

Abstract
The Nav1.2 and Nav1.3 voltage-gated sodium channel isoforms demonstrate distinct differences in their kinetics and voltage dependence of fast inactivation when expressed in Xenopus oocytes. Co-expression of the auxiliary β1 subunit accelerated inactivation of both the Nav1.2 and Nav1.3 isoforms, but it did not eliminate the differences, demonstrating that this property is inherent in the α subunit. By constructing chimeric channels between Nav1.2 and Nav1.3, we demonstrate that the carboxyl terminus is responsible for the differences. The Nav1.2 carboxyl terminus caused faster inactivation in the Nav1.3 backbone, and the Nav1.3 carboxyl terminus caused slower inactivation in the Nav1.2 channel. Through analysis of truncated channels, we identified a homologous 60-amino acid region within the carboxyl terminus of the Nav1.2 and the Nav1.3 channels that is responsible for this modulation of fast inactivation. Site-directed replacement of Nav1.3 lysine 1826 in this region to its Nav1.2 analogue glutamic acid 1880 (K1826E) shifted the voltage dependence of inactivation toward that of Nav1.2. The K1826E mutation also accelerated the inactivation kinetics to a level comparable with that of Nav1.2. The reverse Nav1.2 E1880K mutation exhibited much slower inactivation kinetics and depolarized inactivation voltage dependence. A complementary mutation located within the inactivation linker of Nav1.3 (K1453E) caused inactivation changes mirroring those caused by the K1826E mutation in Nav1.3. Therefore, we have identified a homologous carboxyl-terminal residue that regulates the kinetics and voltage dependence of fast inactivation in sodium channels, possibly via a charge-dependent interaction with the inactivation linker.
Keywords: Ion Channels, Membrane Proteins, Oocyte, Protein Chimeras, Sodium Channels, Carboxyl Terminus, Isoforms, Structure-Function, Voltage Clamp, Voltage-dependent
Introduction
Sodium channels depolarize the resting membrane of many excitable cell types to initiate and propagate action potentials by acting as the molecular pores for sodium ion influx (1). The voltage-dependent activation and inactivation events basic to channel gating are highly regulated to ensure normal channel function, which is vital to skeletal muscle contraction, neuronal signaling, and cardiac conduction. Small changes in channel inactivation lead to diseases such as myotonia, paralysis, epilepsy, long QT syndrome, idiopathic ventricular fibrillation, heart failure, or other disorders (2–14).
There are nine mammalian voltage-gated sodium channel isoforms, all of which have a similar molecular structure. Each channel consists of an ion-conducting α subunit associated with one or more accessory β subunits (15). The α subunit is a 260-kDa four-domain transmembrane protein. Each domain contains six transmembrane segments (S1–S6), with the S4 segments acting as voltage sensors and the hairpin-like S5-S6P loops forming the channel pore. Fast inactivation is mediated by the intracellular loop between DIII and DIV, which occludes the inner pore following channel activation.
Because of their roles in action potential firing, the multiple sodium channel isoforms could contribute to diversity in physiological functions. Numerous studies investigating the mechanisms underlying isoform-specific inactivation and the physiological effects of channelopathies have not yet yielded a consensus model concerning the role of the different isoforms. Chimeras created by the exchange of the carboxyl termini between Nav1.5 and Nav1.4 (16) and between Nav1.5 and Nav1.2 (17) suggested that inactivation is affected by the carboxyl-terminal tails. However, the results were mixed regarding how the carboxyl terminus determined channel-specific inactivation.
In this report, we investigated two temporally distinct isoforms expressed in the central nervous system. Nav1.3 is mainly embryonic, whereas Nav1.2 expression is maintained at a high level throughout all developmental stages (18–22). Biophysically, the Nav1.2 channel shows marked differences compared with the Nav1.3 channel in terms of fast inactivation in Xenopus oocytes (23, 24). Through analysis of chimeric channels, we showed that the carboxyl terminus is required to transfer the inactivation characteristics of the donor channel. Through analysis of truncated channels, we identified a 60-residue region in the carboxyl terminus that was responsible for this modulation. Within this region, a single homologous amino acid residue (Glu1880 in Nav1.2 and Lys1826 in Nav1.3) was both necessary and sufficient for the specific inactivation of each channel.
EXPERIMENTAL PROCEDURES
Molecular Cloning
The wild-type rat Nav1.3 cDNA coding region was cloned in a pLCT2 low copy vector (pNa3T). The wild-type rat Nav1.2 cDNA coding region was cloned in a pLCT1 vector (pNa200). Chimeric channels were constructed by exchanging restriction fragments produced either by existing vector or linker-created restriction sites. The 2233-3C chimera was constructed by swapping the 3.12-kb ClaI-NotI pNa3T fragment into the pNa200 vector digested with BglII and NotI restriction enzymes and modified by adding a ClaI linker to the BglII end. 3322-2C was constructed by ligating the 3.549-kb BglII-NotI pNa200 fragment into the BglII linker-added NotI-ClaI pNa3T vector. 2223-3C and 3332-2C chimeras were cloned by exchanging the BstEII-NotI fragments that span from the beginning of the DIV S1 to the end of the carboxyl terminus. Carboxyl-terminal chimeras 2222-3C and 3333-2C were created by swapping the BsrGI-NotI fragment covering the entire carboxyl terminus and part of the carboxyl region of DIV S6. DIV chimeras 2223-2C and 3332-3C were created by switching a BstEII-BsrGI fragment covering most of the DIV S1 to most of the DIV S6. The DIII chimera 2232-2C was constructed by swapping in the BstEII-NotI fragment from pNa200 into the 2233-3C fragment digested with the same restriction enzymes.
To construct the mutations, PCR-based site-directed mutagenesis was performed on the carboxyl-terminal regions (BstEII-NotI fragments) of the rNav1.3 and rNav1.2 channels in pGem18B. Two consecutive in-frame stop codons (TAATAA) were created immediately after amino acids 1912, 1902, 1882, 1862, 1842, and 1822 to produce carboxyl-terminal truncated Nav1.3 CΔ129, Nav1.3 Δ109, Nav1.3 CΔ89, Nav1.3 CΔ69, Nav1.3 CΔ49, and Nav1.3 CΔ39 channels, respectively. Similarly, Nav1.2 truncated channels were created using two consecutive stop codons after positions 1966, 1956, 1936, 1916, 1896, and 1876. PCR products were sequenced using big dye chemistry on an ABI DNA sequencer (Laguna Scientific, Laguna Niguel, CA) to confirm the presence of the desired mutations and the absence of any additional mutations, and the BstEII-NotI regions were cloned back into the full-length rNav1.3 and rNav1.2 plasmids.
Expression and Electrophysiology
In vitro transcription of NotI-linearized DNA templates using the T7 mMESSAGE mMACHINE kit (Ambion, Austin, TX) was utilized to produce RNA. The quality of the transcribed mRNA was confirmed by glyoxal gel analysis. Stage V oocytes were removed from adult female Xenopus laevis frogs and prepared as described previously (25). Oocytes were incubated in ND-96 medium, which consisted of 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, and 5 mm HEPES, pH 7.5, supplemented with 0.1 mg/ml gentamicin, 0.55 mg/ml pyruvate, and 0.5 mm theophylline. RNA encoding rNav1.2, rNav1.3, and chimeric channels was injected at ∼20–50 ng/oocyte to obtain current amplitudes between 0.8 and 6 μA. When the channels were coexpressed with the β1 or β2 subunits, a 1:10 molar ratio of α to β RNA was injected. Oocytes were incubated at 20 °C for >40 h in ND-96 before voltage clamping.
Sodium currents were recorded using the two-electrode voltage clamp OC-725 (Warner Instruments, Hamden, CT) at room temperature, with a DigiData 1321A interface (Molecular Devices, Sunnyvale, CA) and pCLAMP 8.0 software (Molecular Devices) as described previously (26, 27). Currents were recorded in ND-96 without supplements in the absence and presence of 400 nm tetrodotoxin. Capacitive transients were eliminated by subtraction of currents recorded in tetrodotoxin.
The voltage dependence of activation was analyzed using a step protocol in which oocytes were depolarized for 100 ms from a holding potential of −100 mV to a range of potentials from −95 mV to +50 mV in 5-mV increments. Peak currents were normalized to the maximum peak current and plotted against voltage. To calculate a reversal potential, the resulting I-V curve of each data set was individually fit with the equation,
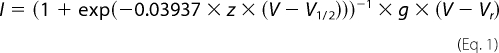 |
where I is the current amplitude, z is the apparent gating charge, V is the potential of the given pulse, V½ is the half-maximal voltage, g is a factor related to the number of open channels during the given pulse, and Vr is the reversal potential. Conductance was then calculated directly using the equation,
where G represents conductance, and I, V, and Vr are as described above. The conductance values were fit with the two-state Boltzmann equation,
where z is the apparent gating charge, V is the potential of the given pulse, and V½ is the potential for half-maximal activation.
Current amplitude at the 50 ms time point was measured by dividing the average current between the 50 and 51 ms times by the peak current during a depolarization to −10 mV.
The voltage dependence of inactivation was determined using a two-step protocol in which a conditioning pulse was applied from a holding potential of −100 mV to a range of potentials from −100 mV to +15 mV in 5-mV increments for 100 ms, immediately followed by a test pulse to −5 mV. The peak current amplitudes during the subsequent test pulses were normalized to the peak current amplitude during the first test pulse, plotted against the potential of the conditioning pulse, and fit with a two-state Boltzmann equation,
where I is equal to the test pulse current amplitude, V is the potential of the conditioning pulse, V½ is the voltage for half-maximal inactivation, and a is the slope factor.
