:673–681. doi: 10.2337/db07-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yu SW, Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem. 2006;281:27251–27263. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- Lillig CH, Berndt C, Vergnolle O, Lonn ME, Hudemann C, Bill E, Holmgren A. Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc Natl Acad Sci U S A. 2005;102:8168–8173. doi: 10.1073/pnas.0500735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Cook NR, Albert C, Zaharris E, Gaziano JM, Van Denburgh M, Buring JE, Manson JE. Vitamins C and E and beta carotene supplementation and cancer risk: a randomized controlled trial. J Natl Cancer Inst. 2009;101:14–23. doi: 10.1093/jnci/djn438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase: disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci U S A. 2004;101:15893–15898. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. 2008;1783:713–727. doi: 10.1016/j.bbamcr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Liu PC, Thiele DJ. Modulation of human heat shock factor trimerization by the linker domain. J Biol Chem. 1999;274:17219–17225. doi: 10.1074/jbc.274.24.17219. [DOI] [PubMed] [Google Scholar]
- Liu RM, Gao L, Choi J, Forman HJ. gamma-glutamylcysteine synthetase: mRNA stabilization and independent subunit transcription by 4-hydroxy-2-nonenal. Am J Physiol. 1998;275:L861–869. doi: 10.1152/ajplung.1998.275.5.L861. [DOI] [PubMed] [Google Scholar]
- Loewen PC, Switala J. Purification and characterization of catalase HPII from Escherichia coli K12. Biochem Cell Biol. 1986;64:638–646. doi: 10.1139/o86-088. [DOI] [PubMed] [Google Scholar]
- Loewen PC, Switala J. Purification and characterization of catalase-1 from Bacillus subtilis. Biochem Cell Biol. 1987;65:939–947. doi: 10.1139/o87-122. [DOI] [PubMed] [Google Scholar]
- Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem. 1999;274:27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- Masuda C, Wanibuchi H, Otori K, Wei M, Yamamoto S, Hiroi T, Imaoka S, Funae Y, Fukushima S. Presence of a no-observed effect level for enhancing effects of development of the alpha-isomer of benzene hexachloride (alpha-BHC) on diethylnitrosamine-initiated hepatic foci in rats. Cancer Lett. 2001;163:179–185. doi: 10.1016/s0304-3835(00)00687-x. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Higashiyama S, Kijima Y, Suzuki K, Kawano K, Akiyama M, Kawata S, Tarui S, Deutsch HF, Taniguchi N. Human liver manganese superoxide dismutase. Purification and crystallization, subunit association and sulfhydryl reactivity. Eur J Biochem. 1990;194:713–720. doi: 10.1111/j.1432-1033.1990.tb19461.x. [DOI] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- Meech R, Mackenzie PI. UDP-glucuronosyltransferase, the role of the amino terminus in dimerization. J Biol Chem. 1997;272:26913–26917. doi: 10.1074/jbc.272.43.26913. [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem. 2005;280:20340–20348. doi: 10.1074/jbc.M412081200. [DOI] [PubMed] [Google Scholar]
- Morgan D, Rebelato E, Abdulkader F, Graciano MF, Oliveira-Emilio HR, Hirata AE, Rocha MS, Bordin S, Curi R, Carpinelli AR. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology. 2009;150:2197–2201. doi: 10.1210/en.2008-1149. [DOI] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer. 2006;6:947–960. doi: 10.1038/nrc2015. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Oblong JE, Gasdaska PY, Sherrill K, Powis G. Purification of human thioredoxin reductase: properties and characterization by absorption and circular dichroism spectroscopy. Biochemistry. 1993;32:7271–7277. doi: 10.1021/bi00079a025. [DOI] [PubMed] [Google Scholar]
- Ochi T. Hydrogen peroxide increases the activity of gamma-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Arch Toxicol. 1995;70:96–103. doi: 10.1007/BF02733669. [DOI] [PubMed] [Google Scholar]
- Ochi T. Menadione causes increases in the level of glutathione and in the activity of gamma-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Toxicology. 1996;112:45–55. doi: 10.1016/0300-483x(96)03348-3. [DOI] [PubMed] [Google Scholar]
- Omenn GS. Chemoprevention of lung cancers: lessons from CARET, the beta-carotene and retinol efficacy trial, and prospects for the future. Eur J Cancer Prev. 2007;16:184–191. doi: 10.1097/01.cej.0000215612.98132.18. [DOI] [PubMed] [Google Scholar]
