

Abstract
High-risk human papillomavirus (HPV) infection is the primary risk factor for cervical cancer. HPVs establish persistent infection by maintaining their genomes as extrachromosomal elements (episomes) that replicate along with host DNA in infected cells. The productive life cycle of HPV is intimately tied to the differentiation program of host squamous epithelium. This review examines the involvement of host chromatin in multiple aspects of the papillomavirus life cycle and the malignant progression of infected host cells. Papillomavirus utilizes host mitotic chromosomes as vehicles for transmitting its genetic materials across the cell cycle. By hitchhiking on host mitotic chromosomes, the virus ensures accurate segregation of the replicated viral episomes to the daughter cells during host cell division. This strategy allows persistent maintenance of the viral episome in the infected cells. In the meantime, the virus subverts the host chromatin-remodeling factors to promote viral transcription and efficient propagation of viral genomes. By associating with the host chromatin, papillomavirus redirects the normal cellular control of chromatin to create a cellular environment conducive to both its own survival and malignant progression of host cells. Comprehensive understanding of HPV-host chromatin interaction will offer new insights into the HPV life cycle as well as chromatin regulation. This virus-host interaction will also provide a paradigm for investigating other episomal DNA tumor viruses that share a similar mechanism for interacting with host chromatin.
Papillomavirus and cancer
Papillomaviruses (PVs) are small DNA tumor viruses that induce a variety of benign and malignant epithelial lesions in the infected hosts [1]. Over 140 HPV types have been identified to date. Depending on the potential for inducing malignant transformation, these viruses are further classified into high-risk and low-risk HPVs. High-risk HPVs are strongly associated with the development of cervical cancer [2], which is the second most common cancer among women worldwide and the leading cause of death from cancer among women in developing countries; approximately 500,000 cases and 275,000 deaths are reported annually. Over 97% of cervical cancers contain a high-risk HPV genome and express the viral oncogenes E6 and E7, providing a direct link between HPV infection and carcinogenesis. Currently available HPV vaccines protect against up to four major types of cancer causing strains. Alternative approaches are needed for curing ongoing infections because HPV can establish latent infections that persist for years or even decades in host cells. The deregulated expression of high-risk HPV oncogenes is a critical event for the oncogenic progression of HPV-positive lesions. In addition, substantial cytogenetic changes are needed for high-risk HPV-infected cells to progress to invasive tumors. These cellular events do not occur until many years after the initial infection, supporting the concept that long-term infection with high-risk HPVs is required for the malignant progression of HPV pathogenic lesions.
The differentiation-dependent papillomavirus life cycle
PVs contain circular double-stranded DNA genomes of approximately 8000 base pairs. The viruses have a specific tropism for squamous epithelial cells and must infect cells within the dividing basal layer. The productive life cycle of HPV is intimately tied to the differentiation status of the host squamous epithelium [1]. Upon infecting basal cells, the viral genomes are established as episomes that replicate along with host DNA. The various phases of the HPV life cycle are controlled through tightly regulated activation of the early and late viral promoters as the infected basal cell migrates towards the epithelial surface [3]. In the basal epithelium where initial stages of the viral life cycle takes place, the early viral promoter is transcribed to express the viral E1, E2, E6 and E7 genes [4, 5]. The papillomavirus E2 is a regulatory protein essential for creating favorable cellular conditions to establish infection and to properly complete the viral life cycle. E2 is a sequence-specific DNA binding protein involved in viral DNA replication, episome maintenance, and viral transcription [3]. It consists of an N-terminal transcriptional activation domain linked to a C-terminal DNA binding/dimerization domain by a flexible hinge [6]. The multiple functions of E2 rely on its sequence-specific recognition of a number of E2 binding sites within papillomavirus genomes. E2 initiates viral genome replication by loading the viral helicase E1 onto the origin of replication [7, 8], allowing the viral episome to be maintained at low copy numbers in the basal epithelium. During mitosis, E2 ensures accurate partitioning of the replicated viral genomes to daughter cells by tethering them to host mitotic chromosomes [9-13]. E2 binds to four E2-binding sites (E2BSs) in the long control region (LCR) of the HPV genome in a cooperative manner that results in either activation (at low levels of E2) or repression (at high concentrations of E2) of E6 and E7 expression from the early promoter [14, 15]. E6 and E7 are the primary viral oncogenes, which promote cell growth through a variety of mechanisms including inactivation of the p53 and pRb tumor suppressors, respectively [16-19]. This strict control of the viral early promoter allows the minimum expression of E6 and E7 that is needed to drive cells into S-phase for viral genome replication, while preventing the inopportune over-expression of these viral oncoproteins that leads to dysplasias and carcinomas. As discussed below, the cellular factors that contribute to the tightly regulated viral transcription are not very well understood. As HPV-positive cells differentiate, the late promoter is activated, leading to the production of the late genes and the high-level amplification of the viral genome [3]. In the highly differentiated suprabasal cells, viral DNA is packaged into newly formed capsids and progeny virions are released from the cell [1].
During the malignant progression of high-risk HPV-positive lesions, the viral DNA often integrates into the host genome, resulting in the disruption of the E2 gene [1, 20, 21]. The loss of E2 expression de-represses the viral oncogene E6 and E7 expression, which in turn stimulates cellular proliferation [18, 22-25]. As such, the disruption of E2 gene has been mechanistically linked to malignant progression of HPV-associated cancers [1].
Papillomavirus chromatin structure
Papillomavirus genomes are organized in the form of specifically positioned nucleosomes packaged into chromatin [26-29]. The chromatin-like structures were first reported for full particles of Bovine papillomavirus (BPV) and HPV purified from bovine cutaneous fibropapillomas and human plantar warts respectively [29]. Under an electron microscope, the BPV and HPV nucleoprotein complexes appear as circular structures composed of nucleosomes interconnected by a naked DNA filament [29]. The further investigation of papillomavirus chromatin was long hampered by the absence of a tissue culture system for vegetative growth of these viruses. BPV1-transformed hamster embryo fibroblasts (HEF), containing 200 to 250 copies of episomal genome per cell, therefore offer a great source of the episome. Using this system, episomal viral chromatin was isolated for the first time [30]. In situ DNase digestion and restriction endonucleases cleavage has identified a major DNase-hypersensitive region in the transcriptional regulatory elements of the BPV1 chromatin [30]. Further analysis of the episomal nucleoprotein complexes of BPV1 using DNase I treatment localized hypersensitive regions to noncoding parts of the genome coinciding with the origin of replication and the 5′ ends of most of the early mRNAs. Additionally, regions of hypersensitivity were identified within the structural genes [31]. Similar to the episomal viral chromatin, nucleosome organization analysis of integrated HPV18 DNA in HeLa cells revealed a nuclease-hypersensitive site within the viral noncoding regulatory region that harbors transcriptional control sequences, which map to the 5′ ends of most of the early mRNAs [32]. Chromatin structure analysis of the nuclei of CaSki cells carrying 500 intrachromosomal copies of HPV16 identified nucleosomes in specific positions within the LCR of HPV16 and the E6 and E7 genes [26]. These nucleosomes function to repress the activity of the E6 promoter during in vitro transcription of HPV16 chromatin. The repression is relieved by the addition of transcription factors AP-1 and Sp1 and it has been shown that the latter has the ability to alter the nucleosomal structure in vitro. The chromatin organization of the HPV16 genome suggests important regulatory roles of nucleosomes in the viral oncogene transcription [26]. In vivo studies are needed to further understand how chromatin dynamics regulates viral transcription during the differentiation-dependent viral life cycle.
