

Abstract
Replication factor C (RFC) catalyzes assembly of circular proliferating cell nuclear antigen clamps around primed DNA, enabling processive synthesis by DNA polymerase during DNA replication and repair. In order to perform this function efficiently, RFC must rapidly recognize primed DNA as the substrate for clamp assembly, particularly during lagging strand synthesis. Earlier reports as well as quantitative DNA binding experiments from this study indicate, however, that RFC interacts with primer-template as well as single- and double-stranded DNA (ssDNA and dsDNA, respectively) with similar high affinity (apparent Kd ≈ 10 nM). How then can RFC distinguish primed DNA sites from excess ssDNA and dsDNA at the replication fork? Further analysis reveals that despite its high affinity for various DNA structures, RFC selects primer-template DNA even in the presence of a 50-fold excess of ssDNA and dsDNA. The interaction between ssDNA or dsDNA and RFC is far less stable than between primed DNA and RFC (koff > 0.2 s−1 versus 0.025 s−1, respectively). We propose that the ability to rapidly bind and release single- and double-stranded DNA coupled with selective, stable binding to primer-template DNA allows RFC to scan DNA efficiently for primed sites where it can pause to initiate clamp assembly.
Replicative DNA polymerases synthesize DNA with high processivity in collaboration with two key accessory proteins: a circular clamp and a clamp loader complex (see Fig. 1; reviewed in Refs. 1 and 2). These accessory proteins are conserved in structure and function among a variety of organisms including bacteriophage (e.g. T4 gp45 clamp and gp44/62 clamp loader), bacteria (e.g. Escherichia coli β clamp and γ complex clamp loader), archaebacteria (e.g. Pyrococcus furiosus proliferating cell nuclear antigen (PCNA)1 clamp and replication factor C (RFC) clamp loader), and eukaryotes such as Saccharomyces cerevisiae and humans (PCNA and RFC) as well (see Ref. 3 for a review of recent crystal structures). Clamps are ring-shaped multisubunit proteins with an inner diameter of ~35 Å that is large enough to encircle duplex DNA (e.g. homotrimeric PCNA) (4). When bound to polymerase and DNA, the clamp forms a sliding tether that stabilizes polymerase on the primer-template and facilitates processive DNA replication (5). A polymerase-clamp complex, such as S. cerevisiae Pol δ·PCNA, can extend DNA with a processivity of hundreds of nucleotides per template binding event, a substantial increase from the 6–12-nucleotide processivity of Pol δ alone (6, 7).
Fig. 1. A model eukaryotic DNA replication fork.

The diagram depicts the minimal protein activity required at a DNA replication fork, including the DNA-unwinding helicase, single-stranded DNA-binding protein (RPA), primase (Pol α), leading strand polymerase (Pol δ), lagging strand polymerase (Pol δ and/or Pol ε), circular sliding clamp (PCNA), and the clamp loader (RFC), which must catalyze PCNA assembly at multiple primed sites during lagging DNA strand synthesis.
The circular clamp is opened and assembled around primer-template DNA by the clamp loader, a multiprotein machine whose action is fueled by ATP binding and hydrolysis. Clamps must be present at primed DNA sites for polymerase to bind them and initiate processive DNA synthesis (7, 8). Efficient clamp assembly is particularly important during lagging DNA synthesis when a clamp is required at the initiation of every Okazaki fragment (Fig. 1). Given an estimated Okazaki fragment size of 100–200 nucleotides in eukaryotes and a Pol δ·PCNA-catalyzed DNA synthesis rate of ~100 nucleotides/s, clamp assembly is required once every 1 or 2 s during lagging strand synthesis (7). The replicative polymerase, Pol III holoenzyme, from the well examined E. coli model system has a similar requirement for timely clamp assembly as it completes a 1000–2000-nucleotide Okazaki fragment every 1–2 s (reviewed in Ref. 2). Thus, clamp loaders have to rapidly recognize primed sites on template DNA and assemble clamps on them for efficient initiation and completion of DNA replication.
The S. cerevisiae clamp loader, RFC, is composed of five proteins: RFC1 (95 kDa), RFC2 (40 kDa), RFC3 (38 kDa), RFC4 (36 kDa), and RFC5 (40 kDa). All five proteins are essential for viability according to deletion analysis (9–14) (reviewed in Ref. 15). The five proteins share sequence homology among each other and with clamp loader proteins from other organisms (e.g. γ and δ′ proteins of E. coli γ complex and gp44 protein of bacteriophage T4 gp44/62; reviewed in Refs. 14 and 16). Human RFC is very similar to the S. cerevisiae RFC complex, with one large subunit (p140) and four small subunits (p37, p36, p40, and p38) that correspond to RFC1, −2, −3, −4, and −5, respectively (15). The homology occurs chiefly in seven regions, RFC boxes II–VIII, of which boxes III and V have the most well defined function as ATP-binding Walker A and B motifs, respectively. The large RFC1 subunit contains an additional box I at the amino terminus, which shares homology with DNA ligases and is known to bind DNA but appears unnecessary for clamp loading (17).