Entry into inactivation was analyzed using a two-pulse protocol in which a conditioning pulse to +10 mV was applied from a holding potential of −100 mV, beginning with a duration of 0 ms and increasing by 2 ms with each consecutive sweep, to elicit inactivation. This was immediately followed by a test pulse to −5 mV to determine the fraction of current that had been inactivated. Current amplitudes recorded during subsequent test pulses were normalized to the initial test pulse and plotted against the time of the conditioning pulse. Data for the Nav1.3, Nav1.3-derived chimeric α subunits alone, Nav1.3 + calmodulin (CaM),2 and Nav1.3 + CaM1234 were fit from the 2 ms time point instead of the 0 ms time point because a slight potentiation of less than 1% was observed in several oocytes. This effect was not observed for Nav1.2 or Nav1.2-derived chimeras or in the presence of the β1 subunit. In the case of α + β1, these data were then fit with the double exponential equation,
where I is the current, AFast and ASlow are the relative proportions of current inactivating with the time constants τFast and τSlow, and t is the period of the conditioning pulse. When α subunits were expressed alone, the data were fit with the single exponential equation,
Recovery from inactivation was analyzed using three separate two-pulse protocols. Each protocol began with a conditioning depolarization from a holding potential of −100 to −5 mV for 50 ms, which inactivated >95% of the channels. This was followed by a decreasing recovery time interval at −100 mV and a test depolarization to −5 mV. The three protocols differed only in the maximum length of recovery time and the time interval by which that recovery period decreased: 25-ms maximum and 1-ms decrements in the short protocol, 200-ms maximum and 5-ms decrements in the intermediate protocol, and 3000-ms maximum and 100-ms decrements in the long protocol. Fractional recovery was calculated by dividing the maximum current amplitude during the test pulse by the maximum current amplitude of the corresponding conditioning pulse. The recovery data were fit with either the double exponential equation,
or the triple exponential equation,
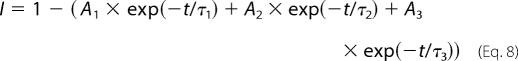 |
in which A1, A2, and A3 are the relative percentages of current that recovered with the time constants τ1, τ2, and τ3, and t is the recovery time.
Use-dependent inactivation was analyzed at a frequency of 39 Hz using 17.5-ms depolarizations to −10 mV from a holding potential of −100 mV. The protocol was carried out for 2.56 s, which was long enough for the current to have reached equilibrium. Peak current amplitudes were normalized to the peak current amplitude during the first depolarization and plotted against pulse number.
The kinetics of fast inactivation were determined from currents elicited during the same step protocol used for the voltage dependence of activation, in which oocytes were depolarized for 100 ms from a holding potential of −100 mV to a range of potentials from −95 to +50 mV in 5-mV increments. Inactivation time constants were determined using the Chebyshev method to fit each trace with either the single exponential equation,
or the double exponential equation,
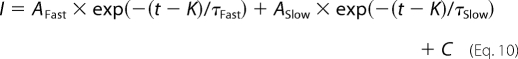 |
where I is the current, AFast and ASlow are the relative proportions of current inactivating with the time constants τFast and τSlow, K is the time shift, and C is the steady-state asymptote. The time shift was selected manually as the point at which the macroscopic current began to inactivate exponentially.
RESULTS
Fast Inactivation Kinetics of Nav1.2 and Nav1.3
We characterized the biophysical differences between the Nav1.2 and Nav1.3 sodium channel isoforms using Xenopus oocytes, a standard and highly controlled heterologous system for studying ion channel function. Wild-type Nav1.2 and Nav1.3 sodium channels were expressed as α subunits alone and in the presence of the auxiliary β1 subunit. Fig. 1A shows sample macroscopic current traces for the Nav1.2, Nav1.3, Nav1.2 plus β1, and Nav1.3 plus β1 channels recorded during a 100-ms depolarization to −10 mV from a holding potential of −100 mV. In the absence of β1, Nav1.3 sodium current showed a significantly slower decaying phase compared with Nav1.2 at all voltages tested. As a result of the slower inactivation, Nav1.3 demonstrated a residual current that was more than 5-fold higher than that for Nav1.2 (19.6 ± 5.2% for Nav1.3 compared with 3.4 ± 2.9% for Nav1.2) at −10 mV (Fig. 1B).
FIGURE 1.

Differences between Nav1.2 and Nav1.3. A, representative sodium currents recorded at room temperature using the two-electrode voltage clamp on Xenopus oocytes expressing either wild-type Nav1.2, Nav1.3, Nav1.2 + β1, or Nav1.3 + β1. Traces shown were elicited by a −10 mV depolarization from a holding potential of −100 mV and normalized to the peak current amplitude. B, the percentage of residual current was determined by dividing the average current during the last 10 ms of each depolarization by the peak current of the corresponding trace. Sample sizes were 6 for Nav1.2, 8 for Nav1.3, 6 for Nav1.2 + β1, and 5 for Nav1.3 + β1. C, the voltage dependences of inactivation (circles) and activation (triangles) for the wild-type Nav1.2 and Nav1.3 channels expressed as α subunits alone. D, the voltage dependences of inactivation (circles) and activation (triangles) for the wild-type Nav1.2 and Nav1.3 channels expressed as α + β1. The data points represent means, and the error bars indicate S.D. values. The data in C and D were fit with a two-state Boltzmann equation as described under “Experimental Procedures,” and the parameters of the fits are shown in Table 1.
Co-expression of β1 accelerated inactivation of both the Nav1.2 and Nav1.3 isoforms, consistent with previous results examining the effect of the β1 subunit on sodium channel currents in oocytes (28–31). Despite this acceleration, Nav1.3 + β1 still inactivated more slowly than Nav1.2 + β1 (Fig. 1A). However, residual currents at the end of the 100-ms depolarization pulse were comparable, with 5.2 ± 2.9% for Nav1.2 + β1 and 5.8 ± 3.0% for Nav1.3 + β1 (Fig. 1B).
To quantify the difference in kinetics between the Nav1.2 and Nav1.3 channels, inactivation time constants were determined by fitting the current traces at −10 mV with either a single exponential equation or a double exponential equation. In the case of Nav1.3, a single exponential equation fit very well, resulting in a single time constant that was termed τSlow. The rapidly inactivating component of the Nav1.2 current required a two-component exponential equation, resulting in both τSlow and τFast. The τSlow for Nav1.3 was significantly slower than that for Nav1.2, as shown in Table 1. Because co-expression of β1 accelerated the kinetics of Nav1.3 inactivation, two time constants were required to fit those currents. Both the τSlow and τFast time constants for Nav1.3 + β1 were significantly slower that those for Nav1.2 + β1 (Table 1).
TABLE 1.
Voltage dependence and inactivation kinetics parameters of Nav1.2, Nav1.3, and chimeric channels
Values presented are means ± S.D.
| Channel | Voltage dependence of fast inactivation |
Voltage dependence of activation |
Kinetics of fast inactivation (at −10 mV) |
Entry into inactivation |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V½ | a | n | V½ | Z | n | τSlow | ASlow | τFast | AFast | n | τSlow | ASlow | τFast | AFast | n | |
| mV | mV | mV | e0 | ms | % | ms | % | ms | % | ms | % | |||||
| Nav1.2 | −36.0 ± 3.2 | 8.0 ± 0.6 | 8 | −13.1 ± 1.4 | 5.0 ± 0.3 | 6 | 17.6 ± 3.0 | 38 ± 4 | 4.2 ± 0.9 | 62 ± 4 | 8 | 4.9 ± 0.5 | 97.2 ± 2.8 | NAa | NA | 7 |
| Nav1.3 | −16.2 ± 0.8 | 4.7 ± 0.5 | 8 | −14.6 ± 2.4 | 6.9 ± 0.6 | 8 | 28.4 ± 4.0b | 100 | NA | NA | 14 | 14.8 ± 1.4b | 100 | NA | NA | 6 |
| Nav1.2 + β1 | −44.9 ± 5.0 | 8.5 ± 1.6 | 8 | −13.6 ± 1.4 | 4.4 ± 0.1 | 6 | 9.3 ± 3.0 | 20 ± 7 | 0.9 ± 0.2 | 80 ± 7 | 5 | 3.4 ± 1.5 | 31.2 ± 2.8 | 0.7 ± 0.4 | 63.2 ± 3.2 | 5 |
| Nav1.3 + β1 | −27.8 ± 2.5c | 8.5 ± 0.7 | 12 | −16.2 ± 0.8 | 5.8 ± 0.6 | 5 | 27.5 ± 3.7c | 43 ± 9c | 2.2 ± 0.5c | 57 ± 9c | 9 | 10.6 ± 3.0c | 88.8 ± 8.0 | NA | NA | 8 |
| 2233-3C | −22.3 ± 2.0 | 5.1 ± 0.8 | 12 | −16.3 ± 2.3 | 6.1 ± 0.4 | 6 | 24.1 ± 2.9 | 100 | NA | NA | 7 | 12.2 ± 0.9 | 100 | NA | NA | 5 |
| 2232-2C | −37.6 ± 2.6 | 9.0 ± 0.5 | 8 | −15.2 ± 1.9 | 5.2 ± 0.4 | 7 | 21.6 ± 4.2 | 26 ± 4 | 5.8 ± 0.9 | 74 ± 4 | 8 | 5.4 ± 0.7 | 98.8 ± 2.2 | NA | NA | 9 |
| 2223-3C | −20.6 ± 2.0 | 4.6 ± 0.4 | 10 | −17.2 ± 2.5 | 6.6 ± 0.6 | 9 | 23.0 ± 2.6 | 100 | NA | NA | 6 | 10.8 ± 3.0 | 100 | NA | NA | 5 |
| 2223-2C | −31.5 ± 0.8 | 7.2 ± 0.4 | 8 | −12.5 ± 2.0 | 5.9 ± 0.8 | 8 | 22.8 ± 2.4 | 38 ± 6 | 6.5 ± 1.2 | 62 ± 6 | 8 | 5.4 ± 0.4 | 100 | NA | NA | 8 |
| 2222-3C | −22.3 ± 1.0 | 5.4 ± 0.4 | 11 | −16.4 ± 1.6 | 5.8 ± 0.4 | 8 | 27.4 ± 4.5 | 100 | NA | NA | 5 | 9.9 ± 1.5 | 100 | NA | NA | 11 |
| 3322-2C | −34.2 ± 1.1 | 7.8 ± 0.8 | 5 | −11.8 ± 1.5 | 5.4 ± 0.4 | 8 | 21.5 ± 1.6 | 42 ± 7 | 5.7 ± 0.7 | 58 ± 7 | 5 | 6.3 ± 0.8 | 100 | NA | NA | 6 |
| 3332-2C | −34.6 ± 2.6 | 9.4 ± 0.5 | 8 | −14.4 ± 2.0 | 5.4 ± 0.4 | 8 | 27.4 ± 4.9 | 24 ± 4 | 6.5 ± 1.9 | 76 ± 4 | 5 | 6.8 ± 0.8 | 100 | NA | NA | 8 |
| 3333-2C | −37.4 ± 2.6 | 9.9 ± 1.7 | 7 | −16.2 ± 2.7 | 5.6 ± 0.6 | 5 | 27.6 ± 5.8 | 18 ± 13 | 7.5 ± 1.8 | 82 ± 13 | 5 | 8.2 ± 0.5 | 100 | NA | NA | 8 |
| 3332-3C | −22.6 ± 1.4 | 7.4 ± 0.8 | 11 | −14.3 ± 1.2 | 5.4 ± 0.3 | 9 | 37.7 ± 11.0 | 100 | NA | NA | 6 | 5.2 ± 0.2 | 100 | NA | NA | 5 |
a NA, not applicable because current trace was efficiently fitted with a single exponential equation.