Throughout the PV life cycle, the viral chromatin structure is tightly regulated at different stages of epithelial differentiation to control the viral early and late gene expression. DNase I hypersensitivity analysis of HPV 31 chromatin in cell lines that maintain viral genomes extrachromosomally reveals a major shift in nuclease sensitivity upon differentiation [27]. In undifferentiated cells, hypersensitive regions were detected in the upstream regulatory region (URR) involved in the early gene transcription. Upon differentiation, the late transcript initiation region becomes accessible to DNase I, indicating a differentiation-induced chromatin remodeling around the late promoter [27]. The state of histone modifications as well as the spectrum of transcription factors bound to the early and late HPV 31 viral promoters were further examined in both undifferentiated and differentiated cells using chromatin immunoprecipitation assays [33]. In undifferentiated cells, the chromatin surrounding both early and late viral promoter regions reveals an open, transcriptionally active state with dimethylated forms of histone H3 K4 as well as acetylated histone H3 and H4. Upon differentiation, the levels of dimethylated H3K4 and acetylated H3 are increased around both the early and late promoter regions, while the acetylated H4 is increased at the early promoter [33]. This observation suggests that nucleosomes of both early and late promoter regions are further activated through histone modifications to coordinate the HPV transcription during differentiation [33].
Differentiation-associated regulation of viral transcription is also thought to involve changes in the binding of host cell transcription factors to the viral promoter and regulatory regions. A dynamic change in the binding of transcription factors to the keratinocyte enhancer (KE)/early promoter region in the upstream regulatory region and to the late promoter sequences was observed throughout differentiation [33]. It appears that the combinatorial binding of multiple transcription factors to the early and late promoter region contributes to the viral gene transcription during host epithelial differentiation [33]. Along this line, Carson and Khan used Panomics TransSignal protein/DNA array to identify changes in the levels of cellular transcription factors bound to the HPV16 promoter/regulatory sequences during the differentiation of W12 cells which contain HPV16 episomes. They identified approximately 30 transcription factors that specifically bind to HPV16 sequences and may be involved in regulating HPV16 transcription during differentiation [34]. In most of the cases, the transcription factor binding to HPV16 promoters is increased during epithelial differentiation [34]. How these transcription factors contribute to regulation of the differentiation-dependent viral transcription remained to be investigated.
Papillomavirus episome maintenance
During latent infection, HPVs maintain the viral genome as autonomous replicating episomes in proliferating basal epithelial cells [1]. The functions of the high-risk HPV E6 and E7 oncoproteins are essential for the viral genome to be stably maintained as an episome in the infected cells [35]. It was postulated that these oncoproteins may create a suitable cellular environment for HPV maintenance through abrogating the cellular checkpoints that block the long-term retention of viral episomes [36]. In addition, to establish persistence in host cells, PVs have evolved a strategy for hitchhiking on cellular mitotic chromosomes (Fig. 1A). This mechanism ensures that the replicated viral episomes are retained inside the nuclei of dividing host cells and faithfully partition to the daughter cells during mitosis (Fig. 1A). For many papillomaviruses, this non-covalent association of viral DNA with chromosomes is mediated by the viral E2 protein (Fig. 1B). BPV1 genomes persist as stable episomes in transformed rodent cells, and have been used as a model system to establish E2's role in viral genome maintenance [9-11, 13, 37]. E2 and BPV1 episomes are closely associated with mitotic chromatin in dividing cells [10, 11, 13]. Long-term maintenance of viral episomes requires E2 binding to the multiple E2 binding sites within a cis-minichromosome maintenance element (MME) of the viral genome [12]. These studies demonstrate that E2 facilitates viral genome segregation by interacting simultaneously with condensed mitotic chromatin and viral genomes. However, the DNA binding function of E2 is not necessary for its association with mitotic chromosomes [13]. A cellular protein was therefore postulated to link E2 to cellular mitotic chromosomes.
Fig. 1.
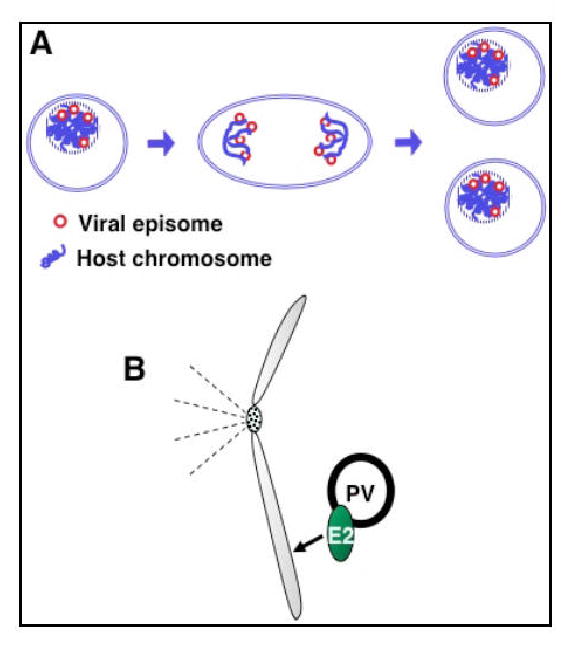
Papillomaviruses establish persistent infection by maintaining viral genomes as episomes in host cells. A. During persistent infection, some of the PVs hitchhike on cellular mitotic chromosomes to ensure that the replicated viral episomes are retained inside the nuclei of dividing host cells and faithfully partition to the daughter cells during mitosis. B. For many papillomaviruses, this non-covalent association of viral DNA with chromosomes is mediated by the viral E2 protein. The viral E2 facilitates viral genome segregation by interacting simultaneously with condensed mitotic chromatin and viral genomes.