Recent crystal structures of the E. coli γ complex clamp loader, γ3δδ′ (18, 19), and the small RFC subunit from archae-bacterium P. furiosus (20) reveal striking structural similarities as well among clamp loaders from different organisms. Each subunit is composed of three domains, a helical carboxyl-terminal domain III and amino-terminal domains I and II, connected by flexible linker regions. The complete E. coli γ3δδ′ clamp loader structure reveals five subunits arranged in a circle that is closed by interactions among the C-terminal domains but open at the N-terminal domains (the RFC sketch in Fig. 1 is modeled after the γ3δδ′ structure). The δ subunit contains a clamp-binding element in N-terminal domain I that contacts a hydrophobic pocket on the clamp and triggers clamp opening. The crystal structure of a δ·β clamp complex indicates that the clamp fits “flat” up against the clamp loader with its C-terminal face adjacent to the N-terminal face of the clamp loader (3, 21). Fig. 1 shows a sketch of RFC·PCNA derived from the proposed γ3δδ′·β complex structure. Presumably, the primer-template DNA enters the circular clamp through the open interface; however, it is not clear where or how the clamp loader binds DNA to facilitate interaction between the clamp and DNA. The P. furiosus small RFC subunit has the same overall fold as the E. coli γ complex subunits (20), and electron microscopic images of the human RFC complex (22) and archaeal small RFC subunit (23) show the subunits in a circular arrangement, indicating that the eukaryotic/archaeal clamp loaders adopt a similar quaternary structure as the bacterial clamp loader and may therefore employ similar mechanisms of action.
The mechanism of clamp assembly has been examined to varying degrees of detail for E. coli, phage T4, and human/S. cerevisiae clamp loaders. In the E. coli γ complex, ATP binding to the γ subunits induces a change in conformation that exposes δ, allowing it to bind and open the β clamp (24, 25). A recent study of the interactions between γ complex and its substrates indicates that the clamp loader binds β and then primer-template DNA with high affinity, and DNA binding specifically triggers ATP hydrolysis and release of β linked topologically to DNA (26). Notably, the γ complex also binds single-stranded DNA with high affinity in the presence or absence of the clamp (26), raising questions about how the clamp loader recognizes primed DNA but not ssDNA as the substrate for clamp assembly. In the case of bacteriophage T4 clamp loader, gp44/62, the proposed clamp loading mechanism includes ATP binding to the four gp44 subunits in the complex, followed by interaction between gp44/62 and gp45 clamp, hydrolysis of two ATP molecules, interaction with DNA, hydrolysis of the remaining two ATP, and finally release of the clamp·DNA complex (27). A more recent study suggests that only one ATP molecule is hydrolyzed during gp45 assembly on DNA; thus, the precise mechanism of how gp44/62 couples ATP binding and hydrolysis to clamp assembly on DNA is not yet clear (28). Additionally, there is not enough information available on the interaction between gp44/62 and different DNA structures to clarify how the clamp loader selects primer-template DNA as the site for clamp assembly.
In the case of eukaryotic clamp loaders, questions about DNA selection are even more pertinent, given the numerous reports of human/S. cerevisiae RFC binding to double-stranded DNA (29–32), primer-template DNA with either a 3′ primer-template junction (33, 34) or a 5′ phosphorylated primer-template junction (35), and single-stranded DNA (33, 36, 37). These results suggest a relatively low specificity of RFC binding to primer-template DNA, and since the concentration of primed sites is expected to be much lower than that of single-and double-stranded regions in replicating DNA, how does the clamp loader rapidly recognize a primed DNA site for clamp assembly? An early footprinting experiment with S. cerevisiae RFC and a hairpin DNA substrate with a 3′ recessed end demonstrated convincingly that the clamp loader binds at the primer-template junction (33), but the same study also found that RFC binds ssDNA and to some extent dsDNA as well (gel mobility shift experiments). The addition of single-stranded binding protein, RPA, to the reaction reduced the interaction between an RFC·PCNA complex and ssDNA (33). A more recent examination of RFC-DNA interaction by surface plasmon resonance revealed similar inhibition of RFC binding to ssDNA by RPA (38). However, the extent of RPA binding to single-stranded template DNA during replication in vivo is not known, and it may not be sufficient to facilitate highly specific interaction between RFC and primer-template DNA. Other studies have shown that RFC binds dsDNA and that its ATPase activity is stimulated by dsDNA (31, 39), although this result is contradicted by reports that RFC binding to the 3′ primer-template junction is not competed by dsDNA and that the large subunit of RFC in fact binds ssDNA (37). Single-stranded DNA binding activity of human and Drosophila melanogaster RFC has also been observed by electron microscopy, and in this study RFC appeared to have no preference for 3′ or 5′ primer-template junctions (36). Given the variety of DNAs and techniques utilized by different research groups to examine RFC·DNA complexes, a study of the literature does not yield adequate data for a quantitative comparison of clamp loader interactions with different DNA structures. Thus, the question of how RFC selects only primed DNA for clamp assembly although it apparently binds single- and double-stranded DNA remains to be answered.
Efficient assembly and function of a DNA replicase depends to a large extent on the ability of replication proteins to correctly and rapidly recognize DNA structures relevant to their activity. Here we have addressed the question of how the RFC clamp loader distinguishes between binding to primed DNA and single- or double-stranded DNA during PCNA clamp assembly. A quantitative analysis reveals that RFC binds all three DNA structures with high affinity and yet selects primed DNA almost exclusively in the presence of excess single- or double-stranded DNA, possibly via a primed DNA-specific change in its conformation/activity that is integral to the clamp assembly mechanism.
