

Abstract
Human immunodeficiency virus type 1 (HIV-1) can disseminate between CD4+ T cells via diffusion-limited cell-free viral spread or by directed cell-cell transfer using virally induced structures termed virological synapses. Although T-cell virological synapses have been well characterized, it is unclear whether this mode of viral spread is susceptible to inhibition by neutralizing antibodies and entry inhibitors. We show here that both cell-cell and cell-free viral spread are equivalently sensitive to entry inhibition. Fluorescence imaging analysis measuring virological synapse lifetimes and inhibitor time-of-addition studies implied that inhibitors can access preformed virological synapses and interfere with HIV-1 cell-cell infection. This concept was supported by electron tomography that revealed the T-cell virological synapse to be a relatively permeable structure. Virological synapse-mediated HIV-1 spread is thus efficient but is not an immune or entry inhibitor evasion mechanism, a result that is encouraging for vaccine and drug design.
As with enveloped viruses from several viral families, the human immunodeficiency virus type 1 (HIV-1) can disseminate both by fluid-phase diffusion of viral particles and by directed cell-cell transfer (39). The primary target cell for HIV-1 replication in vivo is the CD4+ T-cell (13), which is infectible by CCR5-tropic (R5) and CXCR4-tropic (X4) viral variants (29). R5 HIV-1 is the major transmitted viral phenotype and dominates the global pandemic, whereas X4 virus is found later in infection in ca. 50% of infected individuals, and its presence indicates a poor disease progression prognosis (23). Cell-cell HIV-1 transfer between T cells is more efficient than diffusion-limited spread (8, 16, 32, 38), although recent estimates for the differential range from approximately 1 (42) to 4 (6) orders of magnitude. Two structures have been proposed to support contact-mediated intercellular movement of HIV-1 between T cells: membrane nanotubes (33, 43) and macromolecular adhesive contacts termed virological synapses (VS) (15, 17, 33). VS appear to be the dominant structure involved in T-cell-T-cell spread (33), and both X4 (17) and R5 HIV-1 (6, 15, 42) can spread between T cells via this mechanism.
VS assembly and function are dependent on HIV-1 envelope glycoprotein (Env) engaging its primary cellular receptor CD4 (2, 6, 17). This interaction recruits more CD4 and coreceptor to the site of cell-cell contact in an actin-dependent manner (17). Adhesion molecules cluster at the intercellular junction and are thought to stabilize the VS (18). In parallel, viral Env and Gag are recruited to the interface by a microtubule-dependent mechanism (19), where polarized viral budding may release virions into the synaptic space across which the target cell is infected (17). The precise mechanism by which HIV-1 subsequently enters the target T-cell cytoplasm remains unclear: by fusion directly at the plasma membrane, fusion from within an endosomal compartment, or both (4, 6, 15, 25, 34).
Viruses from diverse families including herpesviruses (9), poxviruses (22) and hepatitis C virus (44) evade neutralizing antibody attack by direct cell-cell spread, since the tight junctions across which the these viruses move are antibody impermeable. It has been speculated that transfer of HIV-1 across VS may promote evasion from immune or therapeutic intervention with the inference that the junctions formed in retroviral VS may be nonpermissive to antibody entry (39). However, available evidence regarding whether neutralizing antibodies (NAb) and other entry inhibitors can inhibit HIV-1 cell-cell spread is inconsistent (25). An early analysis suggested that HIV-1 T-cell-T-cell spread is relatively resistant to neutralizing monoclonal antibodies (NMAb) (12). A later study agreed with this conclusion by demonstrating a lack of permissivity of HIV-1 T-cell-T-cell spread, measured by transfer of viral Gag, to interference with viral fusion using a gp41-specific NMAb and a peptidic fusion inhibitor (6). In contrast, another analysis reported that anti-gp41-specific NMAb interfered effectively with HIV-1 spread between T cells (26). Inhibitors of the HIV-1 surface glycoprotein (gp120)-CD4 or gp120-CXCR4 interaction reduced X4 HIV-1 VS assembly and viral transfer if applied prior to mixing of infected and receptor-expressing target cells (17, 19), but the effect of these inhibitors has not been tested on preformed VS. Thus, the field is currently unclear on whether direct T-cell-T-cell infectious HIV-1 spread is susceptible or not to antibody and entry inhibitor-mediated disruption of VS assembly, and the related question, whether the VS is permeable to viral entry inhibitors, including NAb. Addressing these questions is of central importance to understanding HIV-1 pathogenesis and informing future drug and vaccine design.
Since estimates reported in the literature of the relative efficiency of direct HIV-1 T-cell-T-cell spread compared to cell-free spread vary by approximately 3 orders of magnitude (6, 38, 42), and the evidence for the activity of viral entry inhibitors on cell-cell spread is conflicting, we set out to quantify the efficiency of infection across the T-cell VS and analyze the susceptibility of this structure to NAb and viral entry inhibitors. Assays reporting on events proximal to productive infection show that the R5 HIV-1 T-cell VS is approximately 1 order of magnitude more efficient than cell-free virus infection, and imaging analyses reveal that the VS assembled by HIV-1 is most likely permeable to inhibitors both during, and subsequent to, VS assembly. Thus, we conclude that the T-cell VS does not provide a privileged environment allowing HIV-1 escape from entry inhibition.
MATERIALS AND METHODS
Cell culture and virus infection.
The cell lines Jurkat CE6.1, Jurkat. Tat.CCR5, A201, A301.R5, and ACH-2 were obtained from the National Institute for Biological Standards and Controls (NIBSC) Centre for AIDS Reagents (CFAR), Potters Bar, United Kingdom. A301.R5 cells were derived by retroviral transduction and cultured as previously described (7). Cells were cultured in complete medium (CM) composed of RPMI (Gibco/Invitrogen), 10% fetal calf serum (FCS), 100 U of penicillin/ml, and 100 μg of streptomycin (PAA, Yeovil, United Kingdom)/ml. Unless otherwise stated, cells were cultured at 37°C in 5% CO2. Jurkat.Tat.CCR5 and A301.R5 were maintained on CM supplemented with 1 mg of G418 (Sigma, Poole, United Kingdom)/ml to maintain expression of CCR5. Peripheral blood mononuclear cells (PBMC) were isolated from whole blood or buffy coats (from the National Blood Transfusion Service, Bristol, United Kingdom) by Ficoll-Hypaque (Sigma-Aldrich) gradient centrifugation. A MACS CD4 T-Cell Kit-II (Miltenyi UK, Inc.) was used to negatively isolate CD4+ T cells that were routinely >99% pure. For experiments with the R5 HIV-1BaL isolate, primary CD4+ T cells were activated with CM supplemented with 5 μg of PHA-L (Sigma)/ml and 10 U of interleukin-2 (IL-2; NIBSC)/ml for 3 days and then CM with IL-2 alone until use on day 7 of culture. For experiments with the X4 HIV-1IIIB isolate, primary CD4+ T cells were purified from PBMC and used immediately. Cells (107) were infected in a volume of 1 ml with IIIB or BaL (NISBC) virus-containing supernatant overnight at a multiplicity of infection (MOI) of ∼0.01 and then cultured in 50 ml of CM and typically used on days 7 to 9 of infection. At this time point the surface expression of Env was highest, as detected by anti-gp120 antibody 2G12 (NIBSC) and anti-human IgG-phycoerythrin conjugate (Jackson Laboratories), and analyzed by flow cytometry on a FACSCalibur using Flow-Jo software (Becton Dickinson UK).
Inhibitors.
The following NMAbs with relatively broad neutralizing activity were used: 2G12 (CFAR, NIBSC), glycan-specific, binds to mannose groups on gp120 (37, 40); IgG1b12 (B12; D. Burton, Scripps Institute), binds the CD4 binding surface on gp120 (5); and 2F5 (D. Katinger, Polymun, Inc.) that binds the membrane-proximal extracellular region (MPER) of gp41 and inhibits virus-cell fusion (30). Q4120 is a CD4 domain 1-specific MAb that efficiently blocks the CD4-gp120 interaction and viral entry (14), T20 (enfuvirtide) is a broadly active synthetic peptide inhibitor of gp41-mediated fusion licensed for human use (28), TAK779 (NIH AIDS Reagent Repository) is a small molecule inhibitor of the gp120-CCR5 interaction (1), and PRO 2000 (Indevus, Inc.) is a polyanionic candidate topical microbicide that interferes with viral entry, at least in part by binding to elements of the coreceptor binding surface (36).