Brd4 is a host chromatin adaptor for papillomavirus
Using proteomic tandem affinity purification (TAP) analysis of E2 functional partners in host cells, the cellular bromodomain protein Brd4 was identified as a major binding protein for BPV1 E2 [38]. Brd4 is a member of the BET family proteins that harbor double bromodomains for association with mitotic chromosomes [39]. It binds the N-terminal transactivation domain of E2 and colocalizes with E2 on mitotic chromosomes [38]. E2 links Brd4 to viral episomes. The E2 binding site was mapped to the C-terminal domain (CTD) of Brd4 [38]. The Brd4 CTD functions as a dominant negative inhibitor to competitively bind to E2 and dissociate the E2/viral episome complex from Brd4 on mitotic chromosomes (Fig. 2) [38]. Disruption of the E2-Brd4 interaction by the Brd4 CTD abolishes viral episome maintenance in host cells (Fig. 1)[40] and inhibits the transformation of rodent fibroblasts by BPV1 DNA [38]. These functional studies establish that mitotic chromosome-associated Brd4 represents the previously unidentified cellular receptor that tethers the BPV1 E2/viral episome complex to host mitotic chromosomes (Fig. 1) [38, 40], providing a molecular mechanism for BPV1 E2-mediated papillomavirus maintenance in infected cells.
Fig. 2.
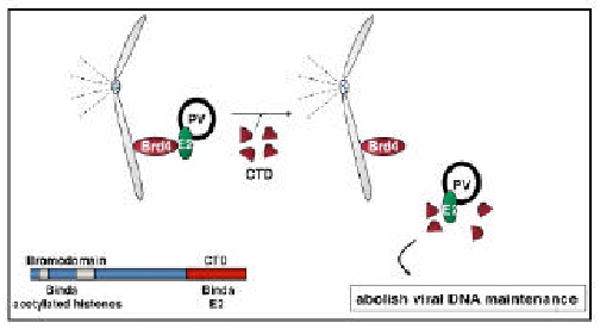
Proposed molecular mechanism for E2/Brd4 mediated episome maintenance. Brd4 functions as a receptor for PV E2/DNA complex on mitotic chromosomes. The exogenously expressed Brd4 CTD prevents the association of Brd4 with E2, abrogating the tethering of the E2/viral episome complex to mitotic chromosomes. The domain structures of Brd4 are also shown. The Figure was modified from [38].
In host cells, Brd4 plays a central role in cellular growth control [41, 42] and cell cycle progression [39, 41, 43-45]. It binds to acetylated histone H3 and H4 through its bromodomains [46]. The Brd4 knockout in mice results in early embryonic lethality. In humans, the Brd4 gene is the target of translocation t(15;19) that defines a highly lethal carcinoma [47]. Brd4 activation in human breast carcinomas induces a gene expression signature that efficiently predicts survival, establishing that dysregulation of Brd4-associated pathways may play an important role in breast cancer progression [48]. Brd4 knockdown in human cells leads to highly decondensed chromatin structure, which results in mitotic catastrophe [49].
The x-ray crystallography structure of the N-terminal domain of HPV16 E2 in complex with the C-terminus of Brd4 shows that E2 interacts with only the last 25 amino acids of Brd4 through its N-terminal transactivation domain [50]. Sequence alignment of this section in Brd4 homologs from different species shows it to be highly conserved throughout evolution [50]. It has been suggested that an important cellular factor(s) may bind to this region to contribute to Brd4's cellular function [50]. Expression of HPV E2 in cells triggers similar chromosomal structural defects and disrupted mitosis as observed in the Brd4 knockdown cells (You et al., unpublished data), indicating that E2 binding to the C-terminal domain of Brd4 may block other important cellular factors from accessing this region, leading to abnormal Brd4 function.
The Brd4-E2 interaction bridges many of the animal and human PV episomes onto mitotic chromosomes for viral genome maintenance [50-54], establishing this complex as a potential therapeutic target for PV infections. The structure of the N-terminal domain of HPV16 E2 in complex with the C-terminus of Brd4 has offered a molecular platform for developing antiviral compounds to block HPV latent infection [50].
Brd4's function in papillomavirus transcriptional regulation
Besides tethering the viral episome to mitotic chromosomes, E2-Brd4 interaction also plays important though as yet not well understood roles in E2-mediated transcriptional activation and repression of the viral oncogenes. HPV E2 transactivates heterologous promoters containing multimerized E2 binding sites [55]. On the other hand, E2 represses the promoter directing the E6 and E7 viral oncogenes in the cancer-associated HPV16 and HPV18 genomes [56-61]. In HPV-positive cells containing integrated viral DNA and a disrupted E2 gene, the dysregulated expression of E6 and E7 results in reduced p53 and pRb expression and increased cellular proliferation [22]. Re-expression of E2 in these cells represses the HPV18 E6/E7 promoter and reverses cellular immortality [62-66]. It has been suggested that E2 represses the E6/E7 promoter by binding its cognate sites proximal to the promoter and displacing other cellular transcription factors [67-69]. In addition, E2 could function as an active repressor by preventing the assembly of a functional preinitiation complex on the viral chromatin template [60].
By analyzing a series of alanine-scanning substitution mutants of the HPV16 E2 N-terminal transactivation domain, it was discovered that the amino acids involved in Brd4 binding are also required for both E2 transcriptional activation and repression of the viral E6/E7 promoter [70-72]. This observation suggests that interaction with Brd4 is crucial for both E2 transactivation and repression functions. Functional studies subsequently demonstrate that Brd4 also functions as a coactivator for E2-mediated transcription activation [70, 73-75]. In addition, Brd4 was identified as a component of the low-risk HPV11 E2 transcriptional silencing complex implicated in repression of the E6/E7 promoter [76]. Brd4 recruits E2 to its cognate binding sites within the viral promoter chromatin template in a bromodomain-dependent manner [77]. Recent findings indicate that Brd4 accomplishes the co-repressor function by recruiting E2 to block the assembly of functional preinitiation complex on the viral promoter chromatin template [60, 77]. On the other hand, Brd4-independent mechanism of E2-mediated repression was also proposed [78].