EXPERIMENTAL PROCEDURES
Proteins, DNA, and Buffers
Overexpression and purification of the complete five-subunit S. cerevisiae RFC complex, as well as truncated RFC complex (RFC1:Δ2–283) and PCNA (gifts from Dr. Mike O’Donnell, Rockefeller University, New York) are described here briefly; the detailed procedure will be published elsewhere.2 Genes for all five RFC subunits, RFC1, -2, -3, -4, and -5, were cloned from S. cerevisiae genomic DNA into a single pET vector, each under the control of a T7 promoter. The proteins were overexpressed in E. coli cells that also produced tRNAs for arginine, isoleucine, and leucine to correct for codon bias and purified by ion exchange chromatography over Q Sepharose and SP Sepharose columns. PCNA was cloned similarly into a pET vector, except with an amino-terminal His tag and cAMP-dependent protein kinase tag, which allowed single-step purification by nickel column chromatography and 32P labeling of PCNA, respectively. E. coli SSB was a gift from Dr. Mike O’Donnell (40). Protein concentrations were measured by the Bradford assay and by absorbance at 280 nm in 6 M guanidinium hydrochloride, using calculated extinction coefficients: RFC, 163520; trRFC, 162120; PCNA, 5600. Trypsin was purchased from Sigma, and T4 polynucleotide kinase and cAMP-dependent protein kinase were purchased from New England Biolabs. Bacteriophage T7 DNA polymerase (5′–3′ exonuclease mutant) was a gift from Dr. Smita Patel (Robert Wood Johnson Medical School, New Jersey).
Oligodeoxyribonucleotides were synthesized by Integrated DNA Technologies and purified by denaturing polyacrylamide gel electrophoresis. The sequences are as follows: 30-mer, 5′-CTG GTA ATA TCC AGA ACA ATA TTA CCG CCA-3′; 30A-mer, 30-mer plus 3′ A; 30cmpT, 5′-T TGG CGG TAA TAT TGT TCT GGA TAT TAC CAG-3′; 81-mer, 5′-GAG CGT TTT TTC CTG TTG CAA ACG ATT GGC GGT AAT ATT GTT CTG GAT ATT ACC AGC AAG GCC GAT AGT TTG AGT TCT TCT-3′; 56-mer, 5′-GAG CGT TTT TTC CTG TTG CAA ACG ATT GGC GGT AAT ATT GTT CTG GAT ATT ACC AG-3′; 56-mer-5p, 5′-TTG GCG GTA ATA TTG TTC TGG ATA TTA CCA GCA AGG CCG ATA GTT TGA GTT CTT CT-3′. Primer-template and duplex DNAs were prepared by mixing 30-mer or 30A-mer with the requisite complementary strand at a ratio of 1.1:1, respectively, in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, followed by heating to 95 °C (2 min) and slow cooling for >6 h. Single-stranded M13 mp18 ssDNA was purified and annealed with a 30-nucleotide primer as described (41). ATP and ATPγS were purchased from Sigma; [α-32P]ATP, [γ-32P]ATP, and [α-32P]dATP were purchased from PerkinElmer Life Sciences. Nitrocellulose membranes and PEI-cellulose TLC plates were purchased from Schleicher and Schuell and EM Science, respectively. The buffers are as follows: buffer H, 30 mM HEPES, pH 7.5, 4 mM MgCl2, 5% glycerol; gel filtration buffer, buffer H plus 0.1 mg/ml bovine serum albumin, 1 mM DTT, 0.1 mM EDTA, 100 mM NaCl; protease buffer, buffer H plus 0.1 mM EDTA, 2 mM DTT; T7 Pol buffer, 40 mM Tris-HCl, pH 7.5, 14 mM MgCl2, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1 mg/ml bovine serum albumin; SDS gel-loading buffer, 50 mM Tris-HCl, pH 7.5, 100 mM DTT, 2% SDS, 0.1% bromphenol blue, 10% glycerol.
PCNA Loading Assays
PCNA (2 μM) was labeled at an N-terminal kinase recognition site in a 100-μl reaction with 20 μCi of [γ-32P]ATP and 50 units of cAMP-dependent protein kinase in NEB kinase buffer for 1 h at 37 °C. Excess [γ-32P]ATP was removed by filtration through Centricon-10 (Millipore Corp.). In the loading reaction, 0.03 μM 32P-PCNA was mixed with 0.03 μM DNA (primed or unprimed M13mp18 ssDNA coated with 8.5 μM SSB or duplex pBluescript plasmid) and 0.5 mM ATP in 60 μl of gel filtration buffer. The reaction was initiated with 0.02 μM RFC or trRFC (RFC1:Δ2–283), incubated at 30 °C for 5 min, and then filtered over a 5-ml Bio-Gel A-15m column equilibrated in gel filtration buffer. 200-μl fractions were collected, and aliquots were quantitated by scintillation counting.
Quantitative DNA Binding Assays
The DNA used in nitrocellulose membrane binding assays was 32P-labeled as follows: 0.05 μM annealed 30-mer/30cmpT was incubated in a 100-μl reaction with 10 μCi of [α-32P]dATP and 0.2 μM T7 DNA polymerase (premixed with a 5-fold molar excess of thioredoxin and 30 mM freshly prepared DTT) in T7 Pol buffer for 5 min at 37 °C. Next, 100 μM dATP was added to the reaction, and after 5 min the reaction was quenched with 30 mM EDTA and 5 min at 90 °C. The DNA was separated from free nucleotide by spinning through a Bio-Gel P-30 column (Bio-Rad). The radiolabeled 3032P-A-mer/30cmpT DNA (0.03 μM) was mixed with appropriate unlabeled DNAs to prepare 30 μM stocks of single-stranded 30A-mer, primer-templates 30A-mer/81-mer, 30A-mer/56-mer (3′ primer-junction), and 30A-mer/56-mer-5p (5′ primer-junction) as well as duplex 30A-mer/30cmpT DNA, as described above.
