

Abstract
dinP is an Escherichia coli gene recently identified at 5.5 min of the genetic map, whose product shows a similarity in amino acid sequence to the E. coli UmuC protein involved in DNA damage-induced mutagenesis. In this paper we show that the gene is identical to dinB, an SOS gene previously localized near the lac locus at 8 min, the function of which was shown to be required for mutagenesis of nonirradiated λ phage infecting UV-preirradiated bacterial cells (termed λUTM for λ untargeted mutagenesis). A newly constructed dinP null mutant exhibited the same defect for λUTM as observed previously with a dinB::Mu mutant, and the defect was complemented by plasmids carrying dinP as the only intact bacterial gene. Furthermore, merely increasing the dinP gene expression, without UV irradiation or any other DNA-damaging treatment, resulted in a strong enhancement of mutagenesis in F′lac plasmids; at most, 800-fold increase in the G6-to-G5 change. The enhanced mutagenesis did not depend on recA, uvrA, or umuDC. Thus, our results establish that E. coli has at least two distinct pathways for SOS-induced mutagenesis: one dependent on umuDC and the other on dinB/P.
Keywords: SOS response, untargeted mutagenesis, frameshift mutations, inducible evolution
Escherichia coli and related bacteria have a genetic network called the SOS regulon that enables them to cope with the consequences of DNA damage in various ways (refs. 1–2 and references therein). Most of the genes belonging to this regulon are repressed by LexA and are induced when LexA is self-cleaved upon association with activated RecA molecules. RecA is activated when it binds to single-stranded DNA that results from DNA damage. The term coprotease is coined for the activity of the activated RecA to promote self-cleavage of LexA and some other proteins. The SOS-induced functions include an activity that allows the DNA polymerase stalled at DNA lesions to bypass them, frequently generating mutations as a result of the translesion synthesis [called targeted mutagenesis (3)]. This mutagenesis primarily depends on the function of the umuDC operon (1, 4). The RecA coprotease activity is required also for converting UmuD to an active form for mutagenesis, UmuD′ (5–7). Furthermore, RecA is believed to play another, more direct, role during the translesion synthesis in concert with UmuD′C complex and DNA polymerase III holoenzyme (7–11).
In addition, the SOS-induced functions have been known to include an activity that enhances mutagenesis in DNA that has not been exposed to an exogenous DNA-damaging agent. This form of mutagenesis [called untargeted mutagenesis (3)] has been studied mainly in two different experimental systems. In one system, mutations on the chromosome or on an F′ plasmid were measured in SOS-constitutive mutants such as recA441 or recA730 (3, 12–14). In these cases, the mutagenesis was dependent on the umuC function (13, 14), and probably the umuD function as well. Most of the mutations observed within the lacI gene on F′ plasmid were base-substitutions (mostly G⋅C to T⋅A, with fewer A⋅T to T⋅A transversions) with some site specificity (12). One plausible explanation for the observed results was that mutations resulted from endogenous forms of DNA damage, such as apurinic sites, which are cryptic if the SOS response is not induced (15). However, the fact that part of recA730-induced mutation is eliminated by mismatch repair suggests that, under SOS-induced conditions in a recA730 mutant, some of untargeted mutations are produced through correctable replication errors (14). In the other system, mutations in nonirradiated λ bacteriophages grown in UV-preirradiated bacterial cells were measured (16–21). This form of mutagenesis of unirradiated λ phages (called λ untargeted mutagenesis (λUTM for short), or alternatively indirect (18) or nontargeted (20) mutagenesis) was independent of the umuC function (20, 21), in contrast to the first system and to mutagenesis of UV-irradiated λ phages infecting UV-preirradiated bacterial cells [Weigle mutagenesis (1)]. λUTM occurred in recA lexA(Def) double-mutant cells, still requiring UV preirradiation (20, 22), but it did not occur in uvr− and polA− mutant strains (17, 23) or in a strain with an insertion of MudI(Ap, lac) phage in the dinB gene (21), an SOS gene that had been localized near the lac locus at 8 min (24). The predominant mutations observed within the λ cI gene were frameshift mutations that mostly occurred at runs of identical bases (20). It has been argued that λUTM results from a transient decrease in DNA replication fidelity, which is caused by an induced SOS function(s) (19, 20). This hypothesis was supported by the fact that λUTM is increased in mismatch repair-deficient strains (19), but the molecular mechanism of λUTM has remained obscure.
Recently, a hypothetical gene designated dinP was identified near the 5.5-min region of the E. coli genetic map during a genomic sequencing project (25). The gene has a sequence similar to the consensus sequence for LexA-binding site (SOS-box) in its putative promoter region. Furthermore, the predicted sequence of the gene product shows weak similarities to the UmuC protein and its homologs involved in DNA damage-induced mutagenesis, which include the Saccharomyces cerevisiae REV1 protein. It also shows a strong similarity to the Caenorhabditis elegans hypothetical protein F22B7.6 of unknown function. No gene similar to umuD was found in the nearby region of dinP. In this paper we show that dinP is identical with the aforementioned dinB gene and that, when overexpressed, it exhibits a potent mutagenic activity without any exogenous agent to damage DNA.
MATERIALS AND METHODS
Media.