In host cells, Brd4 recruits the positive transcription elongation factor b (P-TEFb) to stimulate transcriptional elongation through phosphorylation of the C-terminal domain of the RNA polymerase II (RNAPII) [79, 80]. This complex thus contributes to cellular gene transcription as well as expression of human immunodeficiency virus and human T-lymphotropic virus genomes [81, 82]. The P-TEFb binding site in Brd4 largely overlaps with the E2 binding site [81]. In fact, E2 expressed in mammalian cells competitively inhibits the association between P-TEFb and Brd4 (Yan et al., manuscript in revision). Our recent studies establish that E2 actively represses viral E6/E7 transcription by blocking formation of the active P-TEFb-Brd4 complex on viral chromatin template (Yan et al., manuscript in revision).
Overall, Brd4 functions as a molecular adapter with its bromodomains targeting acetylated chromatin, while the C-terminal domain recruits the viral E2 protein. The multiple functions of E2 rely on its ability to interact with Brd4 and associate with cellular chromatin. Recent studies have established Brd4 as a key chromatin receptor that contributes to multiple processes of PV life cycle through interactions with E2 proteins [60, 70, 73, 74, 83]. In future studies, it will be important to understand the roles of Brd4 in the differentiation-dependent viral life cycle.
Different modes of papillomavirus anchoring on mitotic chromosomes
A wide range of papillomaviruses have adopted the strategy of tethering their genomes to host chromosomes, but individual viruses appear to use different chromosomal targets. A recent localization analysis of 13 different animal and human E2 proteins from seven papillomavirus genera have shown that E2-mediated tethering of viral genomes to mitotic chromosomes is a common strategy shared by these papillomaviruses. However, different viruses bind to different regions of the host chromosomes during mitosis. Unlike BPV1 E2 that binds to all mitotic chromosomes randomly as small speckles in complex with Brd4, several of the E2 proteins appear to localize to more specific regions of mitotic chromosomes [84]. For instance, the alpha-papillomavirus E2 proteins only closely associate with prophase and telophase chromosomes while binding to the pericentromeric region of metaphase chromosomes [84]. In contrast, the HPV8 E2 binds as large speckles at the pericentromeric regions of chromosomes [84]. Further analysis indicates that the HPV8 E2 protein targets the short arms of acrocentric mitotic chromosomes and interacts with the repeated ribosomal DNA loci of host mitotic chromosomes. The novel HPV 8 E2 tethering protein that mediates its association with specific regions of mitotic chromosomes remains to be identified [85]. In addition to mitotic chromosomes, the HPV E2 proteins have been shown to associate with mitotic spindles to enable the partitioning of HPV origin-containing plasmids as minichromosomes in transfected cells [86, 87].
The molecular mechanism involved in maintaining E2 and papillomavirus episome persistence in dividing host cells appears complex. Using a yeast two-hybrid screen, a number of additional mitotic cellular factors have been identified as interacting proteins for BPV1 E2. Among them, ChlR1, a DNA helicase that functions in sister chromatid cohesion, has been postulated as essential for loading E2 onto mitotic chromosomes [87]. Mutagenesis analysis indicates that E2 interaction with ChlR1 is required for episome maintenance in cell culture. ChlR1 associates with mitotic chromosomes during prophase; at this stage of mitosis, E2 colocalizes with ChlR1 on condensing chromatin. As cells progress to metaphase, ChlR1 dissociates from the chromatid arms and relocates to the spindle poles, while the E2 protein remains associated with the chromosomes until the completion of mitosis [87]. This temporal interaction of ChlR1 and E2 on the prophase chromosome suggests that ChlR1 is required for loading the papillomavirus E2 protein onto mitotic chromosomes during early mitosis but not for the maintenance of E2 attachment to mitotic chromosomes [87]. Since the E2 and Brd4 interaction persists throughout mitosis, it may ensure the tethering of E2 to chromosomes until the completion of mitosis [87]. Another protein identified in the BPV1 E2 yeast two-hybrid screen is MKlp2, a kinesin-like motor protein of the central mitotic spindle required for completion of cytokinesis [88]. A subpopulation of E2 was found to colocalize with MKlp2 in the midbody/midplate during late mitosis. The viral genomes also associate with MKlp2 within this stage of cell cycle. It is therefore likely that E2 interacts with this motor protein to ensure viral genome partitioning during cytokinesis [88]. HPV16 E2 was found to colocalize with TopBP1, a cellular protein essential for the initiation of cellular DNA replication, on the chromatin and centrosomes during late telophase, suggesting that TopBP1 could be the mitotic chromatin receptor for HPV16 E2 at this stage of the cell cycle [89]. Since all of the tested E2s have been shown to interact with the mitotic chromosome-associated Brd4 [60, 70, 73, 74, 83], it is conceivable that Brd4 and the identified additional cellular receptors may interact either sequentially or simultaneously with E2 and the viral episome to constitute a tethering cascade/complex for viral genome maintenance and segregation in latently infected cells. The molecular mechanisms by which HPVs adopt cellular machinery to maintain persistent infection in host cells will be an interesting topic for future research.
Cellular factors involved in viral chromatin remodeling
Host cellular factors have been shown to regulate the papillomavirus chromatin conformation, transcription and, consequently, the viral life cycle. The host cell protein nucleolin binds the HPV18 enhancer and controls the opening of the chromatin structure at the HPV18 enhancer [90]. Through this mechanism, nucleolin may function as a regulator of HPV18 oncogene transcription in cervical cancer cells [90]. BRCA1, a breast and ovarian cancer-related tumor suppressor that has been linked to chromatin remodeling, is another host factor that interacts with the HPV18 E2 protein. Recruitment of BRCA1 by E2 onto the E2 binding sites within viral promoter enhances HPV18 E2-dependent transcription in vivo [91]. Within the HPV18 genome, a strong epithelial specific enhancer drives transcription of the viral oncogenes. A higher-order nucleoprotein complex (enhanceosome) involving the sequence-specific JunB/Fra2 transcription factor and the HMG-I(Y) chromatin remodeling factor has been shown to stimulate the transcription activity of the HPV18 enhancer sequences [92]. The CBP/p300 chromatin modulator is further recruited by the HPV18 enhanceosome to stimulate the promoter activity [92]. The activation of the HPV18 P105 promoter by the coactivator CBP directly correlates with the induction of the nucleosomal histone H3 (K14) acetylation on the HPV18 Early Promoter by CBP [93]. Brm is a chromatin remodeling protein associated with SWI/SNF complexes, which are ATP-dependent chromatin remodeling enzymes. Brm association with E2 enhances promoter occupancy by E2 and stimulates E2 transcriptional activation in an episomal context [94].