Cell-cell and cell-free infection assays.
Three different methods were used to separate infected and target cells to measure cell-free infection, which was then compared to samples where the cells were directly cultured together to allow cell-cell viral transfer. In each case, 5 × 105 infected cells (JktBaL) and 5 × 105 target cells (A301.R5) were used, and A201 (CD4− CCR5−) were used as negative control target cells. For infection of target cells by cell-free supernatants, the JktBaL cells were cultured alone in 150 μl of CM for the 6 or 12 h to produce virus. Cells were centrifuged at 2,000 × g for 5 min, and the supernatant was carefully aspirated and immediately added to a culture of 5 × 105 target cells in 150 μl of CM. Target cells were further cultured to achieve a total incubation time of 12 h and then centrifuged and harvested for DNA extraction. For infection of target cells with or without transwells, cells were either cultured directly together or separated by a 3-μm 24-well transwells, allowing full diffusion of virus but not migration of cells [data not shown]). A total of 5 × 105 infected cells were suspended in 50 μl of CM and placed in the upper well, and 5 × 105 target cells were suspended in 250 μl in the lower well. Target cells were harvested by removing transwells and lysed and processed for DNA extraction. For the agitated culture method, we used an adaptation of a procedure described previously (42). Briefly, 5 × 105 of each cell type were resuspended in 1 ml of CM in a six-well plate. Paired samples were either left static or rocked at 70 rpm in 5% CO2. At the relevant time point, the culture was harvested and processed for DNA extraction for quantitative PCR (qPCR).
Cell-free and cell-cell infection inhibition assays.
To measure inhibition of cell-free or cell-cell infection, 5 × 105 A301.R5 target cells were either cultured with an equal number of JktBaL cells or 100 ng of PBMC-grown cell-free BaL virus. At 0 h the inhibitors were titrated onto the cells, starting at 100 μg/ml in a fivefold dilution series for all inhibitors except TAK, which was started at 50 nM. Cells were incubated for 12 h (cell-cell) or 24 h (cell-free), corresponding to a single cycle of viral infection. For B12, inhibition was also measured when the NMAb was added at 10 μg/ml at either 1 or 3 h after mixing. The antibody was diluted in 7.5 μl of CM and carefully added to existing cocultures of 5 × 105 JktBaL and 5 × 105 A301.R5 target cells to avoid disturbing preformed conjugates.
Quantitative real-time PCR for HIV-1 polymerase DNA detection.
Samples were prepared by mixing 5 × 105 infected and 5 × 105 target A301.R5 or control target A201.R5 cells in CM and were incubated at for the required time with or without the addition of inhibitors. Samples were pelleted, supernatant was aspirated, and pellets were frozen at −80°C for later extraction with Qiagen DNeasy kit. Primers and probe to detect HIV-1 DNA were designed by using PrimerExpress3 against a pol sequence common to BaL and HXB2. The forward primer TGGGTTATGAACTCCATCCTGAT and the reverse primer, TGTCATTGACAGTCCAGCTGTCT were used. The probe had the sequence TTCTGGCAGCACTATAGGCTGTACTGTCCATT and was labeled with the fluorophore FAM and the quencher TAMRA. For detection of the β-globin cellular reference gene, the forward primer AACTGGGCATGTGGAGACAGA, the reverse primer CTAAGGGTGGGAAAATAGACCAATAG, and the probe TCTTGGGTTTCTGATAGGCACTGACTCTCTCTG were used. The probe was labeled with VIC and the quencher TAMRA. PCR was performed in a 25-μl total volume, using 1× Eurogentec Mastermix (Eurogentec, Ltd., Southampton, United Kingdom) 0.3 μM concentrations of the primers, 0.1 μM probe, and 2.5 μl of sample DNA. A standard curve was created by titrating DNA from ACH-2 cells. Reactions were performed in single-plex using an ABI 7500 (Applied Biosystems, Warrington, United Kingdom) standard run. For all qPCR measurements, the results of triplicate single-plex reactions detecting pol and β-globin were first averaged to give a mean for each, and then the pol values were divided by the β-globin values to give a relative abundance. For experiments reported in Fig. 1A and B, this was further normalized by dividing the value at each time point by the relevant 0-h result to adjust for the starting level of infection and to enable comparison between cells with differing efficiency of HIV-1 infection. For the experiments whose results are reported in Fig. 1C and D, where the target cell cultures for cell-free infection did not contain any infected cells, the 0-h value is shown in the graph. For all other PCR data presented where target and infected cells were harvested together, the result from a matched sample using the nonpermissive A201 cells as targets was subtracted from the test value at each time point to account for the signal arising from the infected cells. To calculate a percent inhibition of HIV-1 infection, the relative abundance of pol was normalized against A201 signal as described above. This value was subtracted from the no-treatment control value at the relevant time point, and the result divided by the no-treatment control value to give percentage inhibition.
FIG. 1.

R5 HIV-1 spreads more efficiently cell-cell than by cell-free virion diffusion. (A) HIV-1BaL transfer between infected and uninfected T-cell lines. JktBaL was mixed with A301.R5 target cells and, at the time points indicated, the samples were lysed and analyzed for HIV-1 pol by qPCR and then normalized against cellular β-globin. The background signal obtained from nonpermissive A201 cells was subtracted from all values before calculation of percent inhibition compared to no-treatment or no-virus controls. (B) HIV-1 transfer from primary CD4BaL cells to autologous activated primary CD4+ T cells, assayed by qPCR and presented as in panel A over 24 h. The data represent means of eight independent experiments, each carried out in triplicate. (C) JktBaL cells were mixed directly with A301.R5 target cells in a transwell chamber without membrane (□) or were separated by the transwell membrane (▪). At the times shown cultures were lysed and processed for qPCR. The data represent the mean pol copy number relative to β-globin from three independent experiments, each carried out in triplicate. (D) Supernatant was harvested from the lower (containing A301.R5 target cells) chamber of a transwell without (□) or with (▪) a transwell membrane to separate the top well (containing JktBal cells) and assayed for p24 Gag by ELISA. Triplicate wells were sampled from one representative experiment and the bars represent the mean + 1 standard deviation (SD). (E) JktBaL (white bars) or cell-free supernatants harvested from JktBaL cells over a 12-h culture (▪) were mixed with A301.R5 target cells at T = 0. Samples were processed as in panel A, and the data represent the mean pol copy number relative to β-globin from three independent experiments each carried out in triplicate. (F) JktBaL cells were mixed with A301.R5 target cells at T = 0, after which the cultures were either left static (□) or were agitated to prevent stable cell-cell contacts from forming (▪). Samples were processed as in panel A, and the data represent the mean pol copy number relative to β-globin from three independent experiments, each carried out in triplicate. Error bars represent the SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Laser scanning confocal microscopy (LSCM).
Target A301.R5 cells were prelabeled with the nonblocking, CD4 domain 4-specific MAb L120 (14, 17), mixed 1:1 with infected cells (JktBaL or JktIIIB), incubated on poly-l-lysine-coated coverslips for the required time, and then fixed at room temperature with 4% paraformaldehyde (PFA). Samples were quenched with 50 mM NH4Cl and permeabilized with phosphate-buffered saline containing 0.1% Triton-X and 5% FCS. Env was labeled with 10 μg of 2G12 (NIBSC)/ml and Gag with 1:1,000 polyclonal rabbit anti-p24/anti-p17 (NIBSC). Samples were counterstained with anti-mouse-Alexa 488 (Invitrogen UK), anti-human-TRITC, and anti-rabbit-Cy5 (Jackson-Immunotech, United Kingdom) and mounted on glass slides using ProLong Gold (Invitrogen). Images were acquired on a Zeiss Pascal Axiovert 200M by using sequential capture and processed using Adobe Photoshop CS2. Gag polarization and transfer between cells were quantified in 10 randomly acquired low-power fields, each containing an average of approximately 20 conjugates. Conjugate interfaces were defined as contacts between an infected (Env+ and/or Gag+) and a target (CD4+) cell. Polarization of Gag, Env, or CD4 was defined as signal enrichment in the membrane contact zone compared to the noncontact regions quantified using Metamorph software. VS were defined by colocalization of all three markers at the intercellular interface, and percentages were calculated by dividing the number of colocalization events by the number of interfaces. Gag transfer was defined as Gag signal within the target cell in the absence of colocalized Env signal. The total number of these events/field was divided either by the number of VS or by the total number of conjugate interfaces.