L broth (1% Bacto Tryptone/0.5% yeast extract/0.5% NaCl, pH 7.4) was used throughout, unless otherwise indicated. L agar contained 1.5% agar in L broth. If necessary, ampicillin (50 μg/ml), chloramphenicol (20 μg/ml), kanamycin (20 μg/ml), or tetracycline (10 μg/ml) was added. Minimal glucose medium consisted of Vogel–Bonner E medium (26) supplemented with 0.2% glucose. Minimal lactose agar plates contained 0.5% lactose and 1.5% agar in Vogel–Bonner E medium. Top agar contained 0.5% NaCl, 0.6% agar, and 0.2 mg/ml nutrient broth (Difco). The nutrient broth was added to allow bacteria to divide a few times for fixation of mutation.
Bacteria and Bacteriophages.
The original dinB1:: MudI(Ap, lac) strain GW1030 (24) was kindly provided by G. C. Walker (Massachusetts Institute of Technology). MC1061 carrying the ΔlacX74 mutation (27) was used as the host to isolate plasmids containing a dinB-lacZ fusion fragment. CSR603 (28) was used for identifying plasmid-encoded proteins in maxicells. The series of strains (CC101–CC111) was used for detecting base-substitution and frameshift mutations (29, 30). A dinP-null mutant of AB1157 (31) was constructed by replacing the 993-bp SnaBI segment (8697–9690 in Fig. 1) with the kanamycin-resistance (Kmr) gene from pUC4K (obtained from Pharmacia/LKB), as described (32). The gene disruption was confirmed by a Southern blotting analysis, and the resultant strain was designated YG7207. Other strains constructed in this study are listed in Table 1. P1vir was used for general transduction (33).
Figure 1.
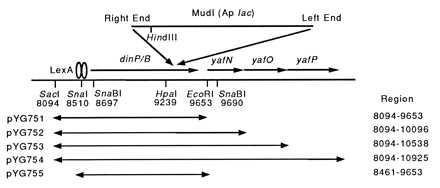
Gene organization around the dinP/B gene. The coding regions are denoted by horizontal lines with a rightward arrowhead. The LexA binding sequence in the dinP/B promoter site and some of the restriction sites referred to in the text are indicated. The numbering of base positions follows that in the DNA database entry D38582. The HpaI (at 9239)-HindIII fragment obtained from the dinB1::MudI(Ap lac) mutant was subjected to DNA sequence analysis. The DNA regions (indicated at the right) carried by the plasmids are denoted by lines with arrowheads at both ends.
Table 1.
Bacterial strains used
| Strain | Relevant genotype | Source or ref. |
|---|---|---|
| GW1030 | dinB1::MudI(Ap, lac) | 24 |
| MC1061 | ΔlacX74 | 27 |
| CSR603 | recA uvrA phr | 28 |
| AB1157 | 31 | |
| YG7207 | ΔdinB::kan derivative of AB1157 | This work |
| CC101–CC111 | 29, 30 | |
| YG2223* | Δ(recA–srlR)306::Tn10 | P1(GW6752, 7) × CC108 |
| YG2224* | ΔumuDC595::cat | P1(RW120, 58) × CC108 |
| YG2226* | uvrA277::Tn10 | P1(CGSC no. 6661†) × CC108 |
| YG2228* | umuC122::Tn5 | P1(GW2100, 59) × CC108 |
The last four strains were constructed by transduction with P1 phage grown in the strain indicated within parentheses and by selecting the respective antibiotic resistance marker.
The strain (CGSC no. 6661, N3055) carrying uvrA277::Tn10 was obtained from the E. coli Genetic Stock Center of Yale University.
Construction of Plasmids.
pTZ19ULC is a derivative of pTZ19U with a lowered copy number (34) and was used as the vector for cloning the dinB-lacZ fusion fragment. To isolate DNA fragments carrying the functional dinP gene and its nearby genes, an approximately 7-kb KpnI fragment was prepared from the Kohara clone 8F9 (35) and inserted into the blunt-ended EcoRI site of pBR322. The resultant plasmid was designated pYG643. Various subfragments from pYG643 were inserted in pBluescript KS+ (pKS, obtained from Stratagene) to construct pYG751–pYG755 for the detection of plasmid-encoded proteins by the maxicell system (see Fig. 1). pYG768, a low-copy-number plasmid carrying dinP and its cognate promoter, was constructed by inserting the SacI–EcoRI DNA fragment (the same as pYG751 in Fig. 1) between the SacI and EcoRI sites of pWSK29, which is a pSC101 derivative carrying the multiple cloning site sequence (MCS) identical to that of pBluescript II SK+ (36). pYG782 is another low-copy-number plasmid carrying dinP without its promoter and operator regions, which was constructed by inserting the 1,087-bp SnaI fragment (8510–9597 in Fig. 1) into the SmaI site of pWKS30, a vector similar to pWSK29 but containing the same MCS in the opposite orientation (36). In pYG782, dinP is transcribed from the vector Plac promoter.
Mutation Assays.
