

Many studies have shown that primary prostate cancers are multifocal1-3, and are composed of multiple genetically distinct cancer cell clones4-6. Whether or not multiclonal primary prostate cancers typically give rise to multiclonal or monoclonal prostate cancer metastases is largely unknown, although studies at single chromosomal loci are consistent with the latter. Here we show through a high-resolution genome-wide SNP and copy number survey that most if not all metastatic prostate cancers have monoclonal origins and maintain a unique signature copy number pattern of the parent cancer cell while also accumulating a variable number of separate subclonally sustained changes. We find no relationship between anatomic site of metastasis and genomic copy number change pattern. Taken together with past animal and cytogenetic studies of metastasis7, and recent single-locus genetic data in prostate and other metastatic cancers8-10, it appears that despite common genomic heterogeneity in primary cancers, most metastatic cancers arise from a single precursor cancer cell. Methodologically, this study establishes that genomic archeology of multiple anatomically separate metastatic cancers in individuals can be used to define the salient genomic features of a parent cancer clone of proven lethal metastatic phenotype.
DNA was isolated from 94 anatomically separate cancer sites in 30 men who died from metastatic prostate cancer (Fig. 1a) and was analyzed by chromosomal metaphase-based comparative genomic hybridization (cCGH) and/or by Affymetrix Genome-Wide Human SNP (single nucleotide polymorphism) Array 6.0 analysis (Affy6).
Figure 1.

a. Metastatic prostate cancer study subjects. Anatomic sample type indicators for 85 cancerous DNA samples studied by CGH and 58 cancerous samples studied by Affy6 are superimposed on posterior bone scan views for each subject. Legend indicates color and number coding of anatomic origin categories. Subjects from whom two or more anatomically distinct prostate cancer samples were studied are denoted with colored symbols upon which the subject’s number is superimposed.
b. Unsupervised hierarchical clustering of cCGH data. Unsupervised hierarchical clustering dendrogram based on SAM-reduced 218 locus metastatic prostate cancer cCGH dataset for 80 samples from 24 subjects in which more than one anatomically separate cancerous DNA sample was available. All samples from an individual subject are color/shape coded using symbols shown (Fig. 1a).
c. Discriminatory Component Analysis of cCGH data. Weighted Fisher criterion based discriminatory component analysis (wFC-DCA) of cCGH data from 80 metastatic prostate cancer samples from 24 subjects projected in 3-D Euclidean space. Samples are identified using color/shape symbols indicated (Fig 1a).
d. Discriminatory Component Analysis of Affy6 data. Weighted Fisher criterion based discriminatory component analysis (wFC-DCA) of Affy6 data from 58 metastatic prostate cancer samples from 14 subjects projected in 3-D Euclidean space. Samples are identified using color/shape symbols indicated (Fig 1a).
e. Unsupervised Hierarchical Clustering of Affy6 copy number data from 58 anatomically separate metastatic prostate cancer sites in 14 subjects. All samples from each of 14 subjects cluster together. Of 3,029,978 total genome segments analyzed in 58 samples studied (52241 per sample), 52.4% show no change in copy number vs subject-specific normal control baseline, 25.4% show gain, and 22.2% show loss.
Eighty-five sites from 29 of the subjects were studied by cCGH. To assess possible clonal relationships of metastasizing cells, two or more anatomically separate cancerous lesions were studied by cCGH in 24 subjects (80 samples, range 2–8 samples/subject). Significance Analysis of Microarrays (SAM)11 was used to detect 218 loci across the genome which were affected by either copy number gain or loss. Copy number data from these 218 loci for the 80 samples were analyzed by unsupervised hierarchical clustering (Fig. 1b). For 15 of 24 subjects (63%) cCGH data from all samples clustered by subject of origin, suggesting a strong clonal relationship of separate metastatic samples in the majority of subjects.
Subject-specific “perfect” clustering of metastatic cCGH copy number data in a substantial number (15/24) of the subjects with multiple anatomically separate samples led us to explore their further association with genomic copy number12 through an unsupervised cluster-subject matching test, a supervised classification-based assessment13, 14, and through distance-based analysis, all of which reject the null hypothesis that observed clustering is random.