Time-lapse LSCM analysis of conjugate lifetimes.
To measure the duration of interaction times between HIV-1-infected and uninfected CD4+ target cells, JktBaL cells were labeled with CellTracker Orange CMRA (Invitrogen) and uninfected primary CD4+ T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen). A total of 104 JktBaL and 104 primary CD4+ T cells were mixed in 50 μl of CM and placed in one well of an Ibidi IbiTreat 24-well angiogenesis slide (Ibidi GmbH, Munich, Germany), and the plastic cover was replaced. The slide was placed within a heated stage and supplemented with humidified 5% CO2. Using a Zeiss Pascal LSM4 with sequential channel capture, images were recorded every 1 min up to 5 h. To quantify infected cell-target cell interaction times, each frame of the time-lapse series was processed in Metamorph to binarize the cell color labels into red (infected) and green (target), the cell profile was dilated to create defined interaction zones (yellow) at cell-cell contact regions, and interaction sites were isolated for quantification. Interaction sites for each x-y frame of the movie were stacked into a three-dimensional box with time along the z axis, allowing each interaction to be tracked over time as an individual object.
Electron microscopy and tomography.
Freshly purified primary CD4+ T cells were mixed 1:1 with JktIIIB cells, or 3-day PHA/IL-2-activated primary CD4+ T cells were mixed with JktBaL cells at 106 cells/ml in RPMI-10% FCS for 3 h at 37°C in 5% CO2, followed by gentle pelleting, aspiration of the supernatant, and fixation in 4% PFA for 10 min at 37°C and 8% PFA for 50 min at room temperature. Samples were left in 2.5% glutaraldehyde in cacodylate buffer (100 mM sodium cacodylate, 50 mM KCl, 2.5 mM CaCl2 [pH 7.2]) overnight, postfixed for 1 h on ice with 1% osmium tetroxide, washed, stained overnight at 4°C with 0.5% uranyl acetate, and dehydrated in a graded ethanol series at room temperature. Samples were embedded in epoxy resin (Roth, Karlsruhe, Germany) according to the manufacturer's instructions. Serial sections (300 nm thick) sections were cut, and dual-axis digital image series were recorded on an FEI Technai TF30 microscope at 300 kV with FEI Eagle 4k charge-coupled device camera (pixel size after binning to 2k 1.99 or 2.53 nm at the specimen level) over a −60° to +60° tilt range (increment 1°) and defocus at −0.2 μm, using the SerialEM software package (27). Tomogram reconstruction, joining of serial tomograms, manual three-dimensional image segmentation and calculation of membrane model point coordinates used IMOD software v3.12.20 (20). Nearest approach points between the two membrane models in each tomogram were calculated and displayed as colored patches using MATLAB (Mathworks, Natick, MA) and UCSF Chimera (31), respectively.
Statistical analyses.
Unless otherwise stated, quantitative data represent the mean values of replicate independent experiments each carried out in triplicate, and error bars represent the standard errors of the mean (SEM). Significance testing throughout was performed using a two-tailed Student t test, with the Bonferroni correction for multiple tests where appropriate.
RESULTS
Efficiency of direct T-cell-T-cell spread of R5 HIV-1.
Since R5 HIV-1 is the virus type that is principally transmitted between individuals, its study is central to understanding viral infection and spread and inhibition of these processes. We therefore adapted a previously established qPCR-based assay for detection of de novo DNA viral transcripts in X4 virus-infected cells (19) for use with R5 viral DNA. The pol region was chosen as a template for primers and probe since this is a late reverse transcript and therefore relevant to infection events proximal to proviral integration and productive replication. Values normalized to β-globin were expressed as copies of HIV-1 DNA/cell, and assay specificity was confirmed by coculturing CD4+ CCR5+ Jurkat.Tat.R5 cells infected with HIV-1BaL (JktBaL) with either the permissive CD4+ CCR5+ T-cell line A301.R5, or the nonpermissive CD4− CCR5− line A201. As previously observed with X4 HIV-1 (18, 19), de novo pol DNA production was not observed in A2.01 target cells (data not shown), whereas A3.01.R5 target cells demonstrated a significant (P < 0.05) increase of ∼5-fold over background in pol DNA over 12 h (Fig. 1A). Because model systems using immortalized T-cell lines may not accurately represent interactions taking place between primary CD4+ T cells, we measured relative R5 HIV-1 pol production after transfer between infected and uninfected primary CD4+ T cells. Activated purified primary CD4+ T cells infected to levels >50% as determined by Gag and Env staining and flow cytometric analysis (data not shown) were mixed with uninfected autologous CD4+ T cells for the times shown, and the samples were processed for qPCR. We observed significant levels of increased pol in mixtures of infected and uninfected primary CD4+ T cells (Fig. 1B) that were similar in magnitude to virus spread between the cell lines, but with the slower kinetics of pol synthesis. This confirmed that the use of T-cell lines appears qualitatively justified for the study of cell-cell spread of R5 HIV-1, with the caveat that it is more rapid than the primary cell system. The HIV-1BaL-infected T-cell line JktBaL was subsequently used in all experiments hereafter for reasons of reproducibility, and A301.R5 cells were used as targets unless otherwise stated.
To quantify differences between cell-free and cell-cell R5 HIV-1 transmission, we used three different assay formats: (i) use of a virus-permeable transwell to separate JktBaL from target cells; (ii) culture of A3.01/R5 target cells with supernatants from JktBaL or direct A3.01/R5 mixing with JktBaL; and (iii) an adaptation of the approach adopted by Sourisseau et al. (42), in which cultures were maintained static for cell-cell interactions to take place or were agitated to prevent stable cell-cell interactions. We confirmed that in each assay infected and target cell viability was maintained at an equivalent level over the time course of the experiment (data not shown). Figure 1C shows that by 12 h after JktBal-target cell mixing there was a robust pol signal detectable that was ∼13-fold greater than the cell-free signal (P < 0.01). Free diffusion of virus across the transwell membrane was determined by sampling supernatant for p24 Gag enzyme-linked immunosorbent assay (ELISA): there was no significant difference in p24 levels above or below the membrane (Fig. 1D). The signal obtained when JktBal-target cells were mixed was 6.5-fold greater than the signal obtained with viral supernatant (Fig. 1E, P < 0.01) and 5.4-fold greater when cells were maintained in a static, as opposed to an agitated, culture (see Fig. 7F, P < 0.01). Taken together, the mean increase in cell-cell compared to cell-free infection at 12 h corresponds to (8.3 ± 3.3)-fold. Although we do not know which assay most precisely mimics the in vivo situation relating to cell-cell compared to cell-free viral spread, we assume that the mean value of approximately 10-fold is broadly representative of relative in vitro efficiency of HIV-1 spread between T cells.
FIG. 7.