The frequency of λ clear plaque mutations from λcI857 was measured as described (21, 22). Host bacteria were grown in L broth at 37°C, centrifuged, and resuspended in 10−2 M MgSO4 for UV irradiation at the dose indicated and for phage adsorption. Host bacteria with phages were incubated for 20 min at 32°C for adsorption and plated with unirradiated AB1157 as indicator lawn. After the plates were incubated for 48 hr at 32°C, infective centers containing clear-plaque mutants were detected, in most cases, as mixed plaques containing c and c+ phages. Mixed plaques were picked up and replated on a lawn of AB1157 to verify the presence of mutants as described (21).
Experiments with the CC strains were done as previously described (37, 38). Briefly, single colonies of the CC strains harboring plasmids were isolated on a minimal glucose medium, inoculated into L broth containing ampicillin in triplicate, and incubated overnight at 37°C. The overnight cultures were washed twice with sodium/potassium phosphate buffer (66 mM, pH 7.2) by centrifugation and resuspended in the same buffer. For the detection of lactose-fermenting (Lac+) revertants, small aliquots (0.1 ml, unless indicated) of the cell suspensions were spread on duplicate minimal lactose agar plates. After appropriate dilution, 0.1-ml portions of the diluted samples were spread on duplicate minimal glucose agar plates for the estimation of viable cells.
Other Methods.
Most procedures for DNA manipulation and bacterial transformation with plasmid DNA followed the standard protocols (33, 39). DNA sequencing was carried out by the Applied Biosystems thermal cycling method with dye-labeled primers, following the procedures recommended by the manufacturer. Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS/PAGE), essentially following the Laemmli method (40), and visualized by fluorography.
RESULTS
Identity of dinP to dinB.
Within its putative promoter region the dinP gene has the sequence 5′-CACTGTATACTTTACCAGTG-3′ (25), which differs by 7 nucleotides from the 20-nucleotide consensus sequence for LexA binding site (5′-TACTGTATATATATACAGTA-3′). To obtain experimental evidence that dinP is indeed inducible by DNA damage, we first examined the possibility that dinP might be identical to one of the SOS genes thus far identified but not well characterized. It seemed that dinB might be a plausible candidate, because the gene was previously localized around the lac locus at 8 min by a mating interruption experiment, a low-resolution mapping procedure (24). In the dinB1::MudI(Ap, lac) mutant, expression of the dinB-fused β-galactosidase gene was shown to be induced by UV irradiation or mitomycin C addition and regulated by both lexA and recA (24).
To determine more accurately the insertion site in the original dinB1::MudI(Ap, lac) strain GW1030 (24), we isolated its chromosomal DNA, digested with BamHI, and ligated with the BamHI-cleaved DNA of pTZ19ULC (34), as described previously (41, 42). The plasmid clones containing the dinB-lacZ fusion were identified from the Lac+ transformants of MC1061 carrying the ΔlacX74 mutation (27). After several steps of subcloning, an approximately 350-bp HpaI–HindIII fragment containing the junction was sequenced by a thermal cycling method. The sequencing result revealed that the Mu insertion had occurred around position 9388 in the dinP sequence (Fig. 1), which corresponds to the 283rd residue of the total 351-amino acid sequence. This assignment agrees well with the previous mapping of some restriction sites near the truncated dinB gene by Kenyon et al. (41). They also showed by in vitro transcription experiment that the Mu insertion had occurred about 900 bp downstream from a LexA-controlled promoter, which is also in good agreement with the distance (870 bp) between the dinP promoter overlapping with an SOS-box sequence and the Mu insertion site determined above. Thus, the identity of dinP to dinB led us to carry out a detailed functional analysis, because dinB had been previously shown to be required for λUTM (22). Hereafter, we use the gene name dinB exclusively.
Detection of Products of the dinB and Downstream Genes.
Prior to the functional analysis, we identified the products of dinB and its downstream genes by using the maxicell system (28). Various DNA fragments around dinB, which had been originally derived from the Kohara clone 8F9 (35), were inserted into the vector plasmid pKS in the direction where dinB is transcribed from its cognate promoter but not from the vector Plac promoter (Fig. 1). The 31-kDa β-lactamase (Bla) served as an internal marker for the analysis by SDS/PAGE (Fig. 2, lane 1). A band of about 42-kDa molecular mass was observed when the dinB coding region was intact (Fig. 2, lanes 2–6), and a band migrating faster than the 42-kDa band appeared when a C-terminal portion of dinB was deleted (data not shown). These results imply that the 42-kDa protein should correspond to the dinB gene product (the calculated molecular mass for the 351-amino acid sequence is 39,516).
Figure 2.
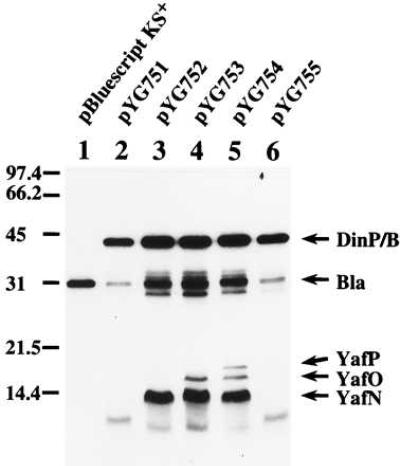
Identification of plasmid-coded proteins. The maxicell method of Sancar et al. (28) was used to label the proteins encoded by plasmids pYG751, pYG752, pYG753, pYG754, and pYG755 in the CSR603 strain with [35S]methionine. Samples were subjected to electrophoresis on an SDS/14% polyacrylamide gel and visualized by fluorography.