To better visualize the relationships of the copy number data among the 80 samples studied, we displayed the full cCGH dataset via the top discriminatory components in 3-D Euclidean space extracted by weighted Fisher criterion-based discriminatory component analysis (wFC-DCA)15, where the overall intrasubject copy number pattern similarity of both clustering cases (for example case 17, the cyan circles) and nonclustering cases (for example case 33, the magenta triangles) is apparent (Fig. 1c). Taken together with the cCGH data clustering results, these data suggest that in the majority of cases, metastatic cells in a given subject may have clonal origins.
To further examine potential clonal origins, we performed Affy6 analysis in a subset of samples from 14 subjects where at least three metastatic deposits were available for analysis. Affy6 genomic position resolution is approximately 5000x cCGH resolution with an average physical distance of ~700 base pairs between a total of over 1.8 million probes. Subject-specific “perfect” clustering is observed for all 58 samples studied from 14 subjects (Fig. 1e). Permutation-based and statistical analyses similar to those performed for the cCGH results showed evidence to reject the null hypothesis of random subject-specific clustering. Interestingly, projection of the Discriminatory Component Analysis results for the Affy6 data (Fig. 1d) show a tighter and more “exclusive” association among anatomically distinct cancer samples from the same subject than that seen in the cCGH data (Fig. 1c), suggesting that misclustering of samples in the cCGH data is likely due to lower assay resolution.
Analysis of the probability of common origins of DNA samples from different individuals is now relatively routine. Proving common origins of different populations of mutant cells from the same individual is not routine, but has long been a topic of inquiry in relation to the origins of metastatic cancer16. Previous analysis of cytogenetic, isoenzyme, and X-chromosome inactivation9, 17, 18 data, as well as cell-line based experimental metastasis studies7, 9, and more recent specific analyses of one or a few genetic loci (including PTEN and TMPRSS2-ETS gene aberrations) have suggested clonal origins of metastatic melanoma and prostate cancers8-10, 19, 20 in at least a substantial percentage of metastatic cancer patients.
As a source of markers of clonality which might be more informative due to their unique genomic position and relative copy number change, we sought to further test the hypothesis of clonal origins of metastatic prostate cancer by examining allele-specific patterns of gain and loss, and regions of the genome affected by homozygous deletion.
Analysis of a representative sample of allele-specific copy number data from two subjects is shown (Figs. 2 and 3). A signature pattern of copy number gains and losses is present in each sample studied, and elements of this signature are present in every anatomically separate cancer DNA sample. Changes present in all samples are here termed omniclonal, and other changes termed subclonal are present in only a subset of samples studied in a given subject. The presence of omniclonal changes unique in chromosomal position and copy number strongly suggests that all metastatic cancer cells in these subjects had a single clonal cancer cell origin, and suggest that studies of multiple metastases in cancer patients can be used to derive a set of changes present in the ultimate parent cancer cell. The presence of omniclonal and subclonal changes depicted (Figs. 2, 3, and 1e) provide a picture of strikingly high-fidelity maintenance of a subject-specific signature set of copy number changes derived from a single parent cancer cell, with variable degrees of additional subclonally maintained changes. Fig. 1e also suggests definable “personalities” of omniclonal and subclonal changes among subjects, with for example a moderate number of medium-sized omniclonal changes and rare subclonal changes in subject 17, relatively sparse omniclonal changes in subject 19 with relatively greater numbers of subclonal changes, and a strikingly high number of relatively small omniclonal changes in subject 33 with a moderate number of subclonal changes.
Figure 2a. Representative sample of allele-specific copy number data from subject 17 (chromosomes 6 and 13).