Electron tomographic reconstruction of the HIV-1 T-cell VS. (A) A single 2-nm-thick digital slice from a tomographic reconstruction of a conjugate between a primary CD4+ T cell (left) and a JktIIIB cell (right) cocultured at a 1:1 ratio for 3 h. (B) Single higher-magnification 5-nm-thick digital slice from a tomographic reconstruction of a conjugate between a primary CD4+ T-cell (left) and a JktBaL cell (right). Arrows indicate viral budding structures on the infected cell; the white asterisk indicates the nucleus of an infected cell. (C) Another single digital slice at a different z height within the same tomogram as in panel A. Arrows indicate budding structures. (D) Same as in panel A but an image from a conjugate between a JktBaL cell and a primary CD4+ T cell. (E) A higher-magnification image represents a diagonal slice through the three-dimensional volume for better visualization of the budding structure (arrow) on the surface of the JktBaL cell. (F) Three-dimensional model of the JktIIIB cell surface (red), the primary CD4+ T-cell surface (green), and virions (red spheres), generated from the same tomogram as in panel A. (G) Three-dimensional model of the JktBaL cell surface (red), the primary CD4+ T-cell surface (green), and virions (red spheres), generated from the same tomogram as in panel D. (H) Higher-power representation of cell membranes shown in the boxed area of panel F. Yellow membrane patches indicate areas of close apposition (<30 nm) of the two membranes. (I) Higher-power representation of cell membranes shown in the boxed area of panel G; the yellow label is as described in panel H.
Cell-free and cell-cell spread of R5 HIV-1 are equally susceptible to entry inhibition.
Having established that three different cell-cell assays for R5 virus spread gave broadly similar results and having compared the efficiency of cell-free and cell-cell spread quantitatively, we next analyzed whether these modes of infection are similarly susceptible to interference with viral entry. We chose the supernatant-based cell-free assay alongside the cell-cell infectivity assay (as shown in Fig. 1E) for these experiments since this was the most technically feasible for this type of inhibition experiment and gave an intermediate result (6.5-fold) in terms of relative viral transfer efficiency. We selected a panel of HIV-1 entry inhibitors based upon their modes of action (receptor blocking or fusion inhibition) their chemical type (neutralizing antibody, peptide, small molecule inhibitor, or polyanion) and their relative molecular mass (0.5 to 150 kDa). Inhibitors were titrated into each system at time of cell-free infectious supernatant or infected cell mixing with target cells. De novo-synthesized viral DNA was measured by qPCR within a time-frame allowing robust pol detection and corresponding to a single round of viral replication in both systems, approximating 12 h in the cell-cell system and 24 h in the cell-free system. Titration curves from combined experiments are shown for each inhibitor tested in Fig. 2A, and the mean 50% inhibitory concentration (IC50) data are summarized in Fig. 2B. All agents showed increasing inhibition of both cell-cell and cell-free infection with increasing concentration, and all achieved ca. 100% inhibition at the higher doses in the range. Mean ID50 values varied across 4 orders of magnitude, representing their relative potency against R5 HIV-1. Considerable biological variation was observed between experiments, particularly with the CCR5 antagonist TAK779, for reasons that are currently unclear. Two of the inhibitors that interfere with gp120-CD4 binding (B12 and Q4120) showed a clear trend at low concentrations toward less effective inhibition of cell-cell viral spread compared to cell-free spread (Fig. 2A). This might reflect a modest selective reduction in inhibition of cell-cell spread as a result of reduced inhibitor access to the VS, increased velocity of gp120 engagement of CD4 on the target cell, or both. Nevertheless, no overall significant difference in inhibitor efficacy as measured at the ID50 was observed between the two systems, whether inhibitors of low (TAK779, ∼0.5 kDa [1]) or high (NAb, ∼150 kDa) molecular mass were tested. This implies that the inhibitors were either acting rapidly to prevent VS assembly and cell-cell virus transmission, or that these molecules enter assembled VS to block viral infection, or both.
FIG. 2.

Cell-free and cell-cell infections are equally susceptible to entry inhibition. (A) A301.R5 target cells were either mixed 1:1 with JktBaL cells or with 12-h cell-free supernatants derived from JktBaL in the presence of titrations of the inhibitors shown. Cells were harvested after 12 h and processed for qPCR of pol and β-globin. Reduction in the pol PCR signal relative to the β-globin signal was expressed for each dilution of each inhibitor. The background signal obtained from nonpermissive A201 cells was subtracted from all of the values before calculation of the percent inhibition compared to no-treatment or no-virus controls. Solid lines represent cell-free virus inhibition, and dashed lines represent cell-cell virus inhibition. (B) Percentage inhibition data from three independent experiments each carried out in triplicate were used to calculate IC50s. □, Cell-free infection inhibition; ▪, cell-cell infection inhibition. Error bars represent the SEM.
Life span of HIV-1 R5 T-cell VS.
To investigate the possibility that inhibitors might interfere with HIV-1 spread across existing VS, we first established the longevity of R5 VS in T cells. For this we assumed that long-lived conjugates formed between HIV-1-infected and uninfected, receptor-expressing CD4+ T cells represented VS, a proportion of which would be functional in cell-cell transfer of HIV-1 infection. Differentially labeled JktBaL cells or control uninfected Jurkat cells were mixed with uninfected primary CD4+ T cells, and their interactions were imaged using time-lapse LSCM. A frame from a representative movie (see Movie S1 in the supplemental material) is shown in Fig. 3A, in which multiple conjugates between infected (red) and uninfected target cells (green) are identified by arrows. To quantify infected cell-target cell interaction times, each frame of the time-lapse series was processed to create binary versions of the two colors and to dilate the cell profile to create defined interaction zones (yellow) at cell-cell contact regions (Fig. 3B). Interaction sites were then isolated for quantification (Fig. 3C). Interaction sites for each x-y frame of the movie were stacked into a 3-dimensional box with time along the z axis, allowing each interaction to be tracked over time as an individual object. Lifetimes of control interactions obtained from uninfected Jurkat and primary CD4+ T cells labeled and processed in the same manner as the test samples were compared to the test, and the data are quantified in Fig. 3D and E. Control and test sample conjugate life spans were similarly represented at times of up to 10 min and are consistent with previously described intravitally measured life spans of normal homotypic T-cell interactions (35). Since ∼100% of the JktBaL cells were infected, this implies that many infected-target cell interactions are similarly transient to uninfected cell-cell interactions: whether these rapid interactions result in viral transfer is unknown. However, none of the control interactions exceeded 10 min, demonstrating that the formation of long-lived T-cell-T-cell contacts is HIV-1-driven. The mean contact time in the test samples was 62 min, and a small number were stable to the end of the assay at 300 min. Since these sustained (>10-min) interactions representing ∼21% of all events approximate the percentage of VS found within T-cell conjugates in other studies with X4 virus (17), we hypothesized that these were likely to represent VS. However, since X4 and R5 HIV-1 VS kinetics of assembly and frequency may vary, we investigated R5 virus VS dynamics in the current system using LSCM of JktBaL-primary CD4+ T-cell conjugates. The percentage of CD4, Gag, and Env capping at different time points was quantified from randomly selected low-power fields as previously described (17). All three markers independently showed increased capping from 1 to 3 h after cell mixing, plateauing between 3 and 6 h (Fig. 4A). Similarly, cocapping of all three markers at a conjugate interface (our definition of a VS) increased significantly between 1 and 3 h but not between 3 and 6 h, reaching a maximum value of ∼30% at 6 h. Gag transfer across the VS measured by distinct localization of Gag within the target cell revealed increasing transfer over the 6-h period. When this was expressed as percentage of total VS, ca. 20% VS showed Gag transfer at 1 h, rising to ∼60 and ∼80% at 3 and 6 h, respectively (Fig. 4B). Expression of these values as a percentage of all conjugate interfaces showed accumulation of target cell-associated Gag to a plateau of ca. 20% between 3 and 6 h (Fig. 4B), a finding consistent with the percentage of long-lived conjugates seen in Fig. 3E. We therefore hypothesize that the majority of the long-lived conjugates forming between HIV-1-infected and CD4+ target cells are likely to contain a VS functional for Gag transfer and that the average functional VS lifetime is probably >1 h.
FIG. 3.