From the DNA sequence (GenBank accession no. D38582), there seemed to exist the three small ORFs downstream of dinB, in the same direction as that of dinB: yafN (97 aa), yafO (132 aa), and yafP (150 aa) (ref. 24; see Fig. 1). An approximately 13-kDa band was observed with the plasmids carrying the intact yafN coding region (Fig. 2, lanes 3–5). Two other faint bands (approximately 16 kDa and 18 kDa) were observed with the plasmids carrying yafO and yafP (Fig. 2, lanes 3 and 4), which probably correspond to YafO and YafP, respectively. Thus, the results by the maxicell analysis are consistent with the prediction from the DNA sequence, accordingly providing a strong support for its correctness. Furthermore, we have recently confirmed the DNA sequence from the amino acid sequence analysis of the purified DinB protein (unpublished results).
Complementation of the Defect for λUTM in a dinB-Null Mutant.
Because the dinB1::MudI(Ap, lac) allele was the only available mutation in the dinB gene, we constructed a dinB-null mutant derivative of AB1157 (designated YG7207), as described in Materials and Methods. The deleted region (8697–9690, see Fig. 1) includes about 90% of the dinB coding region and the first 15 residues of the immediately downstream gene yafN. As shown in Table 2, UV preirradiation of AB1157 increased frequency of incidence of clear mutations in unirradiated λcI857 phages by nearly two orders of magnitude, but the same treatment of YG7207, its dinB derivative, caused only a 2- or 3-fold increase. Similar results were obtained with ES548 (19), a strain used for previous experiments for λUTM, and its ΔdinB derivative constructed by P1 transduction (data not shown). The defect of YG7207 was complemented by pYG768, a pSC101 derivative carrying dinB as the only intact bacterial gene. The yafN gene product seems to play no essential role in the dinB-enhanced mutagenesis even if the gene is cotranscribed with dinB. UV preirradiation of the YG7207 strain carrying pYG768 increased the mutation frequencies up to the same levels as those of AB1157 carrying the dinB+ plasmid, which was about 5-fold higher than that of AB1157 without the plasmid, probably reflecting the increased gene dosage.
Table 2.
Effects of dinP expression on the frequency of λ clear plaque mutations
| Mutation frequency (numbers)
|
||||
|---|---|---|---|---|
| UV dose, J/m2 | AB1157 (dinB+)
|
YG7207 (ΔdinB)
|
||
| No plasmid | pYG768 (dinB+) | No plasmid | pYG768 (dinB+) | |
| 0 | 4.3 × 10−5 | 4.2 ×10−5 | 5.9 × 10−5 | 2.6 × 10−5 |
| (2/45,760) | (2/46,800) | (3/51,040) | (1/38,400) | |
| 70 | 3.3 × 10−3 | 1.8 × 10−2 | 9.5 × 10−5 | 1.2 × 10−2 |
| (57/17,020) | (313/17,550) | (4/42,320) | (216/17,805) | |
| 100 | 3.0 × 10−3 | 1.5 × 10−2 | 1.6 × 10−4 | 1.2 × 10−2 |
| (59/19,700) | (138/9,130) | (5/32,040) | (115/9,550) | |
The mutation frequency was calculated by dividing the numbers of infective centers containing clear-plaque mutants with the total numbers of infective centers. The exact numbers of those counted are shown in parentheses.
Previously, UV preirradiation of the host cell was shown to be required to exhibit high mutagenic activity for nonirradiated λ phage even in recA lexA(Def) double mutants (20, 22). It implied that derepression of the LexA-controlled SOS genes was not sufficient by itself for expressing the λUTM mutagenic pathway, whereas, unlike the UmuDC-dependent mutagenic pathway, activation of RecA was not required. Interestingly, an elevated level of the λUTM activity was observed without UV preirradiation when we employed bacteria harboring another plasmid pYG782 instead of pYG768 to supply DinB proteins in increased amounts (data not shown). The plasmid pYG782 is a pSC101 derivative in which the dinB gene is transcribed from the vector Plac promoter. We interpreted the above observation as indicating that dinB was the only SOS-controlled gene whose expression had to be elevated to exhibit the λUTM activity and, more importantly, that UV preirradiation of the host bacteria was no longer required as long as dinB was overexpressed in the bacterial cells. This interpretation was supported by an independent line of evidence, as described below.
Specificity and Genetic Requirement of the dinB-Enhanced Mutagenesis.
To examine effect(s) of the dinB-null mutation in induced mutagenesis of chromosomal genes, we first compared YG7207 with AB1157 with respect to the reversion frequency of the argE3 (ochre) marker after UV irradiation. UV-induced reversion of the argE3 marker was shown to be abolished in a umuC-defective mutant of AB1157 (43). In contrast to the above case with λUTM, we found no difference between the two strains (data not shown). The UV-induced reversion of argE3 was not enhanced in AB1157 cells carrying pYG751, a high-copy-number plasmid carrying the dinB gene with its cognate regulatory region. In this connection, it was also previously observed that the dinB1::MudI(Ap, lac) strain GW1030 showed no defect in UV-induced mutagenesis of another bacterial gene (measured for rifampicin-resistance mutation in the rpoB gene) or in mutagenesis in UV-irradiated phage λ (22). These facts imply that dinB does not affect mutagenesis at UV-damaged sites.