The stoichiometry of interaction between RFC and primer-template DNA was measured by nitrocellulose membrane binding assays in which a constant amount of 32P-labeled DNA was titrated with increasing concentrations of RFC. Nitrocellulose membranes were pretreated with 0.5 N NaOH for 2 min and then washed thoroughly with H2O and equilibrated in buffer H. The reactions (15-μl total volume) containing 0.5 μM 32P-DNA and 0–3 μM RFC in buffer H were incubated for 10 min at 25 °C, and 10-μl aliquots were filtered through the membrane in a dot-blot assembly (Schleicher and Schuell). The membrane was washed before and after filtration with 120 μl of buffer H. 1-μl aliquots were spotted onto a separate membrane to measure the total DNA in the reaction. Radioactivity on the membrane was quantitated on a PhosphorImager (Amersham Biosciences), and the molar amount of DNA bound to RFC was determined and plotted versus RFC concentration. Experiments measuring equilibrium dissociation constants for the RFC-DNA interaction were performed similarly except with a constant amount of RFC (0.016 μM) or trRFC (0.016 μM) and increasing 32P-DNA (0–0.6 μM) in buffer H in the absence or presence of 0.5 mM ATPγS or ADP; the effect of PCNA on RFC-DNA interaction was measured in similar experiments with 1 mM ATPγS and 1 μM PCNA. The molar amount of DNA bound to RFC was plotted versus DNA concentration. The binding isotherms were fit to a quadratic equation to determine the apparent dissociation constant for the interaction,
| (Eq. 1) |
where D·R represents the amount of DNA bound to RFC, Dt and Rt are total DNA and RFC concentrations, respectively, and Kd is the dissociation constant.
Competitive DNA binding assays were performed by mixing 0.02 μM 32P-labeled substrate DNA with increasing concentrations of unlabeled competitor DNA (0–1 μM) in buffer H (15-μl total volume), followed by the addition of 0.016 μM RFC and filtration through a nitrocellulose membrane after 10 min at 25 °C, as described above; complementary assays were performed with 0.02 μM unlabeled substrate and 0–1 μM labeled competitor. The amount of DNA substrate bound to RFC was plotted versus competitor DNA concentration, and the data were fit to a hyperbola to determine K1/2, the competitor concentration at which half of the substrate remains bound to RFC.
The rate of dissociation of DNA from RFC was measured by incubating 0.016 μM RFC with 0.02 μM 32P-DNA substrate in buffer H for 10 min at 25 °C, followed by the addition of 1 μM unlabeled competitor DNA. At various times, 10-μl aliquots of the reaction were filtered through a nitrocellulose membrane as described above (32P-DNA bound to RFC at time 0 was measured in the absence of competitor). The molar amount of DNA bound to RFC was plotted versus time, and the data were fit to a single exponential function to yield the dissociation rate, koff.
| (Eq. 2) |
Trypsin Digest
Tryptic digestion of RFC was performed at both high and low NaCl concentrations, in the absence and in the presence of various DNA substrates. The 65-μl reaction contained 2 μM RFC and either no DNA or 3 μM DNA in protease buffer at 100 or 30 mM final NaCl concentration. The digest was initiated by the addition of trypsin to a final concentration of 0.4 ng/μl in the reaction and incubation at 37 °C. At varying times (0–15 min), 10-μl aliquots of the reaction were quenched with 10 μl of SDS gel-loading buffer and heated at 90 °C for 5 min prior to analysis on a 12% polyacrylamide gel. The proteolytic products were visualized by staining with Coomassie Blue.
ATPase Assays
Steady-state ATPase activity of RFC was assayed by monitoring hydrolysis of [α-32P]ATP to [α-32P]ADP plus Pi. 0.2 μM RFC was mixed with 1 mM ATP plus [α-32P]ATP, 1 μM PCNA, and 1 μM DNA (when in the reaction) in buffer H plus 0–400 mM NaCl at 25 °C. At varying times, 5-μl aliquots were withdrawn, quenched with 5 μl of 0.5 M EDTA, and analyzed by polyethyleneimine-cellulose thin layer chromatography in 0.6 M potassium phosphate buffer, pH 3.4. The molar amount of [α-32P]ADP formed was quantitated on a PhosphorImager and plotted versus reaction time to yield the ATPase rate constants (kcat), which were then plotted versus NaCl concentration.
RESULTS
Rapid assembly of clamps at primed DNA sites is important to expedite genomic DNA replication, since clamps are essential for processive DNA polymerase activity. In eukaryotes, the RFC clamp loader loads PCNA clamps onto DNA in a reaction fueled by ATP binding and hydrolysis. In an earlier study, RFC was observed to footprint a 3′ primer-template junction, which suggested that the clamp loader specifically recognizes this DNA structure as the site for clamp assembly (33). However, the same report as well as other studies of clamp loaders from S. cerevisiae, D. melanogaster, mice, and humans indicate that RFC binds single-stranded DNA and double-stranded DNA as well (reviewed in Ref. 15). How can RFC distinguish a primed DNA site as the target for clamp assembly from the background of single- and double-stranded DNA at the replication fork? The experiments described below address this question by quantitatively analyzing the interactions between RFC and various DNA substrates.