b Significantly different from Nav1.2, p < 0.0001.
c Significantly different from Nav1.2 + β1, p < 0.001.
Voltage Dependence of Nav1.2 and Nav1.3
The Nav1.2 and Nav1.3 isoforms demonstrated clear differences in the voltage dependence of inactivation when expressed as α subunits alone, with the voltage dependence of Nav1.3 being ∼20 mV more positive compared with Nav1.2 (Fig. 1C and Table 1). The voltage dependence of inactivation was affected by the presence of the β1 subunit. With β1, negative shifts of 8.9 and 11.6 mV were observed for the V½ of inactivation of Nav1.2 and Nav1.3, respectively (Fig. 1D and Table 1). Because the V½ of both channels was shifted in the same direction and by a similar magnitude, the large difference (17.1 mV) in voltage dependence of inactivation between the two isoforms remained. Therefore, although the β1 subunit accelerated fast inactivation, it did not eliminate the inactivation differences between the Nav1.2 and Nav1.3 isoforms.
Because inactivation is coupled to activation in sodium channels (32–34), we tested whether the inactivation differences between Nav1.2 and Nav1.3 could be accounted for by differences in their activation gating. In the absence of β1, the half-activation potentials (V½) and slope values for the normalized conductance curves were comparable for Nav1.2 and Nav1.3 (Fig. 1C and Table 1). In addition, the β1 subunit did not significantly alter the voltage dependence of activation of either the Nav1.2 or Nav1.3 isoform (Fig. 1D and Table 1). Thus, the inactivation differences between Nav1.2 and Nav1.3 did not result from differences in the voltage dependence of activation.
Rates of Entry into and Recovery from Inactivation of Nav1.2 and Nav1.3
Differences in steady-state inactivation may result from gating steps preceding inactivation and/or changes in steps following inactivation. To determine if either of these processes was responsible for the differences in inactivation, we examined the rates of entry into and recovery from inactivation and use-dependent inactivation. Nav1.2 channels entered the inactivated state much faster than Nav1.3 in both the absence and presence of β1 (Fig. 2A and Table 1). β1 accelerated entry rates of both channels without eliminating the differences. As a consequence of the slower rate of entry into inactivation, Nav1.3 required 50 ms to reach ∼85% inactivation with or without β1, whereas Nav1.2 reached 85% inactivation at 5 ms in the presence of β1 and at 11 ms in the absence of β1.
FIGURE 2.

Rates of entry into inactivation, recovery from fast inactivation, and use-dependent inactivation for wild-type Nav1.2 and Nav1.3 channels. A, entry into inactivation was analyzed for wild-type Nav1.2 and Nav1.3 channels expressed as α subunits alone (circles) and as α + β1 subunits (triangles). The symbols represent means, and the error bars indicate S.D. values. The parameters of the fits are shown in Table 1. B, recovery from inactivation was determined using three two-pulse protocols, and the data were fit with either a triple or a double exponential equation, as described under “Experimental Procedures.” The symbols represent means, and the error bars indicate S.D. values. Sample sizes were 5 for Nav1.2, 8 for Nav1.3, 6 for Nav1.2 + β1, and 11 for Nav1.3 + β1. C, use-dependent inactivation was analyzed at 39 Hz for wild-type Nav1.2 and Nav1.3. Currents were elicited during 17.5-ms depolarizations to −10 mV from a holding potential of −100 mV, normalized to the initial peak current amplitude, and plotted against the start time of the first depolarization in the pulse train. The values shown are means, and the error bars show S.D. values. Sample sizes were 8 for Nav1.2, 8 for Nav1.3, 6 for Nav1.2 + β1, and 8 for Nav1.3 + β1.
The Nav1.2 and Nav1.3 isoforms differed slightly in their rates of recovery from inactivation in the absence of β1, but both displayed a slow triphasic pattern (Fig. 2B). The recovery time constants for Nav1.3 were τ1 = 12.4 ± 4.4 ms, τ2 = 92.2 ± 29.5 ms, and τ3 = 491.8 ± 164.2 ms, and the constants for Nav1.2 were τ1 = 4.6 ± 1.2 ms, τ2 = 103.1 ± 56.4 ms, and τ3 = 649.9 ± 280.0 ms. Both the Nav1.2 and Nav1.3 channels exhibited faster biphasic recovery in the presence of β1 (Fig. 2B), consistent with the reported role of β1 in oocytes (28–31). As a result of this acceleration, the differences in recovery between Nav1.2 and Nav1.3 were eliminated, indicating that these two channels do not differ significantly in recovery from inactivation.
An alternative way to evaluate recovery from inactivation is to examine use-dependent inactivation. Because the recovery time courses were similar, we expected that use-dependent inactivation of the Nav1.2 and Nav1.3 isoforms would also be similar. At 39 Hz, both Nav1.2 and Nav1.3 reached equilibrium at about 20% of the initial peak current amplitude. The β1 subunit reduced use-dependent inactivation of both channels to about 80% of the peak current (Fig. 2C), consistent with the acceleration of recovery from inactivation.
The Carboxyl Terminus Is Responsible for the Inactivation Differences between Nav1.2 and Nav1.3
Because inactivation differences between Nav1.2 and Nav1.3 were not eliminated in the presence of β1, we predicted that these differences result from sequence differences within the α subunit. To identify which structural region is responsible for the differences, we swapped domains between Nav1.2 and Nav1.3, as shown in Fig. 3. The chimeric channels were named by the origin of each domain (I–IV) and the carboxyl terminus. For example, 2233-3C consisted of domains I and II from Nav1.2 and domains III and IV and the carboxyl terminus from Nav1.3. All of the chimeric channels expressed comparable levels of current when expressed as α subunits in Xenopus oocytes. They were analyzed with respect to the properties that differed between Nav1.2 and Nav1.3 (voltage dependence of activation, inactivation, and kinetics of inactivation).
FIGURE 3.

Schematic diagram of the mutant channels. A, representation of the different channel domains and the restriction endonucleases that were used to make the chimeras between Nav1.2 and Nav1.3. B, diagram of the chimeras indicating the origin from either Nav1.2 (black) or Nav1.3 (white). C, amino acid sequence alignment of the carboxyl-terminal region for Rattus norvegicus (r), Homo sapiens (h), and Mus musculus (m) Nav1.3 and Nav1.2 sodium channels. Black, dark gray, and gray boxes indicate identical, highly conserved, and conserved residues, respectively. Green boxes illustrate Nav1.3-specific residues, and yellow boxes illustrate Nav1.2-specific residues. Asterisks indicate amino acid differences between rat Nav1.3 and Nav1.2 isoforms, including charge-changing differences (red asterisks). The sites of the truncations are indicated in red.
There was a correlation between inactivation kinetics and the carboxyl terminus. The chimeric channels in which the carboxyl terminus was from Nav1.2 (2232-2C, 2223-2C, 3322-2C, and 3332-2C) exhibited faster kinetics of inactivation like Nav1.2 (Table 1). Even the chimera in which only the carboxyl terminus was from Nav1.2 (3333-2C) displayed inactivation kinetics like Nav1.2 (Table 1). In contrast, chimeric channels with the Nav1.3 carboxyl terminus (2233-3C, 2223-3C, and 2222-3C) exhibited slower kinetics like Nav1.3, with a single time constant of inactivation (Table 1). These results suggest that the carboxyl terminus was responsible for the differences in inactivation kinetics.
The voltage dependence of inactivation was modulated in a similar manner as the kinetics. Chimeras retaining the Nav1.2 carboxyl terminus displayed voltage dependence of inactivation similar to Nav1.2 (Fig. 4, A–C). Chimeras retaining the Nav1.3 carboxyl terminus displayed voltage dependence of inactivation similar to Nav1.3. Substitution of just the Nav1.2 channel carboxyl terminus with that from Nav1.3 (2222-3C) caused a 13.7-mV positive shift in inactivation V½ toward that of Nav1.3 (Fig. 4C and Table 1). The V½ of this chimera was not shifted completely to that of Nav1.3, although the carboxyl terminus of Nav1.2 fully shifted the Nav1.3 voltage dependence of inactivation to that of Nav1.2 in 3333-2C (Fig. 4C and Table 1).
FIGURE 4.