For measuring the dinB activity in untargeted mutagenesis, it seemed necessary to make use of a more general and sensitive assay system. We therefore employed the series of CC101–111 strains that carry various F′lac plasmids, each of which contains a defined sequence alteration in the lacZ gene near the region encoding essential residues in the active site of β-galactosidase (29, 30). To avoid any DNA-damaging treatment for inducing the dinB expression, we transformed each of the CC101–111 strains with pYG751, also in the hope that an increased dinB expression by the multicopy plasmid might enhance mutation highly enough to be detectable even if some portions of the generated mutations were corrected by the mismatch repair system. Each of the transformants was measured for the Lac+ reversion frequency (Table 3). Whereas pYG751 did not affect UV-induced reversion of the argE3 marker as described above, it enhanced various types of spontaneous mutations at greatly differing extents. The most drastic effect, amounting to more than 800-fold increase, was observed with the CC108 transformant, which allows detection of the −1 G change within a G6 cluster sequence. pYG751 enhanced −1 A and +1 G changes, 95- and 44-fold, respectively, with a smaller effect on +1 A change (18-fold increase). Some base-substitution mutations were also increased: G⋅C to T⋅A (96-fold) and A⋅T to T⋅A (28-fold) transversions and G⋅C to A⋅T (48-fold) transitions. Thus, merely increasing the dinB gene dosage resulted in a strong enhancement of frameshift and base-substitution mutations in F′lac plasmid without any exogenous agents to damage DNA.
Table 3.
Increase in mutation frequency by the plasmid-borne dinB gene
| Strain | Change | pKS+ (vector)
|
pYG751 (dinB+)
|
Ratio f/c | ||||
|---|---|---|---|---|---|---|---|---|
| a Inoculum, no. × 10−7 | b Lac+, no./plate | c Mut. freq. × 107 | d Inoculum, no. × 10−7 | e Lac+ no./plate | f Mut. freq. × 107 | |||
| CC101 | A⋅T to C⋅G | 28.4 | 1 | 0.04 | 7.2 | 5 | 0.69 | 17.3 |
| CC102 | G⋅C to A⋅T | 25.2 | 3 | 0.12 | 6.7 | 39 | 5.82 | 48.5 |
| CC103 | G⋅C to C⋅G | 26.4 | 0 | <0.01 | 7.4 | 0.3 | 0.04 | >4.0 |
| CC104 | G⋅C to T⋅A | 26.8 | 3 | 0.11 | 6.2 | 66 | 10.7 | 96.4 |
| CC105 | A⋅T to T⋅A | 25.4 | 3 | 0.12 | 8.8 | 30 | 3.41 | 28.4 |
| CC106 | A⋅T to G⋅C | 26.5 | 0 | <0.01 | 8.5 | 0.3 | 0.04 | >4.0 |
| CC107 | 6G to 7G | 5.8 | 26 | 4.48 | 1.7 | 334 | 196.5 | 43.9 |
| CC108 | 6G to 5G | 5.9 | 16 | 2.71 | 1.7 | 3,698 | 2,175.3 | 802.7 |
| CC109 | 5CG to 4CG | 5.7 | 33 | 5.79 | 2.0 | 24 | 12.0 | 2.1 |
| CC110 | 6A to 7A | 5.0 | 2 | 0.40 | 1.9 | 14 | 7.37 | 18.4 |
| CC111 | 7A to 6A | 6.1 | 12 | 1.97 | 2.2 | 412 | 187.3 | 95.1 |
The numbers indicated in columns a, b, d, and e are averages of three samples, each duplicated for counting colonies.
To study the genetic requirement for the dinB-mediated mutagenesis, we transferred various mutations into CC108 by P1 transduction, and transformed each of such derivatives with pYG782, a dinB+ derivative of pSC101 in which dinB was expressed from the vector Plac promoter. As shown in Table 4, none of the ΔrecA::Tn10, uvrA::Tn10, umuC::Tn5, or ΔumuDC::cat mutations affected the Lac+ reversion frequency. This finding indicates that neither RecA nor UvrA protein (probably other proteins involved in the nucleotide excision repair mechanism as well) is required for the dinB-enhanced mutagenesis to occur in cells with overexpressed dinB, and it also eliminates the possibility that DinB requires either UmuD or UmuD′ to exert its effect.
Table 4.
Effect of various mutations on the dinB-mediated mutagenesis
| Plasmid
|
Lac+ reversion frequency
|
||
|---|---|---|---|
| Strain | Mutation | pWKS30 (vector) | pYG782 (dinB+) |
| CC108 | Wild-type | 9.0 × 10−7 | 7.9 × 10−3 |
| YG2223 | ΔrecA | 5.1 × 10−7 | 6.6 × 10−3 |
| YG2224 | ΔumuDC | 8.3 × 10−7 | 7.1 × 10−3 |
| YG2226 | uvrA::Tn10 | 6.1 × 10−7 | 2.8 × 10−3 |
| YG2228 | umuC::Tn5 | 4.7 × 10−7 | 6.5 × 10−3 |
The Lac+ reversion frequency was measured as described for Table 3, except for that 1-μl aliquots of the suspension of the cells carrying pYG782 were spread on duplicate minimal lactose agar plates.