S. cerevisiae RFC Purified from E. coli Assembles PCNA Clamps Preferentially on Primed DNA Templates
Previous reports on overexpression and purification of S. cerevisiae RFC from E. coli indicated that the RFC subunits are insoluble when expressed individually. However, when genes for the small RFC subunits were co-expressed on a single plasmid in S. cerevisiae, a soluble complex was detectable (42). More recently, a truncated version of the RFC complex with 273 amino acids deleted (of 861 total) from the amino terminus of the RFC1 subunit has been expressed and purified from E. coli (43). This truncated version of RFC is active in catalyzing PCNA clamp assembly on DNA; however, it is not yet known whether all of its properties are identical to those of the wild-type RFC clamp loader. We have purified the full-length, wild-type RFC complex from E. coli with a yield of close to 7 mg of pure protein/liter of cells. Fig. 2A shows an SDS-PAGE analysis of the RFC complex, which has all five subunits present in a 1:1 stoichiometric ratio. (The cloning and purification strategies will be described in detail elsewhere.)2
Fig. 2. E. coli-produced RFC loads PCNA clamps on primed DNA.
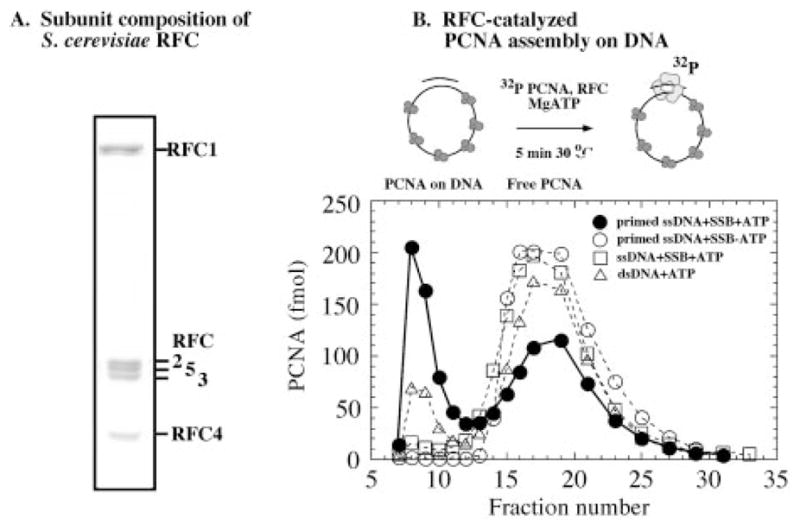
A, an SDS-PAGE analysis of S. cerevisiae RFC complex comprising five subunits in 1:1 stoichiometry. B, PCNA clamp assembly on DNA, assayed by incubating 0.03 μM 32P-PCNA with 0.02 μM RFC and 0.03 μM SSB-coated circular primed M13 ssDNA in the presence (●) or absence (○) of 0.5 mM ATP, unprimed M13 ssDNA with ATP (□), and circular duplex pBluescript DNA with ATP (△) for 5 min at 30 °C, followed by gel filtration as described under “Experimental Procedures.” PCNA loaded on DNA elutes in fractions 7–12 and free PCNA elutes in fractions 14–25.
The clamp loading activity of recombinant RFC was examined by measuring the assembly of 32P-labeled PCNA onto circular DNA (Fig. 2B). When incubated with singly primed, circular M13mp18 ssDNA coated with SSB, RFC loads the clamp onto DNA in the presence of ATP. The large PCNA·DNA complex elutes early from a gel filtration column (fractions 7–12), whereas free PCNA elutes later (fractions 14–25). The reaction is absolutely dependent on the presence of ATP (Fig. 2B) and RFC (data not shown), since no PCNA loading is evident in the absence of either of these reactants. Furthermore, RFC does not load PCNA on ssDNA in the absence of a primer, whether the DNA is coated with SSB (Fig. 2B) or not (data not shown); earlier studies have shown that E. coli SSB and eukaryotic RPA are indistinguishable in this PCNA loading assay (43). Thus, RFC catalyzes PCNA assembly specifically at a primed DNA site, as expected for its function in vivo. Fig. 2B also shows that PCNA is loaded to some extent on supercoiled circular DNA substrates (the same occurs with nicked DNA; data not shown). Clamp assembly on circular duplex DNA may occur at secondary structures with single-stranded/double-stranded DNA junctions that mimic a primer-template, or perhaps S. cerevisiae RFC can load PCNA onto duplex DNA, albeit with much less efficiency than on primed DNA (as suggested for human RFC) (29). The clamp loading assay shows that S. cerevisiae RFC purified from E. coli is active and exhibits a clear preference for loading PCNA onto a primed DNA template.