2b: Changes present in all samples are termed omniclonal (circled in green). 2c: Subclonal changes are present in just one (dark blue), a pair (brown) or three of the samples (light blue) studied. On chromosomes where whole chromosome arm gain or loss appears to have occurred before or after complex intrachromosomal gains and losses took place are difficult to interpret with existing technology and are labeled indeterminate (pink). 2d: Summary of all changes detected. 2e: Depiction of detected changes at each metastatic site studied, superimposed on posterior bone scan view with tan circle representing prostate, black circle representing cancer capable of metastasis. The parent cancer cell arises in the prostate and contains a set of clonally maintained copy number changes depicted in green at the base of each triangle containing labeled changes identified in each metastatic cancer sample. The parent cancer cell divides, maintaining the original set of changes but also giving rise to some new changes that are also maintained subclonally in cells that leave the prostate to populate various metastatic sites which were analyzed in this study. Subclonal diversification likely occurs in both the prostate and sites after initial metastasis, the simplest possible case (subclonal diversification in the prostate) is depicted for discussion purposes only, subclonal diversification likely occurs both in the prostate and at metastatic sites.
Figure 3a. Representative sample of allele-specific copy number data from subject 34 (chromosomes 5 and 8).

3b: Changes present in all samples are termed omniclonal (circled in green). 3c: Subclonal changes are present in just one (dark blue), a pair (brown) or three of the samples (light blue) studied. Indeterminate changes are circled in pink. 3d: Summary of all changes detected. 3e: Depiction of detected changes at each metastatic site studied, superimposed on posterior bone scan view with tan circle representing prostate, black circle representing cancer capable of metastasis.
We found 17 homozygous deletions unique in chromosomal position and unique to one of the 10 study subjects in which they were found (Supplementary Table 8). Fifteen of 17 (88%) of these homozygous deletions were similarly present in all samples studied from a given subject (“omniclonal”), strongly suggesting common cellular clonal origins of the metastatic cancer cell populations in each of these subjects. Genes affected by clonal homozygous deletions include PTEN, BRCA2, TGFBR2, PCAF, PR, FHIT, PPP2R2A, BNIP3L, CDKN2A, and ACVRL1.
The spectrum of anatomic sites affected by metastasis in men with disseminated prostate cancer is variable21, and could possibly be explained by variations at the genomic level. To examine whether specific clonal or subclonal changes are associated with specific anatomic sites of metastasis21, we used permutation-based analyses to compare observed cCGH and Affy6 copy number data from all 85 DNA samples grouped by anatomic location, and find no statistical evidence of copy number pattern similarity on this basis (Supplementary Figs. 4 and 8).
Prostate cancer is more aggressive at every stage in African-Americans than in other racial groups22. We used permutation-based analysis to compare copy number findings in prostate cancer samples from four African-American men represented in the cCGH data, and 2 African-American men represented in the Affy6 data presented. No difference in overall genomic pattern was detected (data not shown), though results are based on a small sample size.
Androgen pathway alterations are thought to play crucial roles in the progression of prostate cancer to a lethal disease, with upregulation of Androgen Receptor (AR) gene expression a consistent finding. With respect to AR copy number, we found that only two subjects (17, 34) show a normal single copy of AR present in all metastatic sites. Seven subjects (3, 12, 19, 22, 31, 32, 33) show gain from 2–8 copies, with most of these falling stably in the 2–3 range. Five subjects (16, 21, 24, 28, 30) show AR gains of 9–40 copies, similar to high level gains found in a minority of cases in previous in situ hybridization based studies23, 24. Interestingly, as average AR copy number in each subject’s set of metastases increases, the variation in copy number among metastatic samples from a given subject also tends to increase, although in most subjects overall AR copy number is relatively stable across metastastic sites for individual subjects, consistent with clonal origins with subclonal variations as seen in the overall copy number analysis (Supplementary Fig. 11).