Life span of the R5 HIV-1 VS. (A) JktBaL cells prestained with CTO (red) and activated primary CD4+ T cells prestained with CFSE (green) were mixed in a 1:1 ratio and seeded onto MatTek poly-d-lysine-coated glass-bottom tissue culture dishes. Cells were imaged by time-lapse LSCM, with one frame acquired every 30 s for 250 min. A representative frame is shown with red, green, and differential interference contrast merged, with cell-cell interactions indicated by yellow arrows. (B) Same two-color image as in panel A after setting the threshold, binarizing the green and red images, and dilating the individual cell profiles as described in Materials and Methods. Cell-cell interaction sites are visible as yellow crescents. (C) The interactions are isolated in x-y two-dimensional frames as discrete white regions. Individual x-y frames were stacked together into a three-dimensional box, allowing tracking of the individual events over time. (D) The test and control series were processed as described in Materials and Methods to give the duration of each cell contact, which were grouped into the temporal categories. (E) The events falling into the >10-min category for the test movie are depicted as individual points, with the red line representing the mean duration in min.
FIG. 4.

Assembly dynamics of the R5 HIV-1 VS. (A) A301.R5 target cells prelabeled with the nonblocking CD4 MAb L120 were mixed with JktBaL cells for the times shown before fixing, permeabilizing, and labeling with the appropriate secondary reagent for CD4 (░⃞) or with the appropriate primary and secondary reagents for Gag (▩) or Env (▪). Labeling and colocalization of all three markers in the same samples was also carried out (□). The data represent the combined percentage values from multiple conjugates counted in randomly selected low-power fields, and error bars represent + 1 SD. *, P < 0.05. (B) Same as in panel A but the percentage of VS in which Gag was observed independently of Env in the target cell was divided by the number of VS (□) or by the total number of conjugate interfaces (▩).
Inhibition of HIV-1 cell-cell spread in preformed VS.
With this knowledge we next addressed the question of whether inhibitors added after conjugate formation interfere with R5 HIV-1-driven VS assembly. Since this concept has not been interrogated for X4 HIV-1 spread across VS, we investigated this in parallel, basing time of addition on the data reported previously (17) that demonstrate X4 HIV-1 VS assembly reaching a plateau in ca. 30% of conjugates by 3 h after infected-target cell mixing. JktBaL- or JktIIIB-A3.01.R5 conjugates were prepared, and inhibitors at a single inhibitory concentration (10 μg/ml for antibodies, 50 μM for PRO 2000, and 1 μM for TAK 779 and AMD3100, which had previously been shown to be close to 100% inhibitory when added at 0 h [Fig. 2A]), were added either at the time of cell mixing or at 1 h after cell mixing when ca. 30% of VS had formed (Fig. 4A). Conjugates were then fixed at 3 h when VS frequency was at a plateau (Fig. 4). Multiple fields were scored for the percentage of conjugates forming VS, and pooled data are shown in Fig. 5. Approximately 30% of JktBaL-A3.01.R5 conjugates formed VS over the 3 h time course in these experiments (Fig. 5A), a finding consistent with the data reported in Fig. 4A, and a very similar number was also observed for JktIIIB-A3.01.R5 conjugates (Fig. 5B). When added at T = 0, R5 HIV-1-mediated VS assembly was most strongly inhibited by blockers of the CD4-gp120 interaction such as B12 (∼60% inhibition) and Q4120 (∼80% inhibition), as previously observed for the X4 VS (17). In contrast, antagonists of the gp120-CCR5 interaction (TAK779 and PRO 2000) were more weakly inhibitory. When added after 1 h of coculture, VS inhibition was significantly reduced for B12 and Q4120, whereas no significant increase in VS number was observed with the other inhibitors. Thus, once formed, the R5 VS appears moderately resistant to disassembly by HIV-1 receptor blockers. In contrast, the effect of CD4-gp120 or CXCR4-gp120 antagonists on the X4 HIV-1 VS was more pronounced. Whether added at 0 or 1 h, all inhibitors were ∼75 to 100% effective at inhibiting VS assembly, suggesting that the preformed X4 VS may be susceptible to disassembly by antagonists of gp120-receptor interactions. Taken together, these data imply that R5 VS assembly is less easily inhibited than the X4 VS by blockers of gp120-receptor interactions, whether applied before or during VS formation. The relative resistance of the R5 VS structure to inhibition may represent an inability of inhibitors to access a preformed VS or, alternatively, may demonstrate that preformed VS structure may be maintained independent of function in terms of HIV-1 cell-cell spread. To investigate these possibilities, we chose a single high-molecular-mass (∼150-kDa) inhibitor, the NMAb B12, for further analysis. Since B12 prevents most VS assembly when introduced at the time of infected-uninfected cell mixing, we hypothesized that the presence of B12 within a VS would most likely signify incorporation of NMAb into a preexisting VS. JktBaL and A3.01 cells were cocultured for 1 h, which results in at least 30% of total VS formation (Fig. 4A), prior to the addition of B12 to the culture, and the cultures were maintained for a further 2 h prior to fixing and labeling for LSCM. Figure 6A shows a representative conjugate in which B12 has robustly labeled the conjugate interface and colocalizes with CD4 on the target cell and viral Gag in the infected cell. These data support the notion that inhibitors of relatively high molecular mass can enter a VS without disassembling the supramolecular structure. To investigate the functional effects of inhibitors on preformed R5 HIV-1 VS, we set up a experiment similar to that described for Fig. 6A with the same concentrations of inhibitors added either at the time of cell mixing or 1 or 3 h later but lysed the cells after a total of 12 h of culture and analyzed them for the relative pol signal. The addition of inhibitors at the time of mixing resulted in 70% inhibition of the relative pol signal (Fig. 6), a finding consistent with the results in Fig. 2. Taken together, the results presented in Fig. 5 and 6 suggest that inhibitors can either prevent VS assembly if present during initial infected cell-target cell contact or if introduced subsequent to VS formation can enter intact VS and inhibit viral transfer into the target cell and subsequent reverse transcription.
FIG. 5.

Time of inhibitor addition effects on VS formation. (A) Target A301.R5 cells prelabeled with the nonblocking CD4 MAb L120 were mixed 1:1 with JktBaL and inhibitor was either added simultaneously (white bars) or after 1 h of cell coculture (black bars). The hatched bars represent VS assembly in the absence of inhibitor (Non-Treated Control, NTC). Cells were cultured for a total of 3 h before fixing, permeabilizing and labeling for Gag and Env. Randomly selected conjugate interfaces were analyzed for colocalization of the three markers defined as VS, as described for Fig. 4. The data are means of values from three independent experiments each carried out in triplicate, and error bars represent SEM. (B) As for (A) except that the infected cells were JktIIIB. P < 0.05.
FIG. 6.