- Paulson KE, Darnell JE, Jr., Rushmore T, Pickett CB. Analysis of the upstream elements of the xenobiotic compound-inducible and positionally regulated glutathione S-transferase Ya gene. Mol Cell Biol. 1990;10:1841–1852. doi: 10.1128/mcb.10.5.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LC, Petrotchenko EV, Negishi M. Crystal structure of SULT2A3, human hydroxysteroid sulfotransferase. FEBS Lett. 2000;475:61–64. doi: 10.1016/s0014-5793(00)01479-4. [DOI] [PubMed] [Google Scholar]
- Pi J, Bai Y, Reece JM, Williams J, Liu D, Freeman ML, Fahl WE, Shugar D, Liu J, Qu W, Collins S, Waalkes MP. Molecular mechanism of human Nrf2 activation and degradation: role of sequential phosphorylation by protein kinase CK2. Free Radic Biol Med. 2007a;42:1797–1806. doi: 10.1016/j.freeradbiomed.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S. Reactive Oxygen Species as a Signal in Glucose-Stimulated Insulin Secretion. Diabetes. 2007b doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–245. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- Pi J, Zhang Q, Woods CG, Wong V, Collins S, Andersen ME. Activation of Nrf2-mediated oxidative stress response in macrophages by hypochlorous acid. Toxicol Appl Pharmacol. 2008;226:236–243. doi: 10.1016/j.taap.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Plummer M, Vivas J, Lopez G, Bravo JC, Peraza S, Carillo E, Cano E, Castro D, Andrade O, Sanchez V, Garcia R, Buiatti E, Aebischer C, Franceschi S, Oliver W, Munoz N. Chemoprevention of precancerous gastric lesions with antioxidant vitamin supplementation: a randomized trial in a high-risk population. J Natl Cancer Inst. 2007;99:137–146. doi: 10.1093/jnci/djk017. [DOI] [PubMed] [Google Scholar]
- Puatanachokchai R, Morimura K, Wanibuchi H, Oka M, Kinoshita A, Mitsuru F, Yamaguchi S, Funae Y, Fukushima S. Alpha-benzene hexachloride exerts hormesis in preneoplastic lesion formation of rat hepatocarcinogenesis with the possible role for hepatic detoxifying enzymes. Cancer Lett. 2006;240:102–113. doi: 10.1016/j.canlet.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Ralser M, Wamelink MM, Kowald A, Gerisch B, Heeren G, Struys EA, Klipp E, Jakobs C, Breitenbach M, Lehrach H, Krobitsch S. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J Biol. 2007;6:10. doi: 10.1186/jbiol61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14:S211–215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- Savageau MA. Biochemical Systems Analysis: A Study of Function and Design in Molecular Biology. Addison-Wesley; Reading, MA: 1976. [Google Scholar]
- Schuppe-Koistinen I, Moldeus P, Bergman T, Cotgreave IA. S-thiolation of human endothelial cell glyceraldehyde-3-phosphate dehydrogenase after hydrogen peroxide treatment. Eur J Biochem. 1994;221:1033–1037. doi: 10.1111/j.1432-1033.1994.tb18821.x. [DOI] [PubMed] [Google Scholar]
- Seelig GF, Simondsen RP, Meister A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J Biol Chem. 1984;259:9345–9347. [PubMed] [Google Scholar]
- Sekhar KR, Long M, Long J, Xu ZQ, Summar ML, Freeman ML. Alteration of transcriptional and post-transcriptional expression of gamma-glutamylcysteine synthetase by diethyl maleate. Radiat Res. 1997;147:592–597. [PubMed] [Google Scholar]
- Shan Y, Lambrecht RW, Ghaziani T, Donohue SE, Bonkovsky HL. Role of Bach-1 in regulation of heme oxygenase-1 in human liver cells: insights from studies with small interfering RNAS. J Biol Chem. 2004;279:51769–51774. doi: 10.1074/jbc.M409463200. [DOI] [PubMed] [Google Scholar]
- Son TG, Camandola S, Mattson MP. Hormetic dietary phytochemicals. Neuromolecular Med. 2008;10:236–246. doi: 10.1007/s12017-008-8037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song IS, Tatebe S, Dai W, Kuo MT. Delayed mechanism for induction of gamma-glutamylcysteine synthetase heavy subunit mRNA stability by oxidative stress involving p38 mitogen-activated protein kinase signaling. J Biol Chem. 2005;280:28230–28240. doi: 10.1074/jbc.M413103200. [DOI] [PubMed] [Google Scholar]
- Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem. 2003;278:2396–2402. doi: 10.1074/jbc.M209195200. [DOI] [PubMed] [Google Scholar]
- Stone JR. An assessment of proposed mechanisms for sensing hydrogen peroxide in mammalian systems. Arch Biochem Biophys. 2004;422:119–124. doi: 10.1016/j.abb.2003.12.029. [DOI] [PubMed] [Google Scholar]
- Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- Sugatani J, Sueyoshi T, Negishi M, Miwa M. Regulation of the human UGT1A1 gene by nuclear receptors constitutive active/androstane receptor, pregnane X receptor, and glucocorticoid receptor. Methods Enzymol. 2005;400:92–104. doi: 10.1016/S0076-6879(05)00006-6. [DOI] [PubMed] [Google Scholar]
- Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- Surh YJ, Kundu JK, Na HK, Lee JS. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr. 2005;135:2993S–3001S. doi: 10.1093/jn/135.12.2993S. [DOI] [PubMed] [Google Scholar]
- Suzukawa K, Miura K, Mitsushita J, Resau J, Hirose K, Crystal R, Kamata T. Nerve growth factor-induced neuronal differentiation requires generation of Rac1-regulated reactive oxygen species. J Biol Chem. 2000;275:13175–13178. doi: 10.1074/jbc.275.18.13175. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol Cell Biol. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z, Anders MW. Identification of an important cysteine residue in human glutamate-cysteine ligase catalytic subunit by site-directed mutagenesis. Biochem J. 1998;336(Pt 3):675–680. doi: 10.1042/bj3360675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M. Compartmentalization of Redox Signaling through NADPH Oxidase-derived ROS. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargo MA, Nguyen L, Colman RF. Subunit interface residues of glutathione S-transferase A1-1 that are important in the monomer-dimer equilibrium. Biochemistry. 2004;43:3327–3335. doi: 10.1021/bi030245z. [DOI] [PubMed] [Google Scholar]
- Weichsel A, Gasdaska JR, Powis G, Montfort WR. Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure. 1996;4:735–751. doi: 10.1016/s0969-2126(96)00079-2. [DOI] [PubMed] [Google Scholar]
- Wiernsperger NF. Oxidative stress as a therapeutic target in diabetes: revisiting the controversy. Diabetes Metab. 2003;29:579–585. doi: 10.1016/s1262-3636(07)70072-1. [DOI] [PubMed] [Google Scholar]
- Woods CG, Fu J, Xue P, Hou Y, Pluta LJ, Yang L, Zhang Q, Thomas RS, Andersen ME, Pi J. Dose-dependent transitions in Nrf2-mediated adaptive response and related stress responses to hypochlorous acid in mouse macrophages. Toxicol Appl Pharmacol. 2009;238:27–36. doi: 10.1016/j.taap.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Ferrell JE., Jr. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature. 2003;426:460–465. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, Khor TO, Li W, Ma J, Kong AN. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol Cancer Ther. 2006;5:1918–1926. doi: 10.1158/1535-7163.MCT-05-0497. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang J, Huang ZZ, Ou X, Lu SC. Cloning and characterization of the 5′-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001a;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang J, Ou X, Huang ZZ, Lu SC. Cloning and analysis of the rat glutamate-cysteine ligase modifier subunit promoter. Biochem Biophys Res Commun. 2001b;285:476–482. doi: 10.1006/bbrc.2001.5190. [DOI] [PubMed] [Google Scholar]
- Yi TM, Huang Y, Simon MI, Doyle J. Robust perfect adaptation in bacterial chemotaxis through integral feedback control. Proc Natl Acad Sci U S A. 2000;97:4649–4653. doi: 10.1073/pnas.97.9.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, Kong AN. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J Biol Chem. 2000;275:39907–39913. doi: 10.1074/jbc.M004037200. [DOI] [PubMed] [Google Scholar]
- Yuan X, Xu C, Pan Z, Keum YS, Kim JH, Shen G, Yu S, Oo KT, Ma J, Kong AN. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol Carcinog. 2006;45:841–850. doi: 10.1002/mc.20234. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Andersen ME. Dose Response Relationship in Anti-Stress Gene Regulatory Networks. PLoS Comput Biol. 2007;3:e24. doi: 10.1371/journal.pcbi.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Pi J, Woods CG, Andersen ME. Phase I to II cross-induction of xenobiotic metabolizing enzymes: A feedforward control mechanism for potential hormetic responses. Toxicol Appl Pharmacol. 2009;237:345–356. doi: 10.1016/j.taap.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–492. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]