Voltage dependence of inactivation and entry into inactivation for the chimeric channels. A, the voltage dependence of inactivation for the Nav1.2-Nav1.3 chimeras. B, the voltage dependence of inactivation for the Nav1.3-Nav1.2 chimeras. C, the voltage dependence of inactivation for the wild-type Nav1.2, wild-type Nav1.3, chimeric 2222--3C, and chimeric 3333-2C channels. D, entry into inactivation for the wild-type Nav1.2, wild-type Nav1.3, chimeric 2222-3C, and chimeric 3333-2C channels. All channels were expressed as α subunits alone. The values shown are means, and the error bars show S.D. values. The parameters of the fits are shown in Table 1.
The rates of entry into inactivation were similarly shifted by swapping just the carboxyl termini of the two isoforms (Fig. 4D and Table 1). In contrast, none of the chimeras showed significant differences in the voltage dependence of activation compared with the parental Nav1.2 and Nav1.3 channels (Table 1). Thus, the carboxyl terminus by itself was responsible for the majority of the differences in inactivation between Nav1.2 and Nav1.3.
60-Residue Region in the Carboxyl Terminus Modulates Fast Inactivation
To identify the specific residues in the carboxyl terminus that were responsible for the modulation of fast inactivation, we constructed successive deletions from the carboxyl terminus. The carboxyl termini of Nav1.2 and Nav1.3 are 229 amino acids, of which the proximal 100 residues are identical in the two isoforms (Fig. 3C). Therefore, we systematically truncated each terminus from the distal end in increments ranging from 39 to 129 residues. All of the truncated channels were functional, and the shortest deletion that caused significant changes in inactivation was 89 residues (Fig. 5, A and B, and Table 2). Larger deletions resulted in faster inactivation kinetics, with the CΔ129 truncated channels showing the fastest inactivation (Fig. 5, A and B). In addition, the voltage dependence of inactivation was shifted in the negative direction with successively larger deletions from 89 to 129 residues in Nav1.3 (Fig. 5C and Table 2). The Nav1.2 CΔ89 and CΔ129 truncated channels showed similar negative shifts, but the Nav1.2 CΔ109 channel demonstrated wild-type voltage dependence (Fig. 5D and Table 2). The voltage dependence of activation was not significantly affected by any of the truncations (Table 2). These results suggest that fast inactivation in both Nav1.3 and Nav1.2 is slowed by a homologous 60-residue region (1823–1883 using Nav1.3 numbering) that modulates fast inactivation.
FIGURE 5.

A, sample sodium current traces for Nav1.3 carboxyl-terminal truncated channels. B, sample sodium current traces for Nav1.2 carboxyl-terminal truncated channels. Shown are current traces for wild-type (black) and truncated CΔ89 (red), CΔ109 (green), and CΔ129 (blue) channels during a −10 mV depolarization from a holding potential of −100 mV, as described under “Experimental Procedures.” The amplitude of each trace was normalized to the peak amplitude, and the traces were shifted manually to align the peak amplitudes. C, the voltage dependence of inactivation curves for truncated CΔ89 (triangles), CΔ109 (squares), and CΔ129 (diamonds) and wild-type (circles) Nav1.3 channels. D, the voltage dependence of inactivation curves for truncated CΔ89 (triangles), CΔ109 (squares), and CΔ129 (diamonds) and wild-type (circles) Nav1.2 channels. Voltage dependence of inactivation was determined using a two-step protocol as described under “Experimental Procedures.” The parameters of the fits and sample sizes are shown in Table 2. Data points indicate means, and error bars show S.D. values.
TABLE 2.
Voltage dependence and inactivation kinetics parameters of truncated channels
Values presented are means ± S.D.
| Channel | Voltage dependence |
Kinetics of inactivation |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Inactivation |
Activation |
||||||||||
| V½ | a | n | V½ | z | n | τSlow | Aslow | τFast | AFast | n | |
| mV | mV | mV | e0 | ms | % | ms | % | ||||
| Nav1.2 | −36.0 ± 3.2 | 8.0 ± 0.6 | 8 | −13.4 ± 1.6 | 5.2 ± 0.4 | 6 | 17.6 ± 3.0 | 38 ± 4 | 4.2 ± 0.9 | 62 ± 4 | 8 |
| Nav1.2CΔ39 | −36.8 ± 1.7 | 8.2 ± 0.6 | 8 | −14.6 ± 0.4 | 4.8 ± 0.4 | 6 | 15.6 ± 1.9 | 40 ± 3 | 5.0 ± 1.0 | 60 ± 3 | 6 |
| Nav1.2CΔ49 | −36.0 ± 2.0 | 8.1 ± 0.6 | 15 | −15.0 ± 1.0 | 4.6 ± 0.4 | 12 | 15.3 ± 1.4 | 44 ± 2 | 3.6 ± 0.7 | 56 ± 2 | 9 |
| Nav1.2CΔ69 | −36.0 ± 1.3 | 8.0 ± 0.4 | 17 | −14.1 ± 0.5 | 4.5 ± 0.6 | 10 | 16.9 ± 1.4 | 34 ± 5 | 4.8 ± 0.5 | 66 ± 5 | 8 |
| Nav1.2CΔ89 | −42.3 ± 1.3a | 7.6 ± 0.4 | 11 | −13.3 ± 2.2 | 3.8 ± 0.6 | 6 | 15.6 ± 1.5 | 17 ± 1 | 3.3 ± 0.2 | 79 ± 1 | 6 |
| Nav1.2CΔ109 | −36.6 ± 1.0 | 7.9 ± 0.4 | 8 | −17.4 ± 1.1 | 5.2 ± 0.6 | 7 | 16.6 ± 1.9 | 23 ± 6 | 4.3 ± 0.4 | 77 ± 6 | 5 |
| Nav1.2CΔ129 | −40.8 ± 1.4b | 7.8 ± 0.6 | 8 | −17.8 ± 2.0 | 5.0 ± 0.2 | 8 | 14.9 ± 0.9 | 16 ± 2 | 4.1 ± 0.4 | 84 ± 2 | 9 |
| Nav1.3 | −16.2 ± 0.8 | 4.7 ± 0.5 | 8 | −14.6 ± 2.4 | 5.2 ± 0.4 | 6 | 28.4 ± 4.0 | 100 | NAc | NA | 14 |
| Nav1.3CΔ39 | −18.2 ± 1.0 | 4.5 ± 0.4 | 9 | −13.4 ± 1.0 | 6.8 ± 0.6 | 8 | 26.5 ± 5.4 | 100 | NA | NA | 6 |
| Nav1.3CΔ49 | −18.9 ± 1.2 | 4.5 ± 0.6 | 12 | −14.9 ± 1.4 | 7.2 ± 1.3 | 11 | 26.6 ± 3.1 | 100 | NA | NA | 5 |
| Nav1.3CΔ69 | −18.5 ± 1.2 | 4.8 ± 0.4 | 17 | −11.7 ± 1.6 | 5.8 ± 0.4 | 11 | 26.2 ± 3.2 | 100 | NA | NA | 7 |
| Nav1.3CΔ89 | −24.2.± 1.8d | 6.5 ± 0.4e | 11 | −12.2 ± 2.2 | 6.2 ± 0.4 | 11 | 35.9 ± 2.7 | 34 ± 2 | 11.6 ± 0.7 | 66 ± 2 | 5 |
| Nav1.3CΔ109 | −27.0 ± 0.8d | 7.3 ± 0.5e | 5 | −10.6 ± 3.0 | 6.2 ± 1.0 | 5 | 34.6 ± 10.5 | 38 ± 11 | 11.2 ± 4.3 | 62 ± 11 | 4 |
| Nav1.3CΔ129 | −30.6 ± 0.7d | 7.2 ± 0.2e | 15 | −11.1 ± 1.3 | 6.0 ± 0.4 | 10 | 27.1 ± 3.4 | 28 ± 5 | 8.0 ± 1.0 | 72 ± 5 | 9 |
a Significantly different from Nav1.2, p < 0.00003.
b Significantly different from Nav1.2, p < 0.003.
c NA, not applicable because current trace was efficiently fitted with a single exponential equation.
d Significantly different from Nav1.3, p < 0.00000001.
e Significantly different from Nav1.3, p < 0.0000002.
A Single Carboxyl-terminal Amino Acid Is Necessary for Nav1.3-type Inactivation
There are 14 amino acid differences between Nav1.3 and Nav1.2 in this 60-residue region (Fig. 3C). To determine which of those residues was important for the inactivation differences, we replaced each of the 14 amino acids that was different in Nav1.3 with the corresponding amino acid from Nav1.2. These experiments were performed using the Nav1.3 background because the effects on inactivation were larger in Nav1.3 than in Nav1.2. The effects of the mutations either individually or in groups are shown in Table 3. The majority of the mutant channels retained the Nav1.3 voltage dependence of inactivation. For example, a channel with substitutions at 1856, 1857, and 1861 was equivalent to Nav1.3 (A1856I/I1857V/N1861A), as was a channel with eight substitutions at 1864, 1870, 1871, 1873, 1874, 1877, 1879, and 1881 (CRLNIKD-E1881D). Including a ninth substitution in that channel (CRLNIKDE-T1882K) resulted is a negative shift of 4 mV in the voltage dependence of inactivation toward that of Nav1.2. However, the most dramatic effect was obtained by replacing a single lysine at position 1826 with glutamic acid (K1826E), which caused a shift of −11.3 mV (Fig. 6A and Table 3). We tested the effects of the K1826E substitution in combination with substitutions at nearby residues (K1826E/D1827E and K1826E/D1827E/A1856I/I1857V), and the results were similar to those for K1826E alone (Table 3). None of the mutations caused a significant change in the voltage dependence of activation. These results suggest that Lys1826 is the major determinant of Nav1.3-like voltage dependence of inactivation.
TABLE 3.