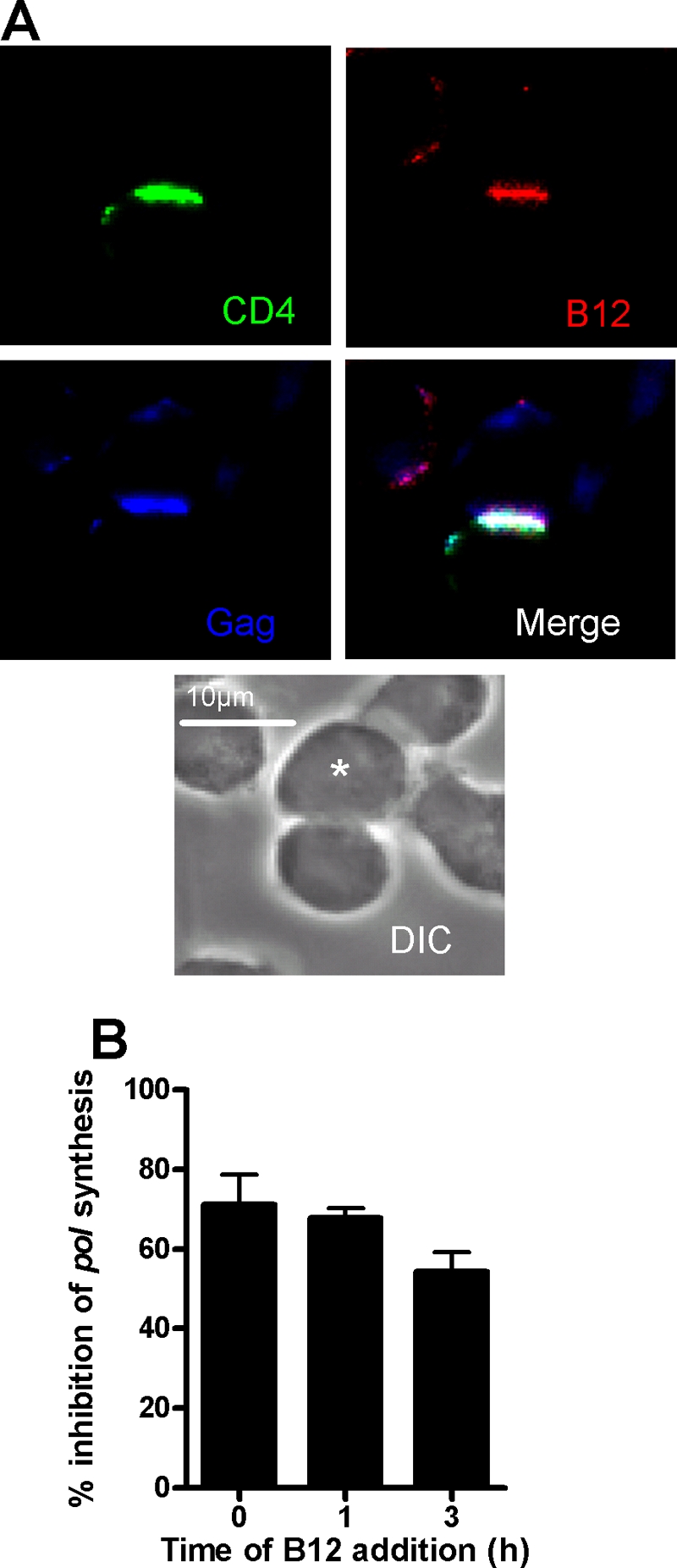
Effects of NMAb B12 on HIV-1 infection across VS. (A) Target A301.R5 cells prelabeled with the nonblocking CD4-specific MAb L120 were mixed with JktBaL cells and cocultured on poly-l-lysine-coated coverslips. After 1 h of coculture, B12 was gently added to yield a final concentration of 10 μg/ml. The cells were cocultured for a further 2 h and then fixed, permeabilized, and labeled for B12 with anti-human IgG-TRITC and for Gag with anti-p24 antiserum, and Gag and CD4 were labeled with the appropriate secondary detection reagents. Coverslips were mounted and analyzed by LSCM. The asterisk labels the infected cell. (B) Target A301.R5 cells were mixed 1:1 with JktBal cells in the presence of 10 μg of B12/ml (T = 0), or the same concentration of B12 was added 1 h or 3 h after cell mixing. Cells were then cultured for a total of 12 h prior to lysis and qPCR for pol and β-globin products. The results are expressed as the percent inhibition of relative pol synthesis compared to no inhibitor or mixtures of JktBaL cells with the nonpermissive A201 target cell control. Each bar represents the mean data from three independent experiments carried out in triplicate, and error bars represent the SEM.
Ultrastructure of the VS.
Although the data shown in Fig. 5 and 6 are suggestive of a VS structure permeable to inhibitors, this is not unequivocal since some inhibitor may conceivably be incorporated into forming VS without necessarily blocking further assembly. We therefore used electron tomography to probe the structure of mature R5 and X4 HIV-1 VS. Conventional thin-section electron microscopy yields ultrastructural details of cellular interactions in two dimensions but is unable to resolve the three-dimensional details of a complex intercellular interface. We therefore carried out electron tomography of serial thick sections spanning ∼1.2 μm total thickness, of epoxy-embedded conjugates between infected Jurkat cells and primary CD4+ T cells fixed 3 h after cell mixing. The associated single-section movies are Movies S2 and S3 in the supplemental material. Figure 7 shows single slices from representative conjugates formed between JktIIIB (Fig. 7A to C) or JktBaL (Fig. 7D and E) and primary CD4+ T target cells. The infected cells were identified based on the presence of budding structures in HIV-1IIIB (Fig. 7B and C)- and HIV-1BaL (Fig. 7E)-infected cells. The plasma membrane interfaces for the two VS are clearly defined, with multiple virions and virus-like vesicles present at the interface between the cells but few points of obvious intracellular contact. Three-dimensional models reconstructed from the tomographic analysis of the serial sections are shown in Fig. 7F and G. It is evident that the VS interface for both virus types is complex with multiple membrane invaginations and projections, and yet the interface between the cells appears loosely structured. A large number of X4 HIV-1 virions are present in gaps between the two cells, many of which are adherent to the target cell plasma membrane, presumably via Env-receptor and/or adhesion interactions. Fewer virions can be seen in the R5 HIV-1-infected VS, and most of these are either free or are associated with the infected cell membrane, possibly reflecting a lower avidity for target cell receptors than the X4 virions. The infected-target cell plasma membranes are relatively distant (>100 nm) over most of the synapse but are punctuated by limited areas of close membrane apposition. Analysis of the distance between the target cell and the nearest infected cell plasma membrane with virions removed to simplify analysis revealed regions that were 30 nm or closer (Fig. 7H and I), a finding consistent with integrin interactions such as those which stabilize the immunological synapse (41) and implicated in X4 HIV-1 VS structure and function (18). These discrete adhesive regions were typically concentrated between interlocking villi surrounded by large areas of noninteracting plasma membrane. We have observed that the plasma membranes are often dramatically ruffled or invaginated (e.g., target cell membrane in Fig. 7D), which might lead to the interpretation, from a single section, of a large internal virus-containing compartment. However, the renderings in Fig. 7G and I imply that this is not a closed compartment but open to the external milieu, and many virions can be seen at the edges of and outside the zone of cell membrane contact. Despite analysis of multiple tomographic reconstructions, we have yet to observe virus within a closed compartment within the target T cell. Nevertheless, since the sections that we have analyzed generally span only a proportion of the entire VS area, we cannot exclude the possibility that some virus is internalized into internal compartments within the target cell. In conclusion, these models reveal a porous intercellular interface containing mature virus particles that would allow access of even high-molecular-weight compounds to virus without the imposition of obvious steric constraints.
DISCUSSION
Enveloped viruses from several different families use direct passage between cells to propagate themselves (39), and HIV-1 is no exception. We found here using three different assays that cell-cell spread is approximately an order of magnitude more efficient than cell-free spread, a figure that agrees with one recent study (42) but disagrees with another (6). We believe that the discrepancy comes from the viral endpoint of cell-cell spread measured: the Sourisseau study assayed for productive infection by measuring Gag release into the supernatant, whereas the Chen study quantified Gag transfer into target cells. Gag transfer does not necessarily correspond to infection, particularly since the authors report that much of the Gag appears to be endocytosed by the target cell, an entry pathway that may result in a high proportion of noninfectious events (2, 4, 6, 15). Like the Sourisseau study, we assayed for a proximal surrogate of productive viral infection, synthesis of late viral DNA product, which we believe is most likely to represent an infectious outcome in the target cell. Divergent experimental conditions may also influence the rate and extent of viral transfer between T cells, including the ratio of infected to uninfected T cells, the chronicity of the infection in the donor cells, and the type of cell, or cell line used.