RFC Binds Single-stranded, Double-stranded, and Primer-Template DNAs with High Affinity
The interaction between RFC and DNA was measured quantitatively using 32P-labeled DNA in nitrocellulose membrane filtration assays. First, we measured the stoichiometry of RFC binding to primer-template DNA, in order to determine the active-site concentration of this recombinant, E. coli-expressed eukaryotic clamp loader. The primer-template is a 31-nucleotide (nt) primer annealed to an 81-nt template to form a 31-nt duplex with 25-nt ssDNA overhangs on either side. The single-stranded DNA substrate is the 31-nt primer, and double-stranded DNA is the primer annealed to its complement. The DNA lengths were chosen to be in excess of the reported RFC footprint of 12 bases on the 5′ single-stranded overhang and 8–15 bases on the duplex region of a primer-template DNA (33). During assay development, we noted that RFC has high affinity for a 5′-phosphate residue on any DNA structure (single-stranded, double-stranded, or primer-template); a similar property of human and D. melanogaster RFC1 amino-terminal domains for recognizing 5′-phosphate on duplex DNA has been reported recently (35). In order to eliminate possible misinterpretation of 5′-phosphate recognition as interaction between RFC and DNAs other than the primer-template, the DNA substrates used in this study were 32P-labeled by incorporation of [α-32P]dATP at the 3′-end by phage T7 DNA polymerase. Fig. 3A shows a titration of 0.5 μM 32P-primer-template DNA with increasing concentrations of RFC. All DNA in the reaction is bound with 0.53 μM RFC in the reaction, indicating that 95% of the protein is active for interaction with DNA.
Fig. 3. RFC binds a variety of DNA substrates with high affinity.

A, 32P-labeled primer-template DNA (0.5 μM) binding to RFC (0–3 μM), assayed by nitrocellulose membrane filtration assays as described under “Experimental Procedures.” The molar amount of bound DNA plotted versus RFC concentration shows a 1:1 stoichiometry for the RFC-DNA interaction. B, a titration of 0.016 μM RFC with 0–0.1 μM different 32P-labeled DNAs yields an apparent Kd of 12 ± 2, 7 ± 1, and 7 ± 1 nM, respectively, for RFC binding to 31/81 primer-template DNA (●), 31-mer ssDNA (□), and 31-mer duplex DNA (△), respectively. C, a similar experiment performed with 0.2 μM RFC. D, the experiment performed with 0.016 μM RFC and primer-template DNA in the presence of 0.5 mM ATPγS (○) and ADP (◇); Kd = 7–9 nM. E, 0.016 μM truncated RFC (missing the N-terminal 283 amino acids of RFC1 that are not essential for clamp assembly) binding primer-template DNA (●), 31-mer ssDNA (□), and 31-mer duplex DNA (△) with high affinity similar to full RFC (apparent Kd = 13, 6, and 5 nM, respectively).
Next, we assayed RFC binding to single-stranded, double-stranded, and primer-template DNAs order to compare quantitatively its affinity for these different DNA structures. A constant amount of RFC (0.016 μM) was titrated with increasing concentrations of 32P-DNA, and the binding isotherm yielded the maximum RFC·DNA complex formed and an apparent Kd for the interaction (Fig. 3B). To our surprise, RFC appears to bind primer-template, ssDNA, and dsDNA to the same extent and with similar high affinity (apparent Kd = 12, 7, and 7 nM, respectively), implying that the clamp loader does not distinguish between these different DNA structures (non-specific DNA binding to the membrane is less than 0.5% of the total DNA bound to RFC).
One possible caveat in the above experiment is that at low nanomolar concentrations, the RFC subunits may not form a stable pentamer. This issue is of particular concern given reports that the large RFC subunit alone can bind single-stranded and double-stranded DNA. Therefore, we performed the same experiment with RFC concentrations up to 0.5 μM (at low micromolar concentrations, RFC migrates as a stable complex in a gel filtration column; data not shown). At all concentrations tested, we observed similar high affinity binding of DNA substrates to RFC (shown in Fig. 3C for 0.2 μM RFC). The DNA binding assays were performed initially in the absence of nucleotide cofactors, and a recent surface plasmon resonance study of the interaction between a truncated version of RFC and primed DNA suggests that the protein·DNA complex is more stable in the presence of ATPγS (38). Thus, DNA binding was measured also in the presence of 0.5 mM ATPγS and ADP (ATPγS is not hydrolyzed by RFC under the assay conditions; data not shown). No significant difference in RFC binding to the three DNAs was detectable in the absence or presence of these nucleotides (shown in Fig. 3D for the primer-template; Kd = 7–9 nM; ssDNA and dsDNA data not shown). The slightly higher amount of RFC·DNA detected in the presence of ATPγS may reflect a subtle effect of the nucleotide on the stability of the complex. Previous reports indicate that the amino-terminal domain of RFC1, which is considered unnecessary for clamp assembly based on deletion analysis, binds double-stranded DNA and possibly single-stranded DNA as well (reviewed in Ref. 15). In order to determine whether the high affinity RFC binding to ssDNA and dsDNA we observe is peculiar to the RFC1 amino-terminal domain or a property of the PCNA loading-active domains, we assayed trRFC (containing a truncated version of RFC1:Δ 2–283) for DNA binding as above; the trRFC complex catalyzes PCNA assembly on primed DNA similar to full-length RFC (data not shown). As observed for full-length RFC, trRFC binds the three DNAs with high affinity (Fig. 3E; Kd = 13, 6, and 5 nM for primer-template, ssDNA, and dsDNA, respectively). The maximum amount of dsDNA bound to trRFC is lower (~50% of total) and may indicate a role for the N-terminal domain of RFC1 in stabilizing the interaction between RFC and dsDNA. Thus, an initial quantitative analysis of the DNA binding activity of RFC does not reveal significant selectivity for primer-template DNA over other DNA structures.