Papillomavirus life cycle involves a variety of changes in the expression of the early and late viral genes and the viral transcription regulation is tightly linked to the host epithelial differentiation. During epithelial differentiation, different cellular factors have displayed distinct roles in regulating the early and late promoters of the papillomavirus. For example, CCAAT/enhancer-binding protein beta (C/EBPbeta), a key transcription factor that induces chromatin opening and the terminal differentiation of keratinocytes, enhances the P670-driven transcription of the HPV16 late genes in the differentiating epithelium, whereas it inhibits the transcription from the early P97 promoter [95].
Modulation of host chromosomal events by papillomavirus
Little is known about how the papillomavirus modulates the host chromatin structure. A histopathological study was conducted to examine chromatin texture in cervical epithelium of 6000 biopsy specimens generated from 2000 patients [96]. The texture image analysis suggests that chromatin condensation may correlate with HPV infection [96]. The overall impact of HPV on the host chromatin texture may reflect the combined effects of individual viral proteins by interacting with their cognate cellular targets. It has been demonstrated that E2 does not simply passively hitchhike on the chromosomal Brd4 protein to segregate the papillomavirus genomes; rather it plays an active role in relocalizing and/or stabilizing the association of Brd4 with both mitotic chromosomes and interphase chromatin [51]. Through interacting with Brd4, E2 becomes associated with transcriptionally active cellular chromatin [97]. It has become apparent that E2-mediated tethering of viral genomes to host chromatin not only drives the partition of the viral genomes to daughter cells but also retains the viral genomes in transcriptionally active regions of the nucleus to escape silencing [97].
Studies on the viral oncogenes E6 and E7 have revealed more direct roles of the viral oncoproteins in modulating host chromatin structure. In vitro studies establish that E6 inhibits p300-mediated acetylation on both p53 and nucleosomal core histones, thereby converting p53-p300 from an activating complex to a chromatin repressor [98]. This study thus provides a mechanism for DNA tumor viruses to repress p53-dependent transcription from cellular chromatin [98]. In human foreskin keratinocytes, HPV16 E7 has been shown to increase histone acetylation in a manner that depends on its binding to both pRB and histone deacetylase. This functional interaction may therefore indirectly create a transcriptionally active chromatin structure to promote expression of genes vital for cell cycle progression [99]. During mitosis, high-risk HPV16 E7 interacts with microtubule-associated nuclear mitotic apparatus protein 1 (NuMA) [100]. The interaction between HPV16 E7 and NuMA correlates with the induction of chromosome alignment defects during prometaphase [100]. It has been proposed that this abrogation of mitotic events by HPV E7 may contribute to viral maintenance and propagation through disrupting the differentiation program of infected epithelia cells [100]. The high-risk HPV E7 protein also interacts with BRG-1, a component of the human SWI/SNF complex that either activates or represses cellular promoters by modulating chromatin structure [101]. This protein-protein interaction deregulates the BRG-1 mediated transcriptional silencing, contributing to the abolishment of host cell cycle control [101].
Perspective and Future Directions
Papillomavirus relies on the association with host chromatin for successful completion of its life cycle. By hitchhiking on the host mitotic chromosomes, the virus ensures accurate segregation of the viral genetic materials to the daughter cells during host cell division, establishing persistent maintenance of viral episome in the infected cells. During this process, the virus subverts host chromatin-remodeling factors to facilitate viral transcription and replication. Through its association with the host chromatin, papillomavirus redirects the normal cellular control of chromatin to benefit its own survival and to induce malignant progression of host cells (Fig. 3).
Fig. 3.
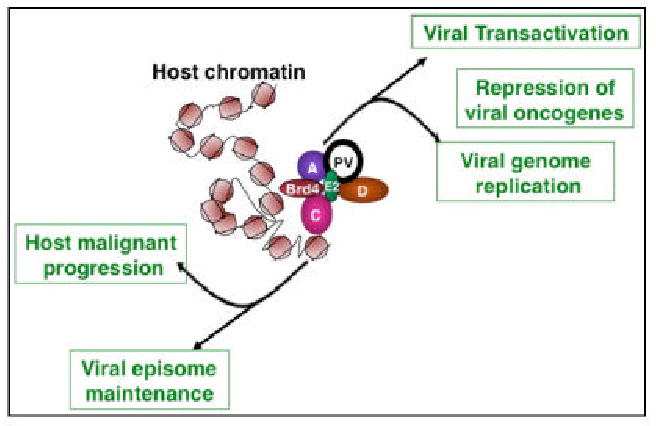
Papillomavirus interaction with cellular chromatin. Through association with the host chromatin, the virus ensures accurate segregation of the viral genetic materials to the daughter cells during host cell division, establishing persistent maintenance of viral episome in the infected cells. Meanwhile, the virus subverts host cellular factors to facilitate viral transcription and replication. Papillomavirus and host chromatin interaction also redirects the normal cellular control of chromatin to induce malignant progression of host cells.
Brd4 has emerged as a central player in bridging the papillomavirus interaction to cellular chromatin. Brd4 tethers the viral genome to host chromosomes to ensure persistent infection. It also plays an important role in regulating the viral oncogene transcription. The molecular mechanism underlying the multiple functions of Brd4 in the papillomavirus life cycle remain to be further investigated in future studies. Notably, all the studies of E2-Brd4 interactions so far have been done using monolayer cell cultures. The functional impact of this virus-host interaction in the context of the differentiation-dependent viral life cycle remains an important question. Brd4 by itself plays an important role in host cellular growth control and cancer development [44, 48, 102]. The important Brd4 activities uncovered in the viral life cycle will serve as a starting point for unraveling the complex cellular functions of Brd4.
The virus-chromatin interaction throughout the papillomavirus life cycle provides an excellent model system to study the HPV life cycle as well as chromatin structure regulation. The new mechanisms identified in these studies will provide a point of departure for developing new compounds to abrogate the virus-host interaction and cure HPV persistent infections. The viral oncogenes E6 and E7 are the main arbitrators of HPV-induced oncogenesis. Insights into mechanisms that repress the viral oncogenes will also offer new strategies to prevent papillomavirus-induced human cancer.
Many other viruses share the same mechanism as papillomaviruses for interacting with host chromatin. For instance, both the Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen (LANA) and the Epstein-Barr virus EBNA1 proteins functionally interact with Brd4 [103] [104] [105]. Besides DNA tumor viruses, Brd4 has also been implicated in the regulation of human immunodeficiency virus transcription [106] and the human cytomegalovirus immediate-early (IE) transcription [107]. Hence, comprehensive understanding of HPV-host chromatin interactions will provide a paradigm for investigating molecular interplays between other pathogenic viruses and their host targets.