Voltage dependence and inactivation kinetics parameters for carboxyl terminal mutants
Values presented are means ± S.D.
| Channela | Voltage dependence |
Kinetics of inactivation |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Inactivation |
Activation |
||||||||||
| V½ | a | n | V½ | z | n | τSlow | ASlow | τFast | AFast | n | |
| mV | mV | mV | e0 | ms | % | ms | % | ||||
| Nav1.3 | −16.2 ± 0.8 | 4.7 ± 0.5 | 8 | −14.6 ± 2.4 | 6.9 ± 0.6 | 8 | 28.4 ± 4.0 | 100 | NAb | NA | 14 |
| K1826E | −27.5 ± 1.6c | 8.3 ± 1.0c | 14 | −14.0 ± 1.0 | 6.2 ± 0.2 | 5 | 42.9 ± 5.8 | 38 ± 4 | 9.8 ± 1.4 | 62 ± 4 | 5 |
| K1826D | −28.6 ± 1.6c | 8.6 ± 0.5c | 10 | −15.4 ± 0.7 | 6.9 ± 0.6 | 9 | 50.8 ± 10.6 | 34 ± 9 | 11.3 ± 1.5 | 66 ± 9 | 9 |
| K1826R | −18.3 ± 1.2 | 4.8 ± 0.9 | 12 | −14.0 ± 1.4 | 5.7 ± 0.8 | 6 | 25.5 ± 3.2 | 100 | NA | NA | 6 |
| K1826A | −25.3 ± 1.3c | 7.1 ± 0.4c | 11 | −12.5 ± 2.2 | 5.4 ± 0.4 | 8 | 38.5 ± 7.2 | 31 ± 6 | 12.0 ± 2.3 | 69 ± 6 | 6 |
| D1827E | −18.2 ± 2.1 | 4.6 ± 0.7 | 8 | −12.3 ± 0.4 | 5.6 ± 0.1 | 5 | 41.0 ± 1.7 | 100 | NA | NA | 5 |
| A1856I | −21.1 ± 1.6c | 5.6 ± 0.9 | 7 | −21.0 ± 1.8c | 8.0 ± 1.1 | 7 | 24.7 ± 3.8 | 100 | NA | NA | 7 |
| A1856I/I1857V | −18.2 ± 0.9 | 4.5 ± 0.5 | 17 | −16.4 ± 1.5 | 8.8 ± 1.6 | 12 | 22.4 ± 1.8 | 100 | NA | NA | 8 |
| N1861A | −17.4 ± 0.7 | 4.8 ± 0.6 | 7 | −13.4 ± 2.7 | 6.2 ± 0.9 | 6 | 29.8 ± 5.9 | 100 | NA | NA | 5 |
| A1856I/I1857V/N1861A | −17.6 ± 1.2 | 3.9 ± 0.4 | 13 | −15.7 ± 2.9 | 8.3 ± 1.8 | 5 | 24.4 ± 3.5 | 100 | NA | NA | 7 |
| C1864R | −18.4 ± 1.3 | 4.6 ± 0.5 | 17 | −15.5 ± 2.0 | 6.8 ± 0.7 | 18 | 25.4 ± 2.5 | 100 | NA | NA | 8 |
| C1864R/R1870K | −16.6 ± 1.4 | 4.6 ± 0.4 | 18 | −14.4 ± 0.6 | 6.9 ± 0.7 | 9 | 26.9 ± 1.5 | 100 | NA | NA | 6 |
| C1864R/R1870K/L1871V | −18.0 ± 0.9 | 4.7 ± 0.4 | 11 | −14.3 ± 1.2 | 6.2 ± 0.4 | 8 | 24.6 ± 2.1 | 100 | NA | NA | 6 |
| CRL-N1873K | −19.1 ± 1.7 | 4.2 ± 0.2 | 9 | −20.0 ± 1.3c | 6.8 ± 2.0 | 9 | 21.4 ± 1.3 | 100 | NA | NA | 5 |
| CRLN-I1874V | −17.2 ± 0.9 | 4.2 ± 0.4 | 6 | −14.5 ± 2.0 | 7.2 ± 0.8 | 6 | 25.2 ± 2.8 | 100 | NA | NA | 5 |
| CRLNI-K1877I | −17.0 ± 1.4 | 4.0 ± 0.2 | 6 | −18.3 ± 0.8 | 7.1 ± 0.8 | 5 | 26.8 ± 2.5 | 100 | NA | NA | 5 |
| CRLNIK-D1879K | −18.2 ± 0.8 | 4.8 ± 0.4 | 6 | −14.2 ± 0.9 | 6.5 ± 0.2 | 8 | 29.6 ± 3.2 | 100 | NA | NA | 8 |
| CRLNIKD-E1881D | −17.8 ± 0.9 | 4.2 ± 0.2 | 11 | −15.6 ± 0.8 | 7.9 ± 0.9 | 8 | 22.4 ± 2.8 | 100 | NA | NA | 7 |
| CRLNIKDE-T1882K | −20.2 ± 1.1c | 4.2 ± 0.2 | 13 | −15.6 ± 1.4 | 7.4 ± 0.9 | 12 | 21.2 ± 2.2 | 100 | NA | NA | 7 |
| K1826E/D1827E | −25.3 ± 0.7c | 8.1 ± 0.4c | 5 | −16.4 ± 2.7 | 10.0 ± 1.8 | 5 | 43.2 ± 3.9 | 25 ± 2 | 8.0 ± 0.4 | 75 ± 2 | 5 |
| D1827E/A1856I/I1857V | −18.2 ± 1.3 | 5.0 ± 0.6 | 6 | −13.8 ± 1.3 | 6.2 ± 0.6 | 5 | 39.1 ± 10.6 | 100 | NA | NA | 5 |
| K1826E/D1827E/A1856I/I1857V | −28.2 ± 0.9c | 8.5 ± 0.8c | 8 | −14.2 ± 2.3 | 6.0 ± 0.6 | 5 | 32.2 ± 2.5 | 35 ± 11 | 6.7 ± 1.3 | 64 ± 11 | 5 |
| Nav1.2 | −36.0 ± 3.2c | 8.0 ± 0.6c | 8 | −13.4 ± 1.6 | 5.2 ± 0.4 | 6 | 17.6 ± 3.0 | 38 ± 4 | 4.2 ± 0.9 | 62 ± 4 | 8 |
| E1880K | −22.8 ± 2.8d | 6.5 ± 0.7d | 13 | −17.4 ± 1.4 | 5.2 ± 0.2 | 9 | 22.7 ± 3.9 | 100 | NA | NA | 9 |
| E1881D | −31.8 ± 1.8d | 7.1 ± 0.4d | 9 | −14.6 ± 0.8 | 5.7 ± 0.3 | 5 | 27.2 ± 5.1 | 38 ± 4 | 7.4 ± 0.4 | 62 ± 4 | 5 |
| E1880K/E1881D | −20.8 ± 1.7d | 5.2 ± 0.3d | 5 | −16.0 ± 1.3 | 5.9 ± 0.2 | 5 | 22.3 ± 3.6 | 100 | NA | NA | 5 |
a Mutant channels are indicated by the amino acid substitution and by the initial amino acid in subsequent lines (e.g. C1864R/R1870K/L1871V is CRL).
b NA, not applicable because current trace was efficiently fitted with a single exponential equation.
c Significantly different from Nav1.3, p < 0.001.
d Significantly different from Nav1.2, p < 0.001.
FIGURE 6.

Fast inactivation properties regulated by a single carboxyl-terminal residue. A, the voltage dependence of inactivation for Nav1.3 K1826E (squares) and Nav1.2 E1880K (triangles) is shown compared with the corresponding Nav1.3 (white circles) and Nav1.2 (black circles) wild-type channels. The parameters of the fits and sample sizes are shown in Table 3. B, normalized current versus the depolarization time is shown for entry into inactivation, as described under “Experimental Procedures.” The parameters of the fits and sample sizes are shown in Table 3. C, recovery from inactivation was determined using three two-pulse protocols and fitted as described under “Experimental Procedures.” Inset, recovery is shown for time points between 200 and 1000 ms on a linear scale. Sample sizes were 9 for Nav1.2, 6 for Nav1.3, 5 for Nav1.2 E1880K, and 11 for Nav1.3 K1826E. D, use-dependent inactivation was analyzed at 39 Hz as described under “Experimental Procedures.” Inset, entry rate is shown for the last two time points in the protocol. Sample sizes were 9 for Nav1.2, 6 for Nav1.3, 5 for Nav1.2 E1880K, and 6 for Nav1.3 K1826E. E, the voltage dependence of inactivation for Nav1.3 K1826D (triangles), Nav1.3 K1826A (squares), Nav1.3 K1826R (diamonds), and wild-type Nav1.3 (white circles) is shown. F, the voltage dependence of inactivation for Nav1.2 E1881D (triangles), Nav1.2 E1880K/E1881D (squares), and wild-type Nav1.2 (black circles) is shown. The parameters of the fits and sample sizes are shown in Table 3. Data points indicate means, and error bars show S.D. values.
Because K1826E was a charge reversal mutation, it seemed likely that charge played a significant role in determining the inactivation properties of Nav1.2 and Nav1.3. To test the effect of charge at this position, we substituted aspartic acid at the same position (K1826D). This mutation caused a negative shift in the voltage dependence of inactivation that was comparable with that of K1826E (Fig. 6E and Table 3). A substitution that neutralized the charge at the same position (K1826A) caused a slightly smaller shift, and a substitution that preserved the charge (K1826R) caused no significant shift in the voltage dependence of inactivation (Fig. 6E and Table 3). These results demonstrate that a positively charged residue at position 1826 was necessary for the Nav1.3 voltage dependence of inactivation.