Fusion transcript formation between TMPRSS2 and ETS family members have been shown to occur in 50% or more of all prostate cancers, with TMPRSS2-ERG fusion transcripts being most commonly found. With regard to TMPRSS2-ERG fusions, we found heterozygous deletion between ERG and TMPRSS2 in 7 of 14 subjects studied by Affy6. When it was present, the same deletion event was found in each metastatic site in a given case, as previously observed by Mehra et al10. We examined TMPRSS2-ERG fusion transcript status in 18 anatomically separate metastatic prostate cancer samples from a subset of these subjects, and it is uniformly present in all 9 samples studied from subjects with ERG deletion, and uniformly absent from all 9 samples studied from subjects without ERG deletion (Supplementary Table 9 and Supplementary Fig. 10). These observations are consistent with this deletion and resulting fusion transcript formation being a common early, pre-metastatic event, although evidently one that is not required for successful tumor cell dissemination. A more thorough cataloging of all ETS family gene fusions will be necessary to understand their role in tumor progression.
Beyond the demonstrated presence of clonal and subclonal changes in each subjects’ set of metastatic samples, overall patterns of genomic change shown (Fig. 1e) vary greatly between subjects. To test whether these overall patterns could be related to therapy received, we compared clonal and subclonal change frequencies (Supplementary Table 10) in 7 subjects having undergone DNA-damaging chemotherapy (cylophosphamide, topotecan, etoposide, and/or carboplatin) vs 7 subjects who did not receive DNA-damaging chemotherapy, and found no statistical differences (Supplementary Table 11 and Supplementary Figs. 12-14).
To our knowledge, the study reported here provides the first full high-resolution genomic overview of copy number changes in multiple metastatic cancers in individual humans, analysis of which adds substantial depth to the clonal origins discussion. This study provides the most comprehensive evidence to date that all or at least the vast majority of patients with metastatic prostate cancer have cancers that originated in a single aberrant cell, a finding likely to extend to other cancers, and demonstrates that it is feasible to use metastatic site comparison to derive the set of changes present in the parent cancer cell in each subject. The findings also demonstrate that there are a substantial but variable number of subclonally maintained changes in metastatic cancer sites in a given subject.
Our findings cast new light on previously published data suggesting that primary prostate cancers are often multifocal1-3and often have multiple separate clonal parent cell origins4-6. Our data show that lethal metastatic prostate cancer cells derive from a common parent cell, and also show that subclonal changes arise and are sustained. Studies of anatomically separate primary cancers from individual subjects using a genome-wide set of loci are indicated to revisit this question and determine whether previous studies were underpowered and detected subclonally maintained differences but missed clonal changes, or whether primary prostate cancer is often truly multifocal as is currently widely believed.
Our recent studies of relative hyper- and hypo- methylation at selected CpG islands in the same subjects’ samples suggest that some hypermethylation changes are “clonal” within a given subject25, while hypomethylation changes are more heterogeneous26. These findings, together with transcript and protein expression studies in a similar set of subjects studied by Shah et al27 suggest that full pathway-based integration of genetic, epigenetic, and protein level data from multiple metastatic samples in a larger series of patients with metastatic cancer may be a uniquely powerful way to establish well-prioritized lists of targets for development of new drug and diagnostic targets.
Several aspects of these data are relevant to tying together what is currently known at the macrogenomic level about metastatic cancer in humans. First is to emphasize that strong evidence of clonal origins does not mean that all cells are genomically identical in a given metastatic cancer site. Cytogenetic and other studies show that a degree of genomic copy number “wobble” exists in metastatic cancer cells18. The data presented here show that despite this “wobble”, a relatively clean, clear, and highly individual-specific pattern of copy number changes occurs in metastatic prostate cancers in the majority of cases, and that this pattern is maintained in aggregate among multiple metastatic sites in individuals with surprising fidelity when compared to cell-line based metastasis studies28.
Second, the findings reported here are based on the aggregate signal from millions of metastatic prostate cancer cell genomes represented in each sample studied. It is possible but seems unlikely that two or more clonal populations dependent upon each other for metastatic “success” could have quite different copy number changes that sum to the data we observe here. Third, these data cannot rule out an alternative hypothesis where clonal-appearing and individually unique copy number patterns observed could be a result of individual subject-specific requirements for successful metastasis. In this alternate scenario, polyclonal, highly genomically unstable cancer cells would “succeed” only if they met very tight copy number gain and loss requirements specific to the subject. It is hard to imagine a feasible biological mechanism through which such specificity could arise from autochthonous cells, so this hypothesis appears unlikely to be correct. Finally, if in most patients metastatic prostate cancer cells have a common clonal origin, this suggests that cancer cells with stem cell properties obtain these properties in the context of a shared set of individual-specific copy number changes, consistent with recent findings29.