Acknowledgments
I thank members of my laboratory for stimulating discussions and valuable comments on the manuscript. I apologize to my colleagues whose relevant work is not cited due to space limitations. JY is supported by HIV-Associated Malignancies Pilot Project Award (National Cancer Institute) and McCABE Fund Pilot Award (University of Pennsylvania).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Howley PM, Lowy DR. Papillomaviruses and their replication. In: Knipe DM, Howley PM, editors. Filelds virology. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2001. p. 2197. [Google Scholar]
- 2.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 3.Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16:83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 4.Hummel M, Hudson JB, Laimins LA. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol. 1992;66:6070–6080. doi: 10.1128/jvi.66.10.6070-6080.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ozbun MA, Meyers C. Human papillomavirus type 31b E1 and E2 transcript expression correlates with vegetative viral genome amplification. Virology. 1998;248:218–230. doi: 10.1006/viro.1998.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McBride AA, Romanczuk H, Howley PM. The papillomavirus E2 regulatory proteins. J Biol Chem. 1991;266:18411–18414. [PubMed] [Google Scholar]
- 7.Berg M, Stenlund A. Functional interactions between papillomavirus E1 and E2 proteins. J Virol. 1997;71:3853–3863. doi: 10.1128/jvi.71.5.3853-3863.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science. 1990;250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 9.Bastien N, McBride AA. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology. 2000;270:124–134. doi: 10.1006/viro.2000.0265. [DOI] [PubMed] [Google Scholar]
- 10.Ilves I, Kivi S, Ustav M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which Is mediated by the viral E2 protein and its binding sites. J Virol. 1999;73:4404–4412. doi: 10.1128/jvi.73.5.4404-4412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehman CW, Botchan MR. Segregation of viral plasmids depends on tethering to chromosomes and is regulated by phosphorylation. Proc Natl Acad Sci U S A. 1998;95:4338–4343. doi: 10.1073/pnas.95.8.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piirsoo M, Ustav E, Mandel T, Stenlund A, Ustav M. Cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. Embo J. 1996;15:1–11. [PMC free article] [PubMed] [Google Scholar]
- 13.Skiadopoulos MH, McBride AA. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J Virol. 1998;72:2079–2088. doi: 10.1128/jvi.72.3.2079-2088.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouvard V, Storey A, Pim D, Banks L. Characterization of the human papillomavirus E2 protein: evidence of trans-activation and trans-repression in cervical keratinocytes. Embo J. 1994;13:5451–5459. doi: 10.1002/j.1460-2075.1994.tb06880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steger G, Corbach S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J Virol. 1997;71:50–58. doi: 10.1128/jvi.71.1.50-58.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. Embo J. 1991;10:4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 18.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 19.Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. Embo J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol. 1995;69:2989–2997. doi: 10.1128/jvi.69.5.2989-2997.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corden SA, Sant-Cassia LJ, Easton AJ, Morris AG. The integration of HPV-18 DNA in cervical carcinoma. Mol Pathol. 1999;52:275–282. doi: 10.1136/mp.52.5.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munger K, Scheffner M, Huibregtse JM, Howley PM. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 1992;12:197–217. [PubMed] [Google Scholar]
- 23.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 25.Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stunkel W, Bernard HU. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J Virol. 1999;73:1918–1930. doi: 10.1128/jvi.73.3.1918-1930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.del Mar Pena LM, Laimins LA. Differentiation-dependent chromatin rearrangement coincides with activation of human papillomavirus type 31 late gene expression. J Virol. 2001;75:10005–10013. doi: 10.1128/JVI.75.20.10005-10013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swindle CS, Engler JA. Association of the human papillomavirus type 11 E1 protein with histone H1. J Virol. 1998;72:1994–2001. doi: 10.1128/jvi.72.3.1994-2001.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Favre M, Breitburd F, Croissant O, Orth G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J Virol. 1977;21:1205–1209. doi: 10.1128/jvi.21.3.1205-1209.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosl F, Waldeck W, Sauer G. Isolation of episomal bovine papillomavirus chromatin and identification of a DNase I-hypersensitive region. J Virol. 1983;46:567–574. doi: 10.1128/jvi.46.2.567-574.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosl F, Waldeck W, Zentgraf H, Sauer G. Properties of intracellular bovine papillomavirus chromatin. J Virol. 1986;58:500–507. doi: 10.1128/jvi.58.2.500-507.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosl F, Westphal EM, zur Hausen H. Chromatin structure and transcriptional regulation of human papillomavirus type 18 DNA in HeLa cells. Mol Carcinog. 1989;2:72–80. doi: 10.1002/mc.2940020205. [DOI] [PubMed] [Google Scholar]
- 33.Wooldridge TR, Laimins LA. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology. 2008;374:371–380. doi: 10.1016/j.virol.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carson A, Khan SA. Characterization of transcription factor binding to human papillomavirus type 16 DNA during cellular differentiation. J Virol. 2006;80:4356–4362. doi: 10.1128/JVI.80.9.4356-4362.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc Natl Acad Sci U S A. 1999;96:8449–8454. doi: 10.1073/pnas.96.15.8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadaja M, Silla T, Ustav E, Ustav M. Papillomavirus DNA replication -from initiation to genomic instability. Virology. 2009;384:360–368. doi: 10.1016/j.virol.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 37.Law MF, Lowy DR, Dvoretzky I, Howley PM. Mouse cells transformed by bovine papillomavirus contain only extrachromosomal viral DNA sequences. Proc Natl Acad Sci U S A. 1981;78:2727–2731. doi: 10.1073/pnas.78.5.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117:349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- 39.Dey A, Ellenberg J, Farina A, Coleman AE, Maruyama T, Sciortino S, Lippincott-Schwartz J, Ozato K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol Cell Biol. 2000;20:6537–6549. doi: 10.1128/mcb.20.17.6537-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.You J, Schweiger MR, Howley PM. Inhibition of E2 binding to Brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J Virol. 2005;79:14956–14961. doi: 10.1128/JVI.79.23.14956-14961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maruyama T, Farina A, Dey A, Cheong J, Bermudez VP, Tamura T, Sciortino S, Shuman J, Hurwitz J, Ozato K. A Mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol Cell Biol. 2002;22:6509–6520. doi: 10.1128/MCB.22.18.6509-6520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RS. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol. 2002;22:3794–3802. doi: 10.1128/MCB.22.11.3794-3802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishiyama A, Dey A, Miyazaki J, Ozato K. Brd4 is required for recovery from antimicrotubule drug-induced mitotic arrest: preservation of acetylated chromatin. Mol Biol Cell. 2006;17:814–823. doi: 10.1091/mbc.E05-08-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mochizuki K, Nishiyama A, Jang MK, Dey A, Ghosh A, Tamura T, Natsume H, Yao H, Ozato K. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J Biol Chem. 2008;283:9040–9048. doi: 10.1074/jbc.M707603200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Z, He N, Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol. 2008;28:967–976. doi: 10.1128/MCB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A. 2003;100:8758–8763. doi: 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.French CA, Miyoshi I, Aster JC, Kubonishi I, Kroll TG, Dal Cin P, Vargas SO, Perez-Atayde AR, Fletcher JA. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19) Am J Pathol. 2001;159:1987–1992. doi: 10.1016/S0002-9440(10)63049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crawford NP, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, Lee MP, Ozato K, Hunter KW. Bromodomain 4 activation predicts breast cancer survival. Proc Natl Acad Sci U S A. 2008;105:6380–6385. doi: 10.1073/pnas.0710331105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.You J, Li Q, Wu C, Kim J, Ottinger M, Howley PM. Regulation of aurora B expression by the bromodomain protein Brd4. Mol Cell Biol. 2009;29:5094–5103. doi: 10.1128/MCB.00299-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abbate EA, Voitenleitner C, Botchan MR. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol Cell. 2006;24:877–889. doi: 10.1016/j.molcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 51.McPhillips MG, Ozato K, McBride AA. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J Virol. 2005;79:8920–8932. doi: 10.1128/JVI.79.14.8920-8932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baxter MK, McPhillips MG, Ozato K, McBride AA. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J Virol. 2005;79:4806–4818. doi: 10.1128/JVI.79.8.4806-4818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brannon AR, Maresca JA, Boeke JD, Basrai MA, McBride AA. Reconstitution of papillomavirus E2-mediated plasmid maintenance in Saccharomyces cerevisiae by the Brd4 bromodomain protein. Proc Natl Acad Sci U S A. 2005;102:2998–3003. doi: 10.1073/pnas.0407818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cardenas-Mora J, Spindler JE, Jang MK, McBride AA. Dimerization of the papillomavirus E2 protein is required for efficient mitotic chromosome association and Brd4 binding. J Virol. 2008;82:7298–7305. doi: 10.1128/JVI.00772-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirochika H, Hirochika R, Broker TR, Chow LT. Functional mapping of the human papillomavirus type 11 transcriptional enhancer and its interaction with the trans-acting E2 proteins. Genes Dev. 1988;2:54–67. doi: 10.1101/gad.2.1.54. [DOI] [PubMed] [Google Scholar]
- 56.Demeret C, Desaintes C, Yaniv M, Thierry F. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J Virol. 1997;71:9343–9349. doi: 10.1128/jvi.71.12.9343-9349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Romanczuk H, Thierry F, Howley PM. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P97 and type 18 P105 promoters. J Virol. 1990;64:2849–2859. doi: 10.1128/jvi.64.6.2849-2859.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan SH, Gloss B, Bernard HU. During negative regulation of the human papillomavirus-16 E6 promoter, the viral E2 protein can displace Sp1 from a proximal promoter element. Nucleic Acids Res. 1992;20:251–256. doi: 10.1093/nar/20.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thierry F, Howley PM. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol. 1991;3:90–100. [PubMed] [Google Scholar]
- 60.Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282:13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 61.Bernard BA, Bailly C, Lenoir MC, Darmon M, Thierry F, Yaniv M. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J Virol. 1989;63:4317–4324. doi: 10.1128/jvi.63.10.4317-4324.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dowhanick JJ, McBride AA, Howley PM. Suppression of cellular proliferation by the papillomavirus E2 protein. J Virol. 1995;69:7791–7799. doi: 10.1128/jvi.69.12.7791-7799.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Francis DA, Schmid SI, Howley PM. Repression of the integrated papillomavirus E6/E7 promoter is required for growth suppression of cervical cancer cells. J Virol. 2000;74:2679–2686. doi: 10.1128/jvi.74.6.2679-2686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hwang ES, Riese DJ, 2nd, Settleman J, Nilson LA, Honig J, Flynn S, DiMaio D. Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J Virol. 1993;67:3720–3729. doi: 10.1128/jvi.67.7.3720-3729.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thierry F, Yaniv M. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. Embo J. 1987;6:3391–3397. doi: 10.1002/j.1460-2075.1987.tb02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–12518. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Demeret C, Yaniv M, Thierry F. The E2 transcriptional repressor can compensate for Sp1 activation of the human papillomavirus type 18 early promoter. J Virol. 1994;68:7075–7082. doi: 10.1128/jvi.68.11.7075-7082.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong G, Broker TR, Chow LT. Human papillomavirus type 11 E2 proteins repress the homologous E6 promoter by interfering with the binding of host transcription factors to adjacent elements. J Virol. 1994;68:1115–1127. doi: 10.1128/jvi.68.2.1115-1127.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan SH, Leong LE, Walker PA, Bernard HU. The human papillomavirus type 16 E2 transcription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J Virol. 1994;68:6411–6420. doi: 10.1128/jvi.68.10.6411-6420.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schweiger MR, You J, Howley PM. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol. 2006;80:4276–4285. doi: 10.1128/JVI.80.9.4276-4285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishimura A, Ono T, Ishimoto A, Dowhanick JJ, Frizzell MA, Howley PM, Sakai H. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J Virol. 2000;74:3752–3760. doi: 10.1128/jvi.74.8.3752-3760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goodwin EC, Naeger LK, Breiding DE, Androphy EJ, DiMaio D. Transactivation-competent bovine papillomavirus E2 protein is specifically required for efficient repression of human papillomavirus oncogene expression and for acute growth inhibition of cervical carcinoma cell lines. J Virol. 1998;72:3925–3934. doi: 10.1128/jvi.72.5.3925-3934.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol. 2006;80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ilves I, Maemets K, Silla T, Janikson K, Ustav M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J Virol. 2006;80:3660–3665. doi: 10.1128/JVI.80.7.3660-3665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Senechal H, Poirier GG, Coulombe B, Laimins LA, Archambault J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology. 