Inactivation of Nav1.3 during a depolarization to −10 mV is slow and can be fit with a single exponential equation, in contrast to the rapidly inactivating Nav1.2 current that is best fit with a double exponential equation (Table 3). The single time constant for Nav1.3 inactivation is slower than the Nav1.2 slow time constant at potentials more negative than 0 mV, although it is faster than the Nav1.2 slow time constant at potentials greater than 0 mV (data not shown). The Nav1.3 mutant K1826E demonstrated significantly faster inactivation compared with wild-type Nav1.3, and the current decay could only be fit with a double exponential equation (Table 3). In addition, a similar fraction of current inactivated with each time constant for Nav1.3 K1826E as for Nav1.2. K1826E was the only substitution to have this effect because none of the other mutations resulted in a second inactivation time constant (Table 3).
An Acidic Residue at the Comparable Position Is Necessary for Nav1.2-type Inactivation
Because the basic lysine at position 1826 was critical for slower inactivation of Nav1.3, we examined the importance of the acidic residue at the comparable position in Nav1.2 (Glu1880) by replacing it with a positively charged lysine (E1880K). The lysine substitution shifted the voltage dependence of Nav1.2 inactivation by 14.8 mV (Fig. 6B and Table 3). In addition, the E1880K mutant demonstrated slow inactivation that was best fit with a single component exponential equation with a time constant comparable with that of Nav1.3 (Table 3).
The role of the carboxyl-terminal residues in Nav1.2 inactivation was both site- and charge-specific, similar to the situation for Nav1.3. The next downstream residue in Nav1.2 is also negative and different from Nav1.3 (Glu1881), but replacing that amino acid with the residue from Nav1.3 (E1881D) did not alter Nav1.2 inactivation (Fig. 6F and Table 3). In addition, E1881D in combination with E1880K had a similar effect as the single E1880K substitution. As was the case for all of the Nav1.3 substitutions, the voltage dependence of activation was not significantly affected by any of the mutations (Table 3).
The Carboxyl-terminal Substitutions Exclusively Affect Fast Inactivation
To more completely determine the roles of Nav1.3 Lys1826 and Nav1.2 Glu1880 in fast inactivation, we determined the rate of entry into inactivation for each of the mutant channels. The rate of entry into inactivation for wild-type Nav1.3 and Nav1.2 differ substantially, as shown in Fig. 6B. Nav1.3 enters the inactivated state significantly more slowly than Nav1.2, with a rate constant of 17.4 ms compared with 4.9 ms for Nav1.2. The Nav1.3 K1826E mutation accelerated the rate of entry ∼2-fold, to 8.7 ms. The Nav1.2 E1880K mutant entered inactivation twice as slowly as the Nav1.2 channel, with an inactivation entry rate of 11.9 ms (Fig. 6B). Therefore, the rates of entry into inactivation depend greatly on the charge of the residue at position 1826 in Nav1.3 or 1880 in Nav1.2.
Once in the inactivated state, sodium channels must transition back to the closed state prior to reactivation by subsequent depolarization. Although the Nav1.2 channel recovery is slightly different from the Nav1.3 channel recovery, both rates were best fit with a triple exponential equation (Fig. 6C). Both the mutant Nav1.2 E1880K and Nav1.3 K1826E channels showed slightly faster recovery compared with the respective wild-type channels. The time to reach 90% recovery was 320 ms for wild-type Nav1.3, 775 ms for wild-type Nav1.2, 240 ms for Nav1.3 K1826E, and 420 ms for Nav1.2 E1880K. Therefore, charge substitutions at this position accelerated recovery slightly but not in a manner specific to either channel.
The ability of a channel to transition out of the inactivated state determines the extent to which it can reopen during repeated activity. Despite their differing recovery rates, both the Nav1.2 and Nav1.3 channels reached complete recovery at the end of the 3-s voltage protocol. Consistent with this result, both the Nav1.2 and Nav1.3 channels displayed similar use-dependent inactivation during 39-Hz stimulation (Fig. 6D). The carboxyl-terminal mutant Nav1.3 K1826E showed no significant differences in use-dependent inactivation. In contrast, the Nav1.2 E1880K mutant showed reduced use-dependent inactivation (∼70%) compared with the other channels, which reached equilibrium at ∼80% (Fig. 6D, inset). Because recovery for this mutant channel was accelerated only slightly more than for the Nav1.3 K1826E mutant, we attribute this reduction in the Nav1.2 E1880K mutant use-dependent inactivation to an alteration in slow inactivation. The 3-s duration of the use-dependent inactivation protocol is most likely long enough for these channels to begin to enter the slow inactivated state. We conclude that the rates of recovery and use-dependent inactivation of the Nav1.3 and Nav1.2 channels are not strongly dependent on the carboxyl-terminal Nav1.3 Lys1826 and Nav1.2 Glu1880 residues.
Role of the III-IV Linker in the Inactivation Differences
One hypothesis for the role of charge in the inactivation differences between Nav1.3 and Nav1.2 is electrostatic repulsion between Nav1.3 Lys1826 in the carboxyl terminus and another residue in the channel. If this hypothesis is correct, then the interacting residue should be charged, and replacement of that residue should mirror the effect of the K1826E replacement in Nav1.3. Because the III-IV linker responsible for sodium channel fast inactivation contains multiple lysine residues, we screened this region for potential interacting residues. We neutralized six lysine residues (1441, 1442, 1453, 1454, 1457, and 1465) near the IFM motif by replacing them with glutamines (Fig. 7A). Loss of the positive charge at each of these positions shifted the voltage dependence of inactivation in the negative direction, although the magnitude of the shift varied greatly (Table 4). The K1453Q mutation caused the largest shift (−11.6 mV), and that mutation was also the only one to significantly shift the voltage dependence of activation (−4.3 mV). In addition, K1453Q accelerated the kinetics of inactivation to a small extent (Fig. 7B).
FIGURE 7.

A, amino acid sequence of the Nav1.3 channel III-IV linker region is shown, with lysine 1453 indicated. The sequence of the Nav1.2 III-IV linker region is identical. B, sample current traces for Nav1.3 lysine 1453 mutants during the 5-mV test depolarization of the inactivation protocol, as described under “Experimental Procedures.” C, voltage dependence of inactivation is shown for Nav1.3 K1453Q (triangles), Nav1.3 K1453E (squares), and Nav1.3 K1453D (diamonds) mutants and wild type Nav1.2 (black circles) and Nav1.3 (white circles) channels. The parameters of the fits and sample sizes are shown in Table 4. Data points indicate means, and error bars show S.D. values.
TABLE 4.
Voltage dependence of activation and inactivation for Nav 1.3 III-IV mutants
Values presented are means ± S.D.
| Channel | Voltage dependence |
|||||
|---|---|---|---|---|---|---|
| Inactivation |
Activation |
|||||
| V½ | a | n | V½ | z | n | |
| mV | mV | mV | e0 | |||
| Nav1.3 | −16.2 ± 0.8 | 4.7 ± 0.5 | 8 | −14.6 ± 2.4 | 6.9 ± 0.6 | 8 |
| K1441Q/K1442Q | −24.4 ± 1.1a | 5.8 ± 0.5b | 9 | −12.9 ± 1.8 | 6.4 ± 0.5 | 6 |
| K1453Q | −27.8 ± 2.2c | 6.6 ± 1.0b | 5 | −18.9 ± 1.6b | 7.2 ± 1.2 | 8 |
| K1454Q | −20.9 ± 1.0c | 4.6 ± 0.4 | 7 | −15.9 ± 1.8 | 7.2 ± 1.0 | 5 |
| K1457Q | −22.2 ± 0.8c | 4.8 ± 0.5 | 5 | −15.6 ± 1.8 | 7.3 ± 0.6 | 3 |
| K1465Q | −18.2 ± 1.0d | 4.9 ± 0.5 | 8 | −14.6 ± 2.2 | 6.9 ± 0.6 | 8 |
| K1453E | −36.4 ± 0.8a | 7.4 ± 0.3a | 10 | −12.8 ± 1.5 | 6.4 ± 0.4 | 9 |
| K1453Q/K1826E | −37.3 ± 1.2a | 7.7 ± 0.8c | 6 | −17.9 ± 2.4 | 8.0 ± 1.2 | 5 |
| K1453E/K1826E | −33.0 ± 2.1a | 7.1 ± 1.4† | 7 | −14.6 ± 1.4 | 6.8 ± 0.4 | 3 |
| K1453D/K1826E | −29.2 ± 1.9a | 6.8 ± 1.0† | 17 | −10.8 ± 1.3 | 7.2 ± 0.6 | 6 |
a Significantly different from Nav1.3, p < 0.000000001.
b Significantly different from Nav1.3, p < 0.001.
c Significantly different from Nav1.3, p < 0.0000005.
d Significantly different from Nav1.3, p < 0.005.
Because K1453Q had the largest effect on the voltage dependence of inactivation, we hypothesized that this residue might be the interacting partner of the carboxyl-terminal residue. We examined the importance of charge at this position by replacing the lysine with either aspartate or glutamate. Both K1453D and K1453E accelerated inactivation of Nav1.3 more prominently than K1453Q (Fig. 7B). In addition, each of the negative charge substitutions caused shifts in the voltage dependence of inactivation that were larger than that of K1453Q (Fig. 7C and Table 4). The shift in voltage dependence of inactivation and the acceleration of inactivation were similar to the effects of the Nav1.3 K1826E mutation in both direction and magnitude, suggesting that the two residues are involved in the same aspect of inactivation. These results are consistent with the hypothesis that the carboxyl-terminal residue at position 1826 in Nav1.3 (1880 in Nav1.2) interacts with Lys1453 in the III-IV linker to mediate isoform-specific inactivation.