Our results show that metastatic prostate cancer deposits in individual men have clonal origins in most if not all cases. Using subject A17 as representative of all subjects in the current study, and considering reports suggesting that prostate cancer cells may lie dormant in the bone marrow for many years30, 31, spread of cancer cells with common clonal origins occurs either in a “Direct Clonal” or “Indirect Clonal” pattern as illustrated in Fig. 5. The large tan circle represents the prostate, and the black circle represents prostate cancers capable of lethal spread. Green circles represent local prostate cancers incapable of spread, and Yellow circles represent nonlethal spreading cancer as suggested by data from Ellis et al30. We found no significant difference in copy number patterns in prostate cancer foci isolated from the prostate at autopsy and metastases from various sites in the 5 subjects where prostate cancer foci were isolated from the prostate at autopsy. “Direct clonal” lethal metastasis provides the simplest explanation of these findings, since “Indirect” metastasis would require that the metastatic prostate cancer metastasize back to the prostate as illustrated by the dashed arrows.
In conclusion, these data suggest that in most if not all metastatic prostate cancer cases, the origins of cancer cells within disparate metastatic prostate cancer deposits can be traced to a single genomically aberrant prostate cell whose macrogenomic copy number changes are relatively stably replicated with each cell division. Upon this relatively stable base of copy number change, additional copy number changes occur and are subclonally sustained. These findings have potentially important implications for treatment of metastatic prostate cancer: understanding and predicting therapeutic success in an individual will likely depend on the degree of clonal uniformity as well as the specific genomic alteration pattern for metastatic lesions in a given patient. Hypothetically, since high clonal diversity should improve cancer cell survival in response to change, the degree of clonality of a given patient’s metastatic prostate cancer cells could have as important an impact on therapeutic response as the specific pattern of genomic changes found in the prostate cancer cells. Additional studies are needed to determine how the macrogenomic monoclonality suggested in the majority of metastatic prostate cancer patients studied here relates to what is found at the microgenomic (individual base pair) level.
Methods Summary
PELICAN Autopsy Study of Lethal Prostate Cancer
Ninety-four cancer samples were studied from 30 men who died of prostate cancer and underwent autopsy as part of the Project to Eliminate Lethal prostate CANcer (PELICAN) rapid autopsy program at the Johns Hopkins Medical Institutions (JHASPC). Initiated in 1994, all JHASPC study subjects gave informed consent to participate as part of a Johns Hopkins Medicine IRB-approved protocol. All subjects underwent androgen-deprivation during the course of their treatment for metastatic prostate cancer, and died between 1995 and 2004. Tissues were snap-frozen and cryostat-microdissected and DNA purified as described previously20. Subject and sample data including distribution of samples studied by cCGH and Affy6 array technology are contained in Supplementary Table 1. Mean estimated cancer sample DNA purity based on hematoxylin and eosin histology is 88% (range 60–99%).
Chromosomal Comparative Genomic Hybridization (cCGH) was performed at resolution of 389 cytogenetic bands (excluding the chromosome Y) in 85 cancer DNA samples from 29 subjects. cCGH data (Supplementary Table 3) are of lower resolution but are highly concordant with array-based CGH (aCGH) results32. CGH was done as described previously33 and as detailed in Supplementary Methods.