The mechanism of enhanced efficiency of cell-cell spread compared to cell-free spread of HIV-1 remains to be formally elucidated. Studies by others (8, 16, 32, 38) and our data presented here and previously published (17), however, suggest that there may be several factors determining efficiency of viral dissemination in these systems. (i) The first factor is the proximity of the target and effector cells. Since the infected and uninfected cells are in relatively close and stable apposition during cell-cell spread, the rate-limiting factor of fluid-phase diffusion is essentially eliminated. (ii) Receptor clustering is an additional factor. The VS is defined as the polarization of CD4 and coreceptor to the site of infected-target cell contact. This would allow de novo budded HIV-1 to engage an optimal number of receptors rapidly without the delay implicit in receptor recruitment required for fusion that is imposed upon individual cell surface-associated virions (21). (iii) Finally, we must consider the MOIs of target cells. The rate of spread of HIV-1 in culture is directly related to the MOI (8). Although we have not been able to quantify the number of infectious virions traversing a VS, the MOI via VS will be much higher than that occurring via cell-free spread because of both the increased total number of particles engaged by the target cell and the reduced time-dependent inactivation of viruses undergoing cell-cell spread compared to cell-free spread. We intend that the assays used here reflect as far as possible the in vivo situation regarding viral spread between lymphocytes. One obvious difference is that our cultures are ∼100-fold less dense than the estimated lymphocyte density in vivo (45). Such a high in vivo density of lymphocytes would increase the chances of both cell-cell and cell-free infection occurring, however, making it difficult to model how this might affect the differential efficiency of viral spread by the two modes.
Our demonstration that a panel of HIV-1 entry inhibitors interferes equivalently with cell-cell and cell-free HIV-1 infection when inhibitors are added at the time of mixing target cells with infected cells or free virus suggests that VS-mediated spread of HIV-1 between T cells is unlikely to be an antibody or drug evasion strategy for the virus. In most scenarios in vivo, the inhibitor (antibody or therapeutic entry inhibitor) would be present prior to addition of virus to cells and so would not have to enter a preformed VS in order to interfere with cell-cell viral spread. However, even if inhibitor was added subsequent to encounter between a virus-infected T cell and an uninfected T cell, our data suggest that such an inhibitor could access the preformed VS and prevent viral cell-cell spread. These results are encouraging for use of prophylactic vaccines designed to elicit neutralizing antibodies and for entry inhibitors applied in a prophylactic or therapeutic setting. Our results showing that all entry inhibitors tested target both X4 and R5 HIV-1 cell-cell spread similarly to diffusion-limited spread are inconsistent with certain of the data obtained in T-cell systems by others (2-4, 6). We consider that the most likely explanation again relates to systems measuring viral antigen uptake by endocytic pathways that are resistant to viral coreceptor antagonists and fusion inhibitors, but their contribution to productive infection is unclear. In this respect the same group that reported an absence of inhibition of VS-mediated Gag transfer by coreceptor antagonists in one study (6) also reported inhibition by the same agent in a later study when a better surrogate of viral infection (transcription of HIV-1 long-terminal-repeat-driven green fluorescent protein) was measured (15). Although when using electron microscopy or tomography we have not observed evidence of endocytic uptake of HIV-1 by primary CD4+ T cells across a VS in the present study or previously (17), our analyses do not exclude this as an outcome of cell-cell transfer of virus. However, we assume that such events may be rare in primary CD4+ T cells compared to the immortalized cell lines used predominantly in other analyses (2-4, 6).
Our measurements of uninfected CD4+ T-cell-T-cell interactions are consistent with previously described intravitally measured life spans of homotypic T-cell interactions (35) and confirm that T cells undergo transient adhesive interactions under normal conditions. A proportion of HIV-1-infected T cells undergo longer interactions, however, and we assume that a percentage of these relates to VS assembly and viral cell-cell transfer. A small minority of events lasted >300 min: these may be very long-lived interactions, perhaps resulting from high-level infection of the donor T-cell leading to enhanced cell-cell interactions, and are consistent with observed conjugate life spans (18 to 32 h) reported earlier (15). Alternatively, these long-lived interactions may represent a small proportion of conjugates undergoing membrane fusion to form syncytia.
Although the electron tomographic analyses carried out here and thin-section electron microscopy carried out previously (17) reveal the T-cell VS to be loosely structured, other cell types may assemble VS-like structures with distinct functional and structural features. We recently described a VS formed between HIV-1-infected macrophages and CD4+ T cells: thin-section electron microscopic analysis revealed a broad interaction surface between the cells that had the appearance of a tight junction but that formed only transiently (11). A study evaluating the ultrastructure of the VS formed between human T cell leukemia virus type 1 and permissive target T cells also found large surfaces of tightly apposed membrane with occasional pockets containing virions (24). Thus, the type of VS assembled is likely to be both cell and virus type specific, and this may influence sensitivity of this mode of viral spread to inhibition. Indeed, in this respect, dendritic cell transfer of HIV-1 to CD4+ T cells in trans across infectious synapses has been demonstrated to be resistant to a variety of NMAb (10, 46). Further comparative analyses of VS formed between different cell types, and their sensitivity to NAb and entry inhibitors are therefore warranted.
Supplementary Material
Acknowledgments
We thank the electron microscopy core facility at EMBL Heidelberg for equipment and assistance with electron tomography; Sylvain Lacomble for assistance with image processing; and Leo Kong, Becky Russell, Will Hillson, and Kate Gartlan for proofreading the manuscript.
This study was supported by the Medical Research Council UK, the International AIDS Vaccine Initiative Neutralizing Antibody Consortium, EUROPRISE, and Fondation Dormeur. S.W. and the electron tomography part of this study were funded by Wellcome Trust grant H5RCYV0 to Stephen Fuller and Deutsche Forschungsgemeinschaft grant SPP1175 to J.A.G.B. Q.J.S. is a Jenner Institute Fellow.
Footnotes
Published ahead of print on 20 January 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Baba, M., O. Nishimura, N. Kanzaki, M. Okamoto, H. Sawada, Y. Iizawa, M. Shiraishi, Y. Aramaki, K. Okonogi, Y. Ogawa, K. Meguro, and M. Fujino. 1999. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. U. S. A. 96:5698-5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanco, J., B. Bosch, M. T. Fernandez-Figueras, J. Barretina, B. Clotet, and J. A. Este. 2004. High level of coreceptor-independent HIV transfer induced by contacts between primary CD4 T cells. J. Biol. Chem. 279:51305-51314. [DOI] [PubMed] [Google Scholar]
- 3.Bosch, B., J. Blanco, E. Pauls, I. Clotet-Codina, M. Armand-Ugon, B. Grigorov, D. Muriaux, B. Clotet, J. L. Darlix, and J. A. Este. 2005. Inhibition of coreceptor-independent cell-to-cell human immunodeficiency virus type 1 transmission by a CD4-immunoglobulin G2 fusion protein. Antimicrob. Agents Chemother. 49:4296-4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosch, B., B. Grigorov, J. Senserrich, B. Clotet, J. L. Darlix, D. Muriaux, and J. A. Este. 2008. A clathrin-dynamin-dependent endocytic pathway for the uptake of HIV-1 by direct T cell-T cell transmission. Antivir. Res. 80:185-193. [DOI] [PubMed] [Google Scholar]
- 5.Burton, D. R., J. Pyati, R. Koduri, S. J. Sharp, G. B. Thornton, P. W. Parren, L. S. Sawyer, R. M. Hendry, N. Dunlop, P. L. Nara, et al. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024-1027. [DOI] [PubMed] [Google Scholar]
- 6.Chen, P., W. Hubner, M. A. Spinelli, and B. K. Chen. 2007. Predominant mode of HIV transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 81:12582-12595. [DOI] [PMC free article] [PubMed]