Based on these results suggesting that the III-IV linker interacts with the carboxyl terminus, a reasonable hypothesis is that charge repulsion between Nav1.3 Lys1826 and III-IV Lys1453 causes the slower inactivation characteristic of Nav1.3 by preventing this interaction. When there is a negative charge at position 1826 in the carboxyl terminus, such as in Nav1.2 or Nav1.3 K1826E, then the carboxyl terminus can interact with the III-IV linker, thus accelerating inactivation. If this hypothesis is correct, it might be possible to restore wild-type Nav1.3 channel inactivation by simultaneously incorporating negative residues in both the carboxyl terminus and the III-IV linker to cause repulsion. To test this possibility, the K1826E mutation was combined with either K1453D or K1453E. However, adding K1826E to either K1453E or K1453D caused relatively small positive shifts in the voltage dependence of inactivation (3.4 and 10.5 mV, respectively), so the combination of negative charges did not restore wild-type Nav1.3-type inactivation (Fig. 8A and Table 4). On the other hand, combining K1826E with the K1453Q charge neutralization caused an even greater negative shift of 9.5 mV in the voltage dependence of inactivation (Fig. 8A and Table 4), consistent with the hypothesis that inactivation is more stable with only a single charge at one of these positions.
FIGURE 8.

Complementary carboxyl-terminal and domain III-IV linker mutants. A, voltage dependence of inactivation is shown for Nav1.3 K1453Q/K1826E (triangles), Nav1.3 K1453E/K1826E (squares), and Nav1.3 K1453D/K1826E (diamonds) double mutants and wild-type Nav1.2 (black circles) and Nav1.3 (white circles) channels. The parameters of the fits and sample sizes are shown in Table 4. B, normalized current versus the depolarization time is shown for entry into inactivation, as described under “Experimental Procedures.” The parameters of the fits and sample sizes are shown in Table 4. C, fraction of inactivating current at the 50 ms time point during a depolarization to −10 mV is plotted for the mutant and wild-type channels. The fraction of the inactivating current was determined as the averaged current during 1 ms at the midpoint of the trace divided by the peak current. Sample sizes were 9 for Nav1.2, 8 for Nav1.3, 5 for Nav1.3 K1453Q/K1826E, 5 for Nav1.3 K1453E/K1826E, and 6 for Nav1.3 K1453D/K1826E. Data points indicate means, and error bars show S.D. values.
Changes in the rates of entry into inactivation paralleled these changes in the voltage dependence of inactivation, with all three double mutants inactivating with faster kinetics compared with Nav1.3 or Nav1.3 K1826E (Fig. 8B). Consistent with this faster rate of inactivation, all three double mutants demonstrated very little residual current at 50 ms (Fig. 8C). These results indicate that charge plays an important role in enabling interaction between the III-IV linker and the carboxyl terminus, which is at least partially responsible for the inactivation differences between Nav1.2 and Nav1.3.
DISCUSSION
A number of previous reports have indicated a role for the sodium channel carboxyl terminus in fast inactivation (14, 16, 17, 35–43). However, a clear mechanism of how the carboxyl terminus is involved in fast inactivation remains elusive. In this report, we have identified a homologous carboxyl-terminal amino acid that is important for fast inactivation differences between the Nav1.2 and Nav1.3 sodium channels. The critical amino acid (Lys1826 in Nav1.3 or Glu1880 in Nav1.2) is both necessary and sufficient for determining the differences in the voltage dependence and kinetics of inactivation.
The carboxyl-terminal region is subdivided into a uniformly structured proximal half and a non-uniform distal half, as shown by structure-function studies of the Nav1.2, Nav1.4, and Nav1.5 channels (16, 44, 45). Within the proximal half, four α helices (I, II, III, and IV) are predicted to form a pair of EF-hands (45–47) that have been shown to stabilize contact between the carboxyl terminus and the domain III-IV linker (45, 48, 49). Mutations that directly alter the EF-hands have been identified in a variety of channelopathies, such as congenital long QT syndrome and Brugada syndrome caused by mutations in the SCN5A gene encoding Nav1.5 channel (38, 40, 41) and inherited epilepsy caused by mutations in the SCN1A gene encoding the Nav1.1 channel (14, 36, 37).
The Nav1.3 Lys1826 and Nav1.2 Glu1880 residues are located in the region of the carboxyl terminus that was designated as the proximal half. However, this position is within the predicted fifth helix and thus not the region involved in formation of the EF-hand motifs (44–47, 50–53). Therefore, alterations at this position are unlikely to directly disrupt the overall secondary structure of the pair of EF-hands that are formed by intermolecular interactions between side chains in the interface of helices I with IV and II with III (44, 49, 54). Furthermore, the Nav1.3 K1826E and Nav1.2 E1880K mutations are unlikely to function as helix breakers because the replacement amino acids have higher helical propensities than the original amino acids (55). Thus, it is unlikely that these mutations disrupt the overall structure of the proximal half of the carboxyl terminus or the EF-hands.
Inactivation stabilization was suggested to occur via a protein-protein interaction between the sixth helix in the carboxyl terminus and a P-rich motif in the III-IV linker (KPQPPIPRP), based on the finding that truncation of the sixth helix and mutations of this motif altered the level of persistent current (48). In addition, stabilization is believed to be mediated by the binding of the calcium-binding CaM protein to an IQ motif located in the sixth helix (47, 51, 52, 56, 57). Disabling the ability of CaM to bind to the IQ motif either via an inactive mutant CaM1234 (51), by a mutant IQ motif (47, 51, 57, 58), or by truncating the IQ motif (44, 56) greatly destabilized inactivation. However, it seems unlikely that Nav1.3 Lys1826 and Nav1.2 Glu1880 residues affect inactivation through CaM in oocytes because neither Nav1.3 nor Nav1.2 inactivation was affected by the presence of CaM in this system (data not shown).
The positive charge at position 1826 is not a common feature in voltage-gated sodium channels, with glutamic acid present in all of the other isoforms (59). In fact, the lysine at position 1826 is unique to this clone of rat Nav1.3, with glutamic acid present in other Nav1.3 clones. For this reason, we believe that the presence of a positive charge at this position is not a normal mechanism of functional variation in sodium channels. However, the dramatic effect on inactivation resulting from the presence of the positive charge indicates that a negative charge at this position plays an important role in determining the normal kinetics and voltage dependence of fast inactivation in sodium channels.
Based on the similar inactivation phenotypes of the Nav1.3 III-IV K1453E and carboxyl-terminal K1826E mutants, one possibility is that the interaction between these two residues is the basis of the inactivation differences. According to this hypothesis, Lys1453 in the III-IV linker and to a lesser extent flanking lysines would repulse Lys1826 in the carboxyl terminus, preventing these two regions from interacting in Nav1.3. Without this interaction, Nav1.3 would inactivate more slowly than Nav1.2. Reversing the charge at either of these positions (K1453E or K1826E) would enable the interaction, resulting in faster inactivation that resembled that of Nav1.2. The effects of the Nav1.2 E1880K mutant are consistent with this proposed mechanism in that it displayed slower inactivation kinetics. However, substituting negative charges in both the carboxyl terminus and III-IV linker of Nav1.3 did not mimic the inactivation properties of wild-type Nav1.3 with positive charges at both positions, suggesting that electrostatic repulsion between these sites cannot fully explain the effects on inactivation. Therefore, an alternative explanation is that the charge substitutions have more local effects within each region of the channel. More detailed information about a direct interaction between these residues is necessary to distinguish between these alternatives to explain the fast inactivation differences between Nav1.2 and Nav1.3.
Acknowledgments
We thank Annie Lee, A. J. Barela, Karoni Dutt, Brian Tanaka, Eric Velazquez, and Lauren Guy for helpful discussions during the course of this work and Joyce Iping and Radit Aur for excellent technical assistance. We thank Dr. John P. Aldeman (Oregon Health and Science University School of Medicine) for generosity in providing the CaM and CaM1234 constructs.
This work was supported, in whole or in part, by National Institutes of Health Grants NS26729 and NS48336.
- CaM
- calmodulin.