Affymetrix Genome-Wide Human SNP array 6.0 analysis
Genome-Wide Human SNP array 6.0 chips (Affy6) were purchased from Affymetrix, Inc. All of the reagents used for the assay were obtained from manufacturers recommended by Affymetrix. We amplified, purified, fragmented and labeled the genomic DNA, hybridized, washed and stained the Affy6 arrays according to the manufacturer’s instructions (Supplementary Methods). We used Partek Genomic Suite (PGS) version 6.4 for allele specific and non-allele specific analyses using default settings (http://www.partek.com/Tutorials) unless otherwise specified. Sixteen subject-paired noncancerous samples from 14 subjects were used to create a copy number baseline (Supplementary Table 2). For each of 58 cancer DNA samples studied by Affy6, we then generated a DNA copy number estimate for all ~1.8 million probes on the Affy6 chip, and then segmented these data into 52221 channels using the PGS Segmentation algorithm. Autosomal and sex-chromosomal segmentation data for the 58 samples was then analyzed in PGS using unsupervised hierarchical clustering (Pearson’s Dissimilarity algorithm) to produce data shown (Fig. 1e). Allele-specific genomic analysis depicted (Figs. 2, 3 and Supplementary Table 8) was performed using the PGS allele-specific analysis algorithm that includes genotype information and allele-specific intensities from paired samples to estimate DNA copy number for each heterozygous SNP, and is further described in Supplementary Methods.
Statistical Analysis
Permutation- Based Classification Analysis. For the cCGH data, considering each metastatic DNA sample with 218 SAM-defined (Supplementary Methods and Supplementary Tables 4 and 5) CGH measures as a vector of 218-elements and the distance between two samples defined as the Euclidean distance of two vectors. For the Affy6 data, we considered each metastatic DNA sample with 52221 measures as a vector of 52221-elements and the distance between two samples defined as the Euclidean distance of two vectors. All the samples were divided into a training set and a testing set. The predicted label for a sample in the testing set is the same as the label of the sample mean of all samples belonging to the same subject in the training set with the smallest distance to the testing sample (nearest mean classifier)13. Leave-One-Out Cross Validation (LOOCV) is then performed utilizing one sample as the test sample and the remaining samples as the training set13. This is repeated such that every sample is used once as testing sample. If the predicted label coincides with the original label, it is correctly classified; otherwise, it is in error. We apply the nearest mean classification method to classify the samples and utilize the LOOCV to estimate the classification error. The error rate is calculated as the percentile of wrongly classified samples over all samples. Statistical tests on cCGH and Affy6 data are one-tailed.
We also tested cCGH and Affy6 data for evidence of clonality by testing the hypothesis that there is no difference between the “between-subject” distance and “within-subject” distance by considering each cCGH sample with 218 CGH measures as a vector of 218-elements and each Affy6 sample with 52221 measures as a vector of 52221 elements. Let Dbm be the average “between-subject” distance over all sample pairs belonging to different subjects and Dbw be the average “within-subject” distance over all sample pairs belonging to the same subject, using the summary statistic Ss = Dbm − Dbw, we compared experimentally observed Ss to the distribution of Ss calculated from 100,000 random permutations of the subject labels34, 35. The experimentally observed cCGH data Ss value is 3.8159, and the maximum value of Ss in the permuted data is 0.8467 (Supplementary Fig. 3), rejecting the null hypothesis with P <0.00001. The experimentally observed Affy6 data Ss value is 110.24, and the maximum value of Ss in the permuted data is 19.62 (Supplementary Fig. 7), rejecting the null hypothesis with P <0.00001. Additional statistical methods details are contained in Supplementary Methods.
Supplementary Material
Figure 4. Potential patterns of metastatic prostate spread.