- 7.Chenine, A., Q. J. Sattentau, and M. Moulard. 2000. Selective HIV-1-induced downmodulation of CD4 and coreceptors. Arch. Virol. 145:455-471. [DOI] [PubMed] [Google Scholar]
- 8.Dimitrov, D. S., R. L. Willey, H. Sato, L. J. Chang, R. Blumenthal, and M. A. Martin. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 67:2182-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Favoreel, H. W., G. Van Minnebruggen, G. R. Van de Walle, J. Ficinska, and H. J. Nauwynck. 2006. Herpesvirus interference with virus-specific antibodies: bridging antibodies, internalizing antibodies, and hiding from antibodies. Vet. Microbiol. 113:257-263. [DOI] [PubMed] [Google Scholar]
- 10.Ganesh, L., K. Leung, K. Lore, R. Levin, A. Panet, O. Schwartz, R. A. Koup, and G. J. Nabel. 2004. Infection of specific dendritic cells by CCR5-tropic human immunodeficiency virus type 1 promotes cell-mediated transmission of virus resistant to broadly neutralizing antibodies. J. Virol. 78:11980-11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groot, F., S. Welsch, and Q. J. Sattentau. 2008. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111:4660-4663. [DOI] [PubMed] [Google Scholar]
- 12.Gupta, P., R. Balachandran, M. Ho, A. Enrico, and C. Rinaldo. 1989. Cell-to-cell transmission of human immunodeficiency virus type 1 in the presence of azidothymidine and neutralizing antibody. J. Virol. 63:2361-2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haase, A. 1999. Population biology of HIV-1 infection: viral and CD4+ T-cell demographics and dynamics in lymphatic tissue. Annu. Rev. Immunol. 17:625-656. [DOI] [PubMed] [Google Scholar]
- 14.Healey, D., L. Dianda, J. P. Moore, J. S. McDougal, M. J. Moore, P. Estess, D. Buck, P. D. Kwong, P. C. L. Beverley, and Q. J. Sattentau. 1990. Novel anti-CD4 monoclonal antibodies separate human immunodeficiency virus infection and fusion of CD4+ cells from virus binding. J. Exp. Med. 172:1233-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubner, W., G. P. McNerney, P. Chen, B. M. Dale, R. E. Gordon, F. Y. Chuang, X. D. Li, D. M. Asmuth, T. Huser, and B. K. Chen. 2009. Quantitative 3D video microscopy of HIV transfer across T-cell virological synapses. Science 323:1743-1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson, D., and M. Huber. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 76:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jolly, C., K. Kashefi, M. Hollinshead, and Q. J. Sattentau. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199:283-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jolly, C., I. Mitar, and Q. J. Sattentau. 2007. Adhesion molecule interactions facilitate human immunodeficiency virus type-1-induced virological synapse formation between T cells. J. Virol. 81:13916-13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jolly, C., I. Mitar, and Q. J. Sattentau. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 81:5547-5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.kremer, J., D. Mastronarde, and M. J. R. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116:71-76. [DOI] [PubMed] [Google Scholar]
- 21.Kuhmann, S. E., E. Platt, S. Kozak, and D. Kabat. 2000. Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J. Virol. 74:7005-7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law, M., R. Hollinshead, and G. L. Smith. 2002. Antibody-sensitive and antibody-resistant cell-to-cell spread by vaccinia virus: role of the A33R protein in antibody-resistant spread. J. Gen. Virol. 83:209-222. [DOI] [PubMed] [Google Scholar]
- 23.Levy, J. A. 2009. HIV pathogenesis: 25 years of progress and persistent challenges. AIDS 23:147-160. [DOI] [PubMed] [Google Scholar]
- 24.Majorovits, E., M. Nejmeddine, Y. Tanaka, G. P. Taylor, S. D. Fuller, and C. R. Bangham. 2008. Human T-lymphotropic virus-1 visualized at the virological synapse by electron tomography. PLoS One 3:e2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin, N., and Q. Sattentau. 2009. Cell-to-cell HIV-1 spread and its implications for immune evasion. Curr. Opin. HIV AIDS 4:143-149. [DOI] [PubMed] [Google Scholar]
- 26.Massanella, M., I. Puigdomenech, C. Cabrera, M. T. Fernandez-Figueras, A. Aucher, G. Gaibelet, D. Hudrisier, E. Garcia, M. Bofill, B. Clotet, and J. Blanco. 2009. Anti-gp41 antibodies fail to block early events of virological synapses but inhibit HIV spread between T cells. AIDS 23:183-188. [DOI] [PubMed] [Google Scholar]
- 27.Mastronarde, D. 2005. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152:36-51. [DOI] [PubMed] [Google Scholar]
- 28.Matthews, T., M. Salgo, M. Greenberg, J. Chung, R. DeMasi, and D. Bolognesi. 2004. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 3:215-225. [DOI] [PubMed] [Google Scholar]
- 29.Moore, J. P., S. G. Kitchen, P. Pugach, and J. A. Zack. 2004. The CCR5 and CXCR4 coreceptors-central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retrovir. 20:111-126. [DOI] [PubMed] [Google Scholar]
- 30.Muster, T., F. Steindl, M. Purtscher, A. Trkola, A. Klima, G. Himmler, F. Ruker, and H. Katinger. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67:6642-6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pettersen, E., T. Goddard, C. Huang, G. Couch, D. Greenblatt, E. Meng, and T. Ferrin. 2004. UCSF Cimera: a visualization system for exploratory research and analysis. J. Comput. Biol. 25:1605-1612. [DOI] [PubMed] [Google Scholar]
- 32.Phillips, D. 1994. The role of cell-to-cell transmission in HIV infection. AIDS 8:719-731. [DOI] [PubMed] [Google Scholar]
- 33.Rudnicka, D., J. Feldmann, F. Porrot, S. Wietgrefe, S. Guadagnini, M. C. Prevost, J. Estaquier, A. T. Haase, N. Sol-Foulon, and O. Schwartz. 2009. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J. Virol. 83:6234-6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggiero, E., R. Bona, C. Muratori, and M. Federico. 2008. Virological consequences of early events following cell-cell contact between human immunodeficiency virus type 1-infected and uninfected CD4+ cells. J. Virol. 82:7773-7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabatos, C. A., J. Doh, S. Chakravarti, R. S. Friedman, P. G. Pandurangi, A. J. Tooley, and M. F. Krummel. 2008. A synaptic basis for paracrine interleukin-2 signaling during homotypic T-cell interaction. Immunity. 29:238-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sachdev, D. D., B. Zerhouni-Layachi, M. Ortigoza, A. T. Profy, M. Tuen, C. E. Hioe, and M. E. Klotman. 2009. The differential binding and activity of PRO 2000 against diverse HIV-1 envelopes. J. Acquir. Immune Defic. Syndr. 51:125-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanders, R. W., M. Venturi, L. Schiffner, R. Kalyanaraman, H. Katinger, K. O. Lloyd, P. D. Kwong, and J. P. Moore. 2002. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 76:7293-7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato, H., J. Orenstein, D. Dimitrov, and M. Martin. 1992. Cell-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 186:712-724. [DOI] [PubMed] [Google Scholar]
- 39.Sattentau, Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815-826. [DOI] [PubMed] [Google Scholar]
- 40.Scanlan, C. N., R. Pantophlet, M. R. Wormald, E. Ollmann Saphire, R. Stanfield, I. A. Wilson, H. Katinger, R. A. Dwek, P. M. Rudd, and D. R. Burton. 2002. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α1→2 mannose residues on the outer face of gp120. J. Virol. 76:7306-7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sims, T. N., and M. L. Dustin. 2002. The immunological synapse: integrins take the stage. Immunol. Rev. 186:100-117. [DOI] [PubMed] [Google Scholar]
- 42.Sourisseau, M., N. Sol-Foulon, F. Porrot, F. Blanchet, and O. Schwartz. 2007. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 81:1000-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sowinski, S., C. Jolly, O. Berninghausen, M. A. Purbhoo, A. Chauveau, K. Kohler, S. Oddos, P. Eissmann, F. M. Brodsky, C. Hopkins, B. Onfelt, Q. Sattentau, and D. M. Davis. 2008. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 10:211-219. [DOI] [PubMed] [Google Scholar]
- 44.Timpe, J. M., Z. Stamataki, A. Jennings, K. Hu, M. J. Farquhar, H. J. Harris, A. Schwarz, I. Desombere, G. L. Roels, P. Balfe, and J. A. McKeating. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17-24. [DOI] [PubMed] [Google Scholar]
- 45.Trepel, F. 1974. Number and distribution of lymphocytes in man: a critical analysis. Klin. Wochenschr. 52:511-515. [DOI] [PubMed] [Google Scholar]
- 46.van Montfort, T., A. A. Nabatov, T. B. Geijtenbeek, G. Pollakis, and W. A. Paxton. 2007. Efficient capture of antibody neutralized HIV-1 by cells expressing DC-SIGN and transfer to CD4+ T lymphocytes. J. Immunol. 178:3177-3185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.