REFERENCES
- 1.Hille B. (2001) Ion Channels of Excitable Membranes, pp. 75–78, 3rd Ed., Sinauer Associates, Inc., Sunderland, MA [Google Scholar]
- 2.Bennett P. B., Yazawa K., Makita N., George A. L., Jr. (1995) Nature 376, 683–685 [DOI] [PubMed] [Google Scholar]
- 3.Wang Q., Shen J., Li Z., Timothy K., Vincent G. M., Priori S. G., Schwartz P. J., Keating M. T. (1995) Hum. Mol. Genet. 4, 1603–1607 [DOI] [PubMed] [Google Scholar]
- 4.Keating M. T., Sanguinetti M. C. (1996) Science 272, 681–685 [DOI] [PubMed] [Google Scholar]
- 5.Chen Q., Kirsch G. E., Zhang D., Brugada R., Brugada J., Brugada P., Potenza D., Moya A., Borggrefe M., Breithardt G., Ortiz-Lopez R., Wang Z., Antzelevitch C., O'Brien R. E., Schulze-Bahr E., Keating M. T., Towbin J. A., Wang Q. (1998) Nature 392, 293–296 [DOI] [PubMed] [Google Scholar]
- 6.Green D. S., George A. L., Jr., Cannon S. C. (1998) J. Physiol. 510, 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bendahhou S., Cummins T. R., Kwiecinski H., Waxman S. G., Ptácek L. J. (1999) J. Physiol. 518, 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makita N., Shirai N., Nagashima M., Matsuoka R., Yamada Y., Tohse N., Kitabatake A. (1998) FEBS Lett. 423, 5–9 [DOI] [PubMed] [Google Scholar]
- 9.Wehrens X. H., Abriel H., Cabo C., Benhorin J., Kass R. S. (2000) Circulation 102, 584–590 [DOI] [PubMed] [Google Scholar]
- 10.Abriel H., Cabo C., Wehrens X. H., Rivolta I., Motoike H. K., Memmi M., Napolitano C., Priori S. G., Kass R. S. (2001) Circ. Res. 88, 740–745 [DOI] [PubMed] [Google Scholar]
- 11.Ufret-Vincenty C. A., Baro D. J., Lederer W. J., Rockman H. A., Quinones L. E., Santana L. F. (2001) J. Biol. Chem. 276, 28197–28203 [DOI] [PubMed] [Google Scholar]
- 12.Wallace R. H., Scheffer I. E., Barnett S., Richards M., Dibbens L., Desai R. R., Lerman-Sagie T., Lev D., Mazarib A., Brand N., Ben-Zeev B., Goikhman I., Singh R., Kremmidiotis G., Gardner A., Sutherland G. R., George A. L., Jr., Mulley J. C., Berkovic S. F. (2001) Am. J. Hum. Genet. 68, 859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Splawski I., Timothy K. W., Tateyama M., Clancy C. E., Malhotra A., Beggs A. H., Cappuccio F. P., Sagnella G. A., Kass R. S., Keating M. T. (2002) Science 297, 1333–1336 [DOI] [PubMed] [Google Scholar]
- 14.Spampanato J., Kearney J. A., de Haan G., McEwen D. P., Escayg A., Aradi I., MacDonald B. T., Levin S. I., Soltesz I., Benna P., Montalenti E., Isom L. L., Goldin A. L., Meisler M. H. (2004) J. Neurosci. 24, 10022–10034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catterall W. A. (2000) Neuron 26, 13–25 [DOI] [PubMed] [Google Scholar]
- 16.Deschênes I., Trottier E., Chahine M. (2001) J. Membr. Biol. 183, 103–114 [DOI] [PubMed] [Google Scholar]
- 17.Mantegazza M., Yu F. H., Catterall W. A., Scheuer T. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 15348–15353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckh S., Noda M., Lübbert H., Numa S. (1989) EMBO J. 8, 3611–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brysch W., Creutzfeldt O. D., Lüno K., Schlingensiepen R., Schlingensiepen K. H. (1991) Exp. Brain Res. 86, 562–567 [DOI] [PubMed] [Google Scholar]
- 20.Furuyama T., Morita Y., Inagaki S., Takagi H. (1993) Mol. Brain Res. 17, 169–173 [DOI] [PubMed] [Google Scholar]
- 21.Felts P. A., Yokoyama S., Dib-Hajj S., Black J. A., Waxman S. G. (1997) Mol. Brain Res. 45, 71–82 [DOI] [PubMed] [Google Scholar]
- 22.Hains B. C., Black J. A., Waxman S. G. (2002) J. Neurosci. Res. 70, 546–552 [DOI] [PubMed] [Google Scholar]
- 23.Auld V. J., Goldin A. L., Krafte D. S., Marshall J., Dunn J. M., Catterall W. A., Lester H. A., Davidson N., Dunn R. J. (1988) Neuron 1, 449–461 [DOI] [PubMed] [Google Scholar]
- 24.Joho R. H., Moorman J. R., VanDongen A. M., Kirsch G. E., Silberberg H., Schuster G., Brown A. M. (1990) Mol. Brain Res. 7, 105–113 [DOI] [PubMed] [Google Scholar]
- 25.Goldin A. L. (1991) Methods Cell Biol. 36, 487–509 [DOI] [PubMed] [Google Scholar]
- 26.Patton D. E., Goldin A. L. (1991) Neuron 7, 637–647 [DOI] [PubMed] [Google Scholar]
- 27.Kontis K. J., Rounaghi A., Goldin A. L. (1997) J. Gen. Physiol. 110, 391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isom L. L., De Jongh K. S., Patton D. E., Reber B. F., Offord J., Charbonneau H., Walsh K., Goldin A. L., Catterall W. A. (1992) Science 256, 839–842 [DOI] [PubMed] [Google Scholar]
- 29.Isom L. L., Ragsdale D. S., De Jongh K. S., Westenbroek R. E., Reber B. F., Scheuer T., Catterall W. A. (1995) Cell 83, 433–442 [DOI] [PubMed] [Google Scholar]
- 30.Morgan K., Stevens E. B., Shah B., Cox P. J., Dixon A. K., Lee K., Pinnock R. D., Hughes J., Richardson P. J., Mizuguchi K., Jackson A. P. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 2308–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qu Y., Curtis R., Lawson D., Gilbride K., Ge P., DiStefano P. S., Silos-Santiago I., Catterall W. A., Scheuer T. (2001) Mol. Cell. Neurosci. 18, 570–580 [DOI] [PubMed] [Google Scholar]
- 32.O'Leary M. E., Chen L. Q., Kallen R. G., Horn R. (1995) J. Gen. Physiol. 106, 641–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kühn F. J., Greeff N. G. (1999) J. Gen. Physiol. 114, 167–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cha A., Ruben P. C., George A. L., Jr., Fujimoto E., Bezanilla F. (1999) Neuron 22, 73–87 [DOI] [PubMed] [Google Scholar]
- 35.Tan B. H., Iturralde-Torres P., Medeiros-Domingo A., Nava S., Tester D. J., Valdivia C. R., Tusié-Luna T., Ackerman M. J., Makielski J. C. (2007) Cardiovasc. Res. 76, 409–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nabbout R., Gennaro E., Dalla Bernardina B., Dulac O., Madia F., Bertini E., Capovilla G., Chiron C., Cristofori G., Elia M., Fontana E., Gaggero R., Granata T., Guerrini R., Loi M., La Selva L., Lispi M. L., Matricardi A., Romeo A., Tzolas V., Valseriati D., Veggiotti P., Vigevano F., Vallée L., Dagna Bricarelli F., Bianchi A., Zara F. (2003) Neurology 60, 1961–1967 [DOI] [PubMed] [Google Scholar]
- 37.Fujiwara T., Sugawara T., Mazaki-Miyazaki E., Takahashi Y., Fukushima K., Watanabe M., Hara K., Morikawa T., Yagi K., Yamakawa K., Inoue Y. (2003) Brain 126, 531–546 [DOI] [PubMed] [Google Scholar]
- 38.Bezzina C., Veldkamp M. W., van Den Berg M. P., Postma A. V., Rook M. B., Viersma J. W., van Langen I. M., Tan-Sindhunata G., Bink-Boelkens M. T., van Der Hout A. H., Mannens M. M., Wilde A. A. (1999) Circ. Res. 85, 1206–1213 [DOI] [PubMed] [Google Scholar]
- 39.Rivolta I., Abriel H., Tateyama M., Liu H., Memmi M., Vardas P., Napolitano C., Priori S. G., Kass R. S. (2001) J. Biol. Chem. 276, 30623–30630 [DOI] [PubMed] [Google Scholar]
- 40.An R. H., Wang X. L., Kerem B., Benhorin J., Medina A., Goldmit M., Kass R. S. (1998) Circ. Res. 83, 141–146 [DOI] [PubMed] [Google Scholar]
- 41.Veldkamp M. W., Viswanathan P. C., Bezzina C., Baartscheer A., Wilde A. A., Balser J. R. (2000) Circ. Res. 86, E91–E97 [DOI] [PubMed] [Google Scholar]
- 42.Deschênes I., Baroudi G., Berthet M., Barde I., Chalvidan T., Denjoy I., Guicheney P., Chahine M. (2000) Cardiovasc. Res. 46, 55–65 [DOI] [PubMed] [Google Scholar]
- 43.Clancy C. E., Rudy Y. (2002) Circulation 105, 1208–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cormier J. W., Rivolta I., Tateyama M., Yang A. S., Kass R. S. (2002) J. Biol. Chem. 277, 9233–9241 [DOI] [PubMed] [Google Scholar]
- 45.Miloushev V. Z., Levine J. A., Arbing M. A., Hunt J. F., Pitt G. S., Palmer A. G., 3rd (2009) J. Biol. Chem. 284, 6446–6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wingo T. L., Shah V. N., Anderson M. E., Lybrand T. P., Chazin W. J., Balser J. R. (2004) Nat. Struct. Mol. Biol. 11, 219–225 [DOI] [PubMed] [Google Scholar]
- 47.Shah V. N., Wingo T. L., Weiss K. L., Williams C. K., Balser J. R., Chazin W. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 3592–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motoike H. K., Liu H., Glaaser I. W., Yang A. S., Tateyama M., Kass R. S. (2004) J. Gen. Physiol. 123, 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim J., Ghosh S., Liu H., Tateyama M., Kass R. S., Pitt G. S. (2004) J. Biol. Chem. 279, 45004–45012 [DOI] [PubMed] [Google Scholar]
- 50.Mori M., Konno T., Morii T., Nagayama K., Imoto K. (2003) Biochem. Biophys. Res. Commun. 307, 290–296 [DOI] [PubMed] [Google Scholar]
- 51.Deschênes I., Neyroud N., DiSilvestre D., Marbán E., Yue D. T., Tomaselli G. F. (2002) Circ. Res. 90, E49–E57 [DOI] [PubMed] [Google Scholar]
- 52.Theoharis N. T., Sorensen B. R., Theisen-Toupal J., Shea M. A. (2008) Biochemistry 47, 112–123 [DOI] [PubMed] [Google Scholar]
- 53.Chagot B., Potet F., Balser J. R., Chazin W. J. (2009) J. Biol. Chem. 284, 6436–6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glaaser I. W., Bankston J. R., Liu H., Tateyama M., Kass R. S. (2006) J. Biol. Chem. 281, 24015–24023 [DOI] [PubMed] [Google Scholar]
- 55.Pace C. N., Scholtz J. M. (1998) Biophys. J. 75, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biswas S., Deschênes I., Disilvestre D., Tian Y., Halperin V. L., Tomaselli G. F. (2008) J. Gen. Physiol. 131, 197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mori M., Konno T., Ozawa T., Murata M., Imoto K., Nagayama K. (2000) Biochemistry 39, 1316–1323 [DOI] [PubMed] [Google Scholar]
- 58.Potet F., Chagot B., Anghelescu M., Viswanathan P. C., Stepanovic S. Z., Kupershmidt S., Chazin W. J., Balser J. R. (2009) J. Biol. Chem. 284, 8846–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldin A. L. (1995) in Ligand- and Voltage-gated Ion Channels (North R. A. ed) pp. 73–112, CRC Press, Inc., Boca Raton, FL [Google Scholar]