DISCUSSION
From the results presented above and many previous studies (1), we conclude that E. coli has at least two distinct pathways for SOS-induced mutagenesis: first, the umuDC-dependent pathway that is essential for mutagenesis by UV and many other DNA-damaging agents, and second, the dinB dependent pathway that can generate mutations in DNA that has not been exposed to a DNA-damaging agent. A potent carcinogen, N-2-acetylaminofluorene (AAF), is known to covalently bind to DNA primarily at the C-8 position of guanine and induce two different types of deletion in E. coli cells (the −1 deletion at G stretch sequences and the −2 deletion at alternating G-C sequences), both dependent on the SOS functions but not on the umuDC function (44). Because both deletion events occurred normally in the dinB1::MudI(Ap, lac) mutant (45) or the newly isolated dinB-null mutant YG7207 (R. P. P. Fuchs, personal communication), E. coli seems to have yet another, third, pathway for SOS-induced mutagenesis [the Npf pathway (45)].
Another major distinction between the dinB- and umuDC-dependent mutagenic pathways lies in the requirement for RecA. The dinB pathway, like the Npf pathway (46), does not require the RecA function once it is induced (refs. 19 and 21 and this work), whereas the umuDC pathway requires RecA at the two later steps after induction, conversion of UmuD to an active form UmuD′ and translesion synthesis. Thus, the mutagenic activity of the umuDC pathway is regulated by multiple post-transcriptional processing (1, 5–7, 47), in addition to a tight transcriptional control due to the high affinity of the umuDC operator sequence to the LexA repressor (48). In contrast, not only does dinB lack a linked umuD-like gene, but its expression is predicted to be less tightly repressed by the LexA repressor than umuDC, because the SOS-box sequence upstream of dinB differs by 7 bases from the consensus sequence, whereas that of umuDC differs by just 2 bases.
How, then, is the activity of the dinB mutagenic pathway regulated? We presume that dinB enhances mutagenesis at DNA sites free of damage. This view is further strengthened by this study, but it is not yet proven because the possibility that some DNA damage of endogenous origin might be involved cannot be eliminated at present. On the above presumption, we infer that the dinB pathway is regulated not at the level of its activity but at that of its products. Because the dinB-mediated untargeted mutagenesis seems to be weak usually when the dinB function is expressed at the single copy on the chromosome, the generated mutations should be correctable by the mismatch repair system if the system is fully active (14, 49). It should be remembered that λ phage, with which the mutagenic activity of dinB was first observed, is less efficiently corrected by the mismatch repair system than is chromosomal DNA (14, 19), probably because it is undermethylated due to the rapid lytic life cycle (50). On the other hand, if dinB enhances mutations on the bacterial chromosome and plasmid DNAs that are fully methylated at least on one strand, they are readily corrected by the mismatch repair as long as the mutation frequency is low, not exceeding the limited capacity of the mismatch correction (51). This reasoning helps us understand why the recA441 (= tif)-induced mutations observed within the lacI gene on F′lac plasmid were confined mostly to G⋅C to T⋅A, and less to A⋅T to T⋅A, transversions that probably occurred at apurinic sites (12). One can argue that such mutations at apurinic sites mediated by the umuDC-dependent pathway were not correctable by the mismatch repair, whereas other mutations at nondamaged sites mediated by the dinB-dependent pathway were mostly corrected by it under the conditions where the mutation frequencies were increased at most severalfold in comparison with noninduced conditions (12).
What is the mechanism by which the DinB protein enhances both frameshift and base-substitution mutations, probably at DNA sites free from damage? The spectrum of the dinB mutator effect shown in Table 3 indicates that a −1G event is 10-fold more frequent than a +1G event and that transversions are also more frequently increased than transitions, largely differing from the spectrum observed with mutH derivatives of the CC strains (30). This finding argues against the possibility that DinB enhances mutagenesis by inactivating the mismatch repair mechanism. Rather, on the basis of the similarity between UmuC and DinB, we speculate that DinB may act, with or without other factor(s), on DNA polymerase III to decrease its fidelity functions. To investigate the mechanism in details, we are currently trying to construct an in vitro mutagenesis system with purified DinB and other proteins, including DNA polymerase III.
Another question to be raised is what advantage E. coli cells might have by possessing the dinB-dependent mutagenic pathway. If DinB indeed enhances mutations in nondamaged DNA rather than in damaged DNA, its role should be by no means for recovering individual cells from DNA damage. We interpret, as previously suggested for SOS-induced mutagenesis in general by Radman et al. (52), that the dinB function expressed under stress conditions may temporarily increase mutation rate so as to generate by chance mutants more adapted to the stressful environments. Although most of mutations generated by any means were deleterious, it is obvious that evolution does not proceed without mutations. It was recently shown that not only DNA damage but also starvation stress lead to induction of the SOS response in E. coli cells (53). In this connection, it seems very intriguing to test whether the dinB function is involved in adaptive mutation (54), because two recent papers (55, 56) indicate that a subpopulation of E. coli cells under starvation conditions become hypermutagenic to both selected and nonselected genes. Our finding described here that dinB functions as a mutator may have some implications for mutagenesis also in other organisms, because DinB homologs exist more ubiquitously than UmuC homologs (57).