Despite Their High Affinity for RFC, ssDNA and dsDNA Cannot Compete with Primed DNA
Given the contrast between the need for RFC to distinguish primer-template DNA from single- and double-stranded DNA and initial evidence that RFC binds all three DNAs with high affinity (Fig. 3B), we continued the investigation further to determine whether the interaction between RFC and primed DNA is somehow different from that between RFC and ssDNA and dsDNA. To measure directly whether RFC exhibits preference for one DNA structure over the others, we assayed RFC binding to primer-template DNA in the presence of increasing concentrations of competitor single- or double-stranded DNA in the reaction.
As shown in Fig. 4A, when RFC (0.016 μM) is incubated with 32P-labeled primer-template (0.02 μM) premixed with unlabeled ssDNA (0–1 μM), nearly all of it forms RFC·primed DNA complex, even in the presence of 1 μM ssDNA (at 3 μM ssDNA competitor, primed DNA binding is reduced to about 40%; data not shown). A complementary experiment with 0.02 μM unlabeled primer-template and 0–1 μM 32P-labeled ssDNA shows very weak binding of ssDNA to RFC under these conditions (K1/2 = 160 nM), relative to ssDNA binding in the absence of primer-template DNA (Kd = 7 nM; Fig. 3B). Similar results are obtained with double-stranded DNA as the competitor. Experiments with 32P-labeled primer-template DNA and 32P-labeled dsDNA, respectively, show RFC binding to primed DNA even in the presence of 1 μM dsDNA and very low affinity of dsDNA for RFC in the presence of the primer-template (Fig. 4B). trRFC exhibits the same profile as full-length RFC in these assays, indicating that resistance of the clamp loader-primed DNA complex to competition with ssDNA and dsDNA is not dependent on the amino-terminal DNA-binding domain of RFC1 (data not shown).
Fig. 4. Single- and double-stranded DNA cannot compete effectively with primed DNA for binding RFC.

A and B, competitive DNA binding assays that measure RFC (0.016 μM) binding to a mixture of 0.02 μM 32P-labeled primer-template plus 0–1 μM unlabeled 31-mer single-stranded DNA (A, ●) or 31-mer double-stranded DNA (B, ●), 0.02 μM unlabeled primer-template plus 0–1 μM 32P-labeled ssDNA (A, ■) or dsDNA (B, ■) or 0–1 μM 32P-labeled ssDNA (A, □) or dsDNA (B, □) alone. C, RFC (0.016 μM) binding to 0.02 μM 32P-primer-template DNA plus 0–1 μM primer-template competitor (●), 0.02 μM 32P-ssDNA plus 0–1 μM ssDNA competitor (□), and 0.02 μM 32P-dsDNA plus 0–1 μM dsDNA competitor (△). The data fit to a hyperbola yield a K1/2 value of 0.051, 0.037, and 0.042 μM, respectively. D, 32P-ssDNA (0–1 μM) binding to RFC (0.016 μM) alone (■) or in the presence of 0.02 μM unlabeled primer-template (○) or dsDNA (△); K1/2 = 0.16 and 0.055 μM for ssDNA binding in the presence of primer-template and dsDNA, respectively. E, 32P-ssDNA (0.02 μM) binding to RFC (0.016 μM) in the presence of 0–0.4 μM unlabeled primer-template DNA (●) or ssDNA (□); K1/2 = 0.003 and 0.037 μM with primer-template and ssDNA competitor, respectively. F, a titration of 0.005 μM RFC with 0–0.1 μM 32P-labeled primer-template alone (●) and in the presence of 0.03 μM unlabeled primer-template (○) or ssDNA (□); K1/2 = 0.045 and 0.009 μM with primer-template and ssDNA competitor, respectively
A series of experiments detailed below test further this surprising lack of competition between DNA substrates with similar affinity for RFC. First, experiments were performed with 0.016 μM RFC, 0.02 μM 32P-labeled ssDNA, dsDNA, or primer-template DNA as substrate and increasing amounts of unlabeled self as competitor, to test whether the membrane filtration assay and experimental design support detection of simple competitive DNA binding to RFC (Fig. 4C). For homologous competition between labeled and unlabeled ligand, K1/2 = [labeled ligand] + Kd, where K1/2 is the competitor concentration at which substrate binding is lowered by 50%. This predicts K1/2 ≈ 0.03 μM under these experimental conditions (Kd = 7–12 nM for the three DNAs) (Fig. 3B). The competitive binding curves in Fig. 4C yield K1/2 = 0.051, 0.037, and 0.042 μM for primer-template, ssDNA, and dsDNA, respectively. Thus, each DNA competes effectively with itself, implicating reasons other than the assay technique for the observed lack of competition between primer-template and ssDNA or dsDNA for binding RFC.
In the next experiment, RFC was titrated with increasing concentrations of 32P-labeled single-stranded DNA, alone and in the presence of 0.02 μM unlabeled double-stranded or primer-template DNA. Given that ssDNA and competitor DNA (double-stranded or primer-template DNA) exhibit the same affinity for RFC, if the two DNAs bind the same site on RFC, simple homologous competition predicts a K1/2 of ~0.03 μM for ssDNA binding with 0.02 μM competitor DNA in the reaction. The binding isotherm for ssDNA in the presence of dsDNA is slightly right-shifted relative to the one with ssDNA alone and yields a K1/2 of 0.055 μM. In contrast, the K1/2 for ssDNA binding in the presence of 0.02 μM primer-template is 0.16 μM. Thus, ssDNA and dsDNA compete effectively with each other for binding RFC, but even a small amount of primer-template in the reaction appears to substantially reduce RFC binding to ssDNA (and dsDNA), indicating a distinctive interaction between RFC and primed DNA.