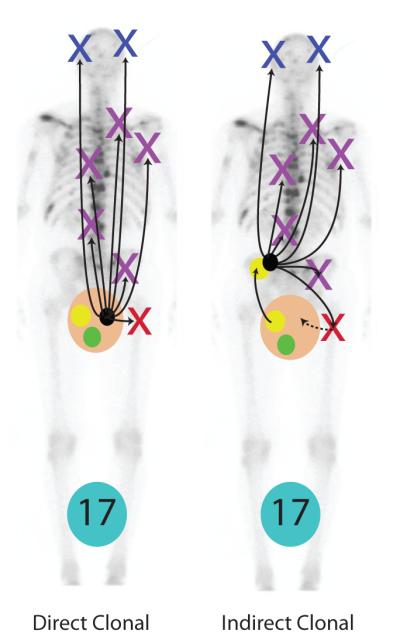
Our results show that most if not all metastatic prostate cancers have clonal origins. Using subject A17 as representative of all subjects, and considering recent data suggesting that in some men prostate cancer cells may lie dormant in the bone marrow for many years30, 31, spread of cancer cells with common clonal origins occurs in a “Direct Clonal” or “Indirect Clonal” pattern shown. The large tan/pink circle represents the prostate, and the black circle represents prostate cancers capable of lethal spread. Green circles represent local prostate cancers incapable of spread, and Yellow circles represent nonlethal spreading cancer as suggested by data from Ellis et al30. We found no significant difference in copy number patterns in prostate cancer foci isolated from the prostate at autopsy and metastases from various sites in the 5 subjects where prostate cancer foci were isolated from the prostate at autopsy. “Direct clonal” lethal metastasis provides the simplest explanation of these findings, since “Indirect” metastasis would require that the metastatic prostate cancer metastasize back to the prostate as illustrated by the dashed arrows.
Acknowledgements
To the participating men and their families who suffered through metastatic prostate cancer and nonetheless gave the gift of participation so that others might benefit. To V. Sinibaldi, T. B. Smyth, and G. J. Mamo for oncologic and urologic clinical support, the Johns Hopkins Pathology Autopsy Service including B. Crain and G. Hutchins, and Ms. A. Alkula, S. Kuivanen, T. Vilkkilä-Qwick, M. Vakkuri, D. Jay, X. Yi, S. H. Hahm, K. Jeffers Keiger, S. H. Chen, P. Powers, M. Taylor, C. Kang, and Partek Customer Support for technical assistance. The work, commenced in 1994, was supported in part by Pirkanmaa Cancer Foundation, Maud Kuistila Foundation, Finnish Medical Foundation, the Medical Research Fund of Tampere University Hospital, Academy of Finland, Cancer Society of Finland, Reino Lahtikari Foundation, Sigrid Juselius Foundation, CaPCURE, John and Kathe Dyson, David Koch, the U.S. National Institutes of Health National Cancer Institute (CA92234), the Prostate Cancer Research and Education Foundation, U.S. Dept of Defense Congressionally Directed Prostate Cancer Research Program, the Grove Foundation, and the American Cancer Society.
Footnotes
Affy6 data used in the study are submitted to GEO, accession number xxxxx (submission in process)
Reference List
- 1.Miller GJ, Cygan JM. Morphology of prostate cancer: the effects of multifocality on histological grade, tumor volume and capsule penetration. Journal of Urology. 1994;152:1709–1713. doi: 10.1016/s0022-5347(17)32368-6. [see comments] [DOI] [PubMed] [Google Scholar]
- 2.Ruijter ET, van de Kaa CA, Schalken JA, Debruyne FM, Ruiter DJ. Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J Pathol. 1996;180:295–299. doi: 10.1002/(SICI)1096-9896(199611)180:3<295::AID-PATH663>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 3.Aihara M, Wheeler TM, Ohori M, Scardino PT. Heterogeneity of prostate cancer in radical prostatectomy specimens. Urology. 1994;43:60–66. doi: 10.1016/s0090-4295(94)80264-5. [DOI] [PubMed] [Google Scholar]
- 4.Cheng L, et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. Journal of the National Cancer Institute. 1998;90:233–237. doi: 10.1093/jnci/90.3.233. [DOI] [PubMed] [Google Scholar]
- 5.Macintosh CA, Stower M, Reid N, Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Research. 1998;58:23–28. [PubMed] [Google Scholar]
- 6.Cheng L, et al. Allelic imbalance in the clonal evolution of prostate carcinoma. Cancer. 1999;85:2017–2022. doi: 10.1002/(sici)1097-0142(19990501)85:9<2017::aid-cncr20>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.Fidler IJ, Talmadge JE. Evidence that intravenously derived murine pulmonary melanoma metastases can originate from the expansion of a single tumor cell. Cancer Res. 1986;46:5167–5171. [PubMed] [Google Scholar]
- 8.Kuukasjärvi T, et al. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597–1604. [PubMed] [Google Scholar]