Acknowledgments
We are very grateful to G. C. Walker (Massachusetts Institute of Technology) for providing us with the GW1030 strain and also for critical reading of the manuscript. We also thank both of the two anonymous referees for their helpful criticisms. One of the authors (H.O.) thanks T. Kogoma, T. Nagata, and H. Shinagawa for reading early manuscripts and providing valuable comments to improve them. This work was supported by grants from the Ministry of Education, Science and Culture, Japan, to T.N. and H.O., from Japan Health Sciences Foundation to T.N., and from the Belgian Fonds National de la Recherche Scientifique to G.M.-M.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: UTM, untargeted mutagenesis; Lac+, lactose-fermenting.
References
- 1.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol. Press; 1995. [Google Scholar]
- 2.Echols H, Goodman M F. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 3.Witkin E M, Wermundsen I E. Cold Spring Harbor Symp Quant Biol. 1978;43:881–886. doi: 10.1101/sqb.1979.043.01.095. [DOI] [PubMed] [Google Scholar]
- 4.Kato T, Shinoura Y. Mol Gen Genet. 1977;156:121–131. doi: 10.1007/BF00283484. [DOI] [PubMed] [Google Scholar]
- 5.Shinagawa H, Iwasaki H, Kato T, Nakata A. Proc Natl Acad Sci USA. 1988;85:1806–1810. doi: 10.1073/pnas.85.6.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burckhardt S E, Woodgate R, Scheuermann R H, Echols H. Proc Natl Acad Sci USA. 1988;85:1811–1815. doi: 10.1073/pnas.85.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nohmi T, Battista J R, Dodson L A, Walker G C. Proc Natl Acad Sci USA. 1988;85:1816–1820. doi: 10.1073/pnas.85.6.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutreix M, Moreau P L, Bailone A, Galibert F, Battista J R, Walker G C, Devoret R. J Bacteriol. 1989;171:2415–2423. doi: 10.1128/jb.171.5.2415-2423.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sweasy J B, Witkin E M, Sinha N, Roegner-Maniscalco J Bacteriol. 1990;172:3030–3036. doi: 10.1128/jb.172.6.3030-3036.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajagopalan M, Lu C, Woodgate R, O’Donnell M, Goodman M F, Echols H. Proc Natl Acad Sci USA. 1992;85:10777–10781. doi: 10.1073/pnas.89.22.10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank E G, Gonzalez M, Ennis D G, Levine A R, Woodgate R. J Bacteriol. 1996;178:3550–3556. doi: 10.1128/jb.178.12.3550-3556.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller J H, Low K B. Cell. 1984;37:675–682. doi: 10.1016/0092-8674(84)90400-8. [DOI] [PubMed] [Google Scholar]
- 13.Witkin E M, Kogoma T. Proc Natl Acad Sci USA. 1984;81:7539–7543. doi: 10.1073/pnas.81.23.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caillet-Fauquet P, Maenhaut-Michel G. Mol Gen Genet. 1988;213:491–498. doi: 10.1007/BF00339621. [DOI] [PubMed] [Google Scholar]
- 15.Schaaper R M, Loeb L A. Proc Natl Acad Sci USA. 1981;78:1773–1777. doi: 10.1073/pnas.78.3.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacob F. C R Hebd Seances Acad Sci. 1954;238:732–734. [PubMed] [Google Scholar]
- 17.Devoret R. C R Hebd Seances Acad Sci. 1965;260:1510–1513. [PubMed] [Google Scholar]
- 18.Ichikawa-Ryo H, Kondo S. J Mol Biol. 1975;97:77–92. doi: 10.1016/s0022-2836(75)80023-4. [DOI] [PubMed] [Google Scholar]
- 19.Caillet-Fauquet P, Maenhaut-Michel G, Radman M. EMBO J. 1984;3:707–712. doi: 10.1002/j.1460-2075.1984.tb01873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood R D, Hutchinson F. J Mol Biol. 1984;173:293–305. doi: 10.1016/0022-2836(84)90122-0. [DOI] [PubMed] [Google Scholar]
- 21.Maenhaut-Michel G, Caillet-Fauquet P. J Mol Biol. 1984;177:181–187. doi: 10.1016/0022-2836(84)90064-0. [DOI] [PubMed] [Google Scholar]
- 22.Brotcorne-Lannoye A, Maenhaut-Michel G. Proc Natl Acad Sci USA. 1986;83:3904–3908. doi: 10.1073/pnas.83.11.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maenhaut-Michel G, Caillet-Fauquet P. Mutat Res. 1990;230:241–254. doi: 10.1016/0027-5107(90)90062-9. [DOI] [PubMed] [Google Scholar]
- 24.Kenyon C J, Walker G C. Proc Natl Acad Sci USA. 1980;77:2819–2823. doi: 10.1073/pnas.77.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohmori H, Hatada E, Qiao Y, Tsuji M, Fukuda R. Mutat Res. 1995;347:1–7. doi: 10.1016/0165-7992(95)90024-1. [DOI] [PubMed] [Google Scholar]
- 26.Vogel H J, Bonner D M. J Biol Chem. 1956;218:97–106. [PubMed] [Google Scholar]