2007;358:10–17. doi: 10.1016/j.virol.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 76.Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee AY, Chiang CM. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J Biol Chem. 2009;284:2778–2786. doi: 10.1074/jbc.M805835200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schweiger MR, Ottinger M, You J, Howley PM. Brd4-independent transcriptional repression function of the papillomavirus e2 proteins. J Virol. 2007;81:9612–9622. doi: 10.1128/JVI.00447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 80.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 81.Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. 2007;104:13690–13695. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho WK, Zhou M, Jang MK, Huang K, Jeong SJ, Ozato K, Brady JN. Modulation of the Brd4/P-TEFb interaction by the human T-lymphotropic virus type 1 tax protein. J Virol. 2007;81:11179–11186. doi: 10.1128/JVI.00408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McBride AA, McPhillips MG, Oliveira JG. Brd4: tethering, segregation and beyond. Trends Microbiol. 2004;12:527–529. doi: 10.1016/j.tim.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 84.Oliveira JG, Colf LA, McBride AA. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc Natl Acad Sci U S A. 2006;103:1047–1052. doi: 10.1073/pnas.0507624103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poddar A, Reed SC, McPhillips MG, Spindler JE, McBride AA. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J Virol. 2009;83:640–650. doi: 10.1128/JVI.01936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Tine BA, Dao LD, Wu SY, Sonbuchner TM, Lin BY, Zou N, Chiang CM, Broker TR, Chow LT. Human papillomavirus (HPV) origin-binding protein associates with mitotic spindles to enable viral DNA partitioning. Proc Natl Acad Sci U S A. 2004;101:4030–4035. doi: 10.1073/pnas.0306848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parish JL, Bean AM, Park RB, Androphy EJ. ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol Cell. 2006;24:867–876. doi: 10.1016/j.molcel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 88.Yu T, Peng YC, Androphy EJ. Mitotic kinesin-like protein 2 binds and colocalizes with papillomavirus E2 during mitosis. J Virol. 2007;81:1736–1745. doi: 10.1128/JVI.01638-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Donaldson MM, Boner W, Morgan IM. TopBP1 regulates human papillomavirus type 16 E2 interaction with chromatin. J Virol. 2007;81:4338–4342. doi: 10.1128/JVI.02353-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grinstein E, Wernet P, Snijders PJ, Rosl F, Weinert I, Jia W, Kraft R, Schewe C, Schwabe M, Hauptmann S, Dietel M, Meijer CJ, Royer HD. Nucleolin as activator of human papillomavirus type 18 oncogene transcription in cervical cancer. J Exp Med. 2002;196:1067–1078. doi: 10.1084/jem.20011053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim J, Lee D, Gwan Hwang S, Hwang ES, Choe J. BRCA1 associates with human papillomavirus type 18 E2 and stimulates E2-dependent transcription. Biochem Biophys Res Commun. 2003;305:1008–1016. doi: 10.1016/s0006-291x(03)00880-5. [DOI] [PubMed] [Google Scholar]
- 92.Bouallaga I, Teissier S, Yaniv M, Thierry F. HMG-I(Y) and the CBP/p300 coactivator are essential for human papillomavirus type 18 enhanceosome transcriptional activity. Mol Cell Biol. 2003;23:2329–2340. doi: 10.1128/MCB.23.7.2329-2340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Valencia-Hernandez A, Cuevas-Bennett C, Garrido E. Transcriptional regulation of human papillomavirus type 18 P105 promoter by the co-activator CBP. Intervirology. 2007;50:418–425. doi: 10.1159/000112917. [DOI] [PubMed] [Google Scholar]
- 94.Kumar RA, Naidu SR, Wang X, Imbalzano AN, Androphy EJ. Interaction of papillomavirus E2 protein with the Brm chromatin remodeling complex leads to enhanced transcriptional activation. J Virol. 2007;81:2213–2220. doi: 10.1128/JVI.01746-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kukimoto I, Takeuchi T, Kanda T. CCAAT/enhancer binding protein beta binds to and activates the P670 promoter of human papillomavirus type 16. Virology. 2006;346:98–107. doi: 10.1016/j.virol.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 96.Guillaud M, Adler-Storthz K, Malpica A, Staerkel G, Matisic J, Van Niekirk D, Cox D, Poulin N, Follen M, Macaulay C. Subvisual chromatin changes in cervical epithelium measured by texture image analysis and correlated with HPV. Gynecol Oncol. 2005;99:S16–23. doi: 10.1016/j.ygyno.2005.07.037. [DOI] [PubMed] [Google Scholar]
- 97.Jang MK, Kwon D, McBride AA. Papillomavirus E2 proteins and the host BRD4 protein associate with transcriptionally active cellular chromatin. J Virol. 2009;83:2592–2600. doi: 10.1128/JVI.02275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomas MC, Chiang CM. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell. 2005;17:251–264. doi: 10.1016/j.molcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 99.Zhang B, Laribee RN, Klemsz MJ, Roman A. Human papillomavirus type 16 E7 protein increases acetylation of histone H3 in human foreskin keratinocytes. Virology. 2004;329:189–198. doi: 10.1016/j.virol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 100.Nguyen CL, Munger K. Human papillomavirus E7 protein deregulates mitosis via an association with nuclear mitotic apparatus protein 1. J Virol. 2009;83:1700–1707. doi: 10.1128/JVI.01971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee D, Lim C, Seo T, Kwon H, Min H, Choe J. The viral oncogene human papillomavirus E7 deregulates transcriptional silencing by Brm-related gene 1 via molecular interactions. J Biol Chem. 2002;277:48842–48848. doi: 10.1074/jbc.M203583200. [DOI] [PubMed] [Google Scholar]
- 102.French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, Fletcher JA, Aster JC. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27:2237–2242. doi: 10.1038/sj.onc.1210852. [DOI] [PubMed] [Google Scholar]
- 103.You J, Srinivasan V, Denis GV, Harrington WJ, Jr, Ballestas ME, Kaye KM, Howley PM. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J Virol. 2006;80:8909–8919. doi: 10.1128/JVI.00502-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ottinger M, Christalla T, Nathan K, Brinkmann MM, Viejo-Borbolla A, Schulz TF. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J Virol. 2006;80:10772–10786. doi: 10.1128/JVI.00804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin A, Wang S, Nguyen T, Shire K, Frappier L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J Virol. 2008;82:12009–12019. doi: 10.1128/JVI.01680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou M, Huang K, Jung KJ, Cho WK, Klase Z, Kashanchi F, Pise-Masison CA, Brady JN. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J Virol. 2009;83:1036–1044. doi: 10.1128/JVI.01316-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kapasi AJ, Spector DH. Inhibition of the cyclin-dependent kinases at the beginning of human cytomegalovirus infection specifically alters the levels and localization of the RNA polymerase II carboxyl-terminal domain kinases cdk9 and cdk7 at the viral transcriptosome. J Virol. 2008;82:394–407. doi: 10.1128/JVI.01681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]