- 9.Sabatino M, et al. Conservation of genetic alterations in recurrent melanoma supports the melanoma stem cell hypothesis. Cancer Res. 2008;68:122–131. doi: 10.1158/0008-5472.CAN-07-1939. [DOI] [PubMed] [Google Scholar]
- 10.Mehra R, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–3590. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y, et al. A ground truth based comparative study on clustering of gene expression data. Front Biosci. 2008;13:3839–3849. doi: 10.2741/2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hastie TR, Tibshirani TR, Friedman J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. Springer; New York: 2001. [Google Scholar]
- 14.Wang Z, et al. Optimized multilayer perceptrons for molecular classification and diagnosis using genomic data. Bioinformatics. 2006;22:755–761. doi: 10.1093/bioinformatics/btk036. [DOI] [PubMed] [Google Scholar]
- 15.Loog M, Duin R, Haeb-Umbach R. Multiclass linear dimension reduction by weighted pairwise fisher criteria. IEEE Transactions on Pattern Analysis and Machine Intelligence. 2001;23:762–766. [Google Scholar]
- 16.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- 17.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 18.Heim S, Mandahl N, Mitelman F. Genetic convergence and divergence in tumor progression. Cancer Res. 1988;48:5911–5916. [PubMed] [Google Scholar]
- 19.Eastham JA, et al. Association of p53 mutations with metastatic prostate cancer. Clin. Cancer Res. 1995;1:1111–1118. [PubMed] [Google Scholar]
- 20.Suzuki H, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204–209. [PubMed] [Google Scholar]
- 21.Bova GS, Chan-Tack K, LeCates WW. Prostate Cancer: Biology, Genetics, and New Therapeutics. Humana Press; 2000. Lethal Metastatic Human Prostate Cancer: A Review of the Literature with Emphasis on Autopsy Studies and Characteristics of Metastases. [Google Scholar]
- 22.Powell IJ. Epidemiology and pathophysiology of prostate cancer in African-American men. J. Urol. 2007;177:444–449. doi: 10.1016/j.juro.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 23.Koivisto P, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–319. [PubMed] [Google Scholar]
- 24.Miyoshi Y, et al. Fluorescence in situ hybridization evaluation of c-myc and androgen receptor gene amplification and chromosomal anomalies in prostate cancer in Japanese patients. Prostate. 2000;43:225–232. doi: 10.1002/(sici)1097-0045(20000515)43:3<225::aid-pros9>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Yegnasubramanian S, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975–1986. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 26.Yegnasubramanian S, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68:8954–8967. doi: 10.1158/0008-5472.CAN-07-6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah RB, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 28.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 29.Vander Griend DJ, et al. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res. 2008;68:9703–9711. doi: 10.1158/0008-5472.CAN-08-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellis WJ, et al. Detection and isolation of prostate cancer cells from peripheral blood and bone marrow. Urology. 2003;61:277–281. doi: 10.1016/s0090-4295(02)02291-4. [DOI] [PubMed] [Google Scholar]
- 31.Vessella RL, Pantel K, Mohla S. Tumor cell dormancy: an NCI workshop report. Cancer Biol. Ther. 2007;6:1496–1504. doi: 10.4161/cbt.6.9.4828. [DOI] [PubMed] [Google Scholar]
- 32.Saramäki OR, Porkka KP, Vessella RL, Visakorpi T. Genetic aberrations in prostate cancer by microarray analysis. Int. J. Cancer. 2006;119:1322–1329. doi: 10.1002/ijc.21976. [DOI] [PubMed] [Google Scholar]
- 33.Kallioniemi A, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 34.Good PI. Permutation, Parametric and Bootstrap Tests of Hypotheses. Springer; 2005. [Google Scholar]
- 35.Kowalski J, Pagano M, DeGruttola V. A Nonparametric Test of Gene Region Heterogeneity Associated With Phenotype. J Am Stat Assoc. 2002;97:398–408. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.