- 27.Casadaban M J, Cohen S N. J Mol Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 28.Sancar A, Hach A M, Rupp W D. J Bacteriol. 1979;137:692–693. doi: 10.1128/jb.137.1.692-693.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cupples C G, Miller J H. Proc Natl Acad Sci USA. 1988;86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cupples C G, Cabrera M, Cruz C, Miller J H. Genetics. 1990;125:275–280. doi: 10.1093/genetics/125.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeWitt S K, Adelberg E A. Genetics. 1962;47:577–586. doi: 10.1093/genetics/47.5.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada M, Hakura A, Sofuni T, Nohmi T. J Bacteriol. 1993;175:5539–5547. doi: 10.1128/jb.175.17.5539-5547.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 34.Yasuda T, Nagata T, Ohmori H. J Bacteriol. 1996;178:3854–3859. doi: 10.1128/jb.178.13.3854-3859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohara Y, Akiyama K, Isono K. Cell. 1987;50:495–508. doi: 10.1016/0092-8674(87)90503-4. [DOI] [PubMed] [Google Scholar]
- 36.Wang R F, Kushner S R. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- 37.Watanabe M, Nohmi T, Ohta T. Mutat Res. 1994;314:27–37. doi: 10.1016/0921-8777(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe M, Nohmi T, Ohta T. Mutat Res. 1994;314:39–49. doi: 10.1016/0921-8777(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 40.Laemmli U K, Farve M. J Mol Biol. 1973;80:575–599. doi: 10.1016/0022-2836(73)90198-8. [DOI] [PubMed] [Google Scholar]
- 41.Kenyon C J, Brent R, Ptashne M, Walker G C. J Mol Biol. 1982;160:445–457. doi: 10.1016/0022-2836(82)90307-2. [DOI] [PubMed] [Google Scholar]
- 42.Ohmori H, Saito M, Yasuda T, Nagata T, Fujii T, Wachi M, Nagai K. J Bacteriol. 1995;177:156–165. doi: 10.1128/jb.177.1.156-165.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elledge S J, Walker G C. J Mol Biol. 1983;164:175–192. doi: 10.1016/0022-2836(83)90074-8. [DOI] [PubMed] [Google Scholar]
- 44.Napolitano R L, Lambert I B, Fuchs R P P. Proc Natl Acad Sci USA. 1997;94:5733–5738. doi: 10.1073/pnas.94.11.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janel-Binz R, Maenhaut-Michel G, Fuchs R P P. Mol Gen Genet. 1994;245:279–285. doi: 10.1007/BF00290107. [DOI] [PubMed] [Google Scholar]
- 46.Maenhaut-Michel G, Janel-Binz R, Fuchs R P P. Mol Gen Genet. 1992;235:373–380. doi: 10.1007/BF00279383. [DOI] [PubMed] [Google Scholar]
- 47.Frank E G, Ennis D G, Gonzalez M, Levine A S, Woodgate R. Proc Natl Acad Sci USA. 1996;93:10291–10296. doi: 10.1073/pnas.93.19.10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis K L, Harlow G R, Gregg-Jolly L A, Mount D W. J Mol Biol. 1994;241:507–523. doi: 10.1006/jmbi.1994.1528. [DOI] [PubMed] [Google Scholar]
- 49.Modrich P. Annu Rev Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- 50.Pukkila P J, Peterson J, Herman G, Modrich P, Meselson M. Genetics. 1983;104:571–582. doi: 10.1093/genetics/104.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schaaper R M. Proc Natl Acad Sci USA. 1988;85:8126–8130. doi: 10.1073/pnas.85.21.8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Radman M, Villani G, Boiteux S, Kinsella A R, Glickman B W, Sparari S. Cold Spring Harbor Symp Quant Biol. 1978;43:937–946. doi: 10.1101/sqb.1979.043.01.103. [DOI] [PubMed] [Google Scholar]
- 53.Taddei F, Matic I, Radman M. Proc Natl Acad Sci USA. 1995;92:11736–11740. doi: 10.1073/pnas.92.25.11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cairns J, Overbaugh J, Miller S. Nature (London) 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- 55.Foster P L. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Torkelson J, Harris R S, Lombardo M-J, Nagendran J, Thulin C, Rosenberg S M. EMBO J. 1997;16:3303–3311. doi: 10.1093/emboj/16.11.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kulaeva O I, Koonin E V, McDonald J P, Randall S K, Rabinovich N, Connaughton J F, Levine A S, Woodgate R. Mutat Res. 1996;357:245–253. doi: 10.1016/0027-5107(96)00164-9. [DOI] [PubMed] [Google Scholar]
- 58.Woodgate R. Mutat Res. 1992;281:221–225. doi: 10.1016/0165-7992(92)90012-7. [DOI] [PubMed] [Google Scholar]
- 59.Marsh L, Nohmi T, Hinton S, Walker G C. Mutat Res. 1991;250:183–197. doi: 10.1016/0027-5107(91)90175-n. [DOI] [PubMed] [Google Scholar]