In complementary experiments, we measured binding of 32P-labeled ssDNA (0.02 μM) to RFC (0.016 μM), in the presence of increasing amounts of unlabeled ssDNA or primer-template DNA as competitor (0–1 μM). Fig. 4E shows that when ssDNA is the competitor, K1/2 for the interaction is 0.037 μM (i.e. at a competitor concentration of Kd plus labeled DNA, the amount of labeled ssDNA bound to RFC is reduced by half (shown also in Fig. 4C)). In contrast, when increasing amounts of primer-template competitor are added to the reaction, the K1/2 is 0.0035 μM (i.e. only 0.0035 μM primed DNA is required to reduce labeled ssDNA (0.02 μM) binding to RFC (0.016 μM) by half). These results confirm that there is no simple competition between ssDNA and primed DNA for RFC and suggest that primed DNA may bind RFC with high affinity whether RFC is free or in the presence of single-stranded (or double-stranded) DNA. In the next experiment, this prediction is tested directly by measuring binding of 32P-labeled primer-template DNA (0–0.1 μM) to RFC (0.005 μM), alone or in the presence of 0.03 μM unlabeled primer-template DNA or ssDNA (Fig. 4F). As expected from the binding isotherm in Fig. 3B, the 32P-labeled primer-template alone binds RFC with high affinity; Kd = 8 nM. With 0.03 μM unlabeled primer-template in the reaction, the binding isotherm shifts to the right and yields a K1/2 of 0.045 μM (expected K1/2 ≈0.04 μM). In striking contrast, however, the primer-template binding isotherm in the presence of 0.03 μM unlabeled ssDNA is virtually identical to that with no other DNA in the reaction and yields a K1/2 of 9 nM (expected K1/2 ≈ 0.04 μM). It appears that the 0.03 mM ssDNA in the reaction is invisible with respect to the interaction between primed DNA and RFC.
The results described above are intriguing because they indicate that the RFC·primed DNA complex is resistant to competition with ssDNA or dsDNA despite similar high affinity of these DNAs for RFC. Possible explanations for these results include the following: (a) the DNAs bind RFC with significantly differing affinity (this explanation is not consistent with the measured apparent equilibrium constants); (b) ssDNA and dsDNA may bind a different site on RFC than primed DNA; (c) the DNAs may bind the same/overlapping site on RFC and the primer-template binds RFC even if the site is occupied with ssDNA or dsDNA, but not vice versa; (d) primed DNA forms a highly stable complex with RFC that is unavailable for interaction with ssDNA or dsDNA; (e) primed DNA induces a conformational change in RFC that lowers its affinity for other DNA structures. Experiments described below examine these possible explanations for the high specificity of primed DNA-RFC interaction.
RFC·Primed DNA Complex Exhibits Higher Stability Relative to Other RFC·DNA Complexes
We utilized a partial tryptic digest assay to detect possible differences between RFC·ssDNA, RFC·dsDNA, and RFC·primed DNA complexes (e.g. the presence of distinct DNA binding sites or conformational changes specific to each DNA substrate). As shown in Fig. 5A, panel 1, the RFC1 subunit is susceptible to tryptic digest over time and is largely degraded within 15 min in the absence of DNA (the smaller RFC subunits appear curiously resistant to proteolysis). When RFC is bound to primer-template DNA, however, the large RFC subunit becomes significantly more resistant to proteolysis, and about half of the original protein remains undigested after 15 min (Fig. 5A, panel 2). Interestingly, the same experiment performed with ssDNA (Fig. 5A, panel 3, 81-mer template; data not shown for 31-mer primer) or dsDNA (Fig. 5A, panel 4) showed no protection of the RFC1 subunit from proteolysis. Initially, we took these data to mean that on binding primer-template DNA, RFC undergoes a conformational change that makes it more resistant to tryptic digest or that the primer-template directly blocks tryptic digestion at its site of interaction. In either case, the effect appeared specific to primer-template DNA.
Fig. 5. Partial tryptic digest and ATPase assays reveal primed DNA-specific changes in RFC.

A, RFC (2 μM) was subjected to proteolysis by trypsin over time, in the absence of DNA (1) or in the presence of 31/81 primer-template (2), 81-mer ssDNA (3), and 31-mer dsDNA (4) in a reaction containing 100 mM NaCl and analyzed by SDS-PAGE as described under “Experimental Procedures.” Similar assays were performed at a lower NaCl concentration of 30 mM, with no DNA (5), primer-template (6), ssDNA (7), and dsDNA (8), in the reaction. B, the ATPase rate of RFC (0.2 μM) at varying NaCl concentrations in the absence of DNA (○) or in the presence of primer-template (●), ssDNA (□), and dsDNA (△). The maximum steady-state ATPase rate is 0.45 s−1 with primed DNA at 150 mM NaCl concentration.
We noted, however, that the high concentration of RFC (2 μM) utilized in the proteolytic digest introduced more NaCl into the reaction than was present in the DNA binding experiments (100 versus ~30 mM, respectively). Therefore, the digest was repeated at 30 mM NaCl (after lowering salt concentration in the RFC preparation by dialysis). As shown in Fig. 5A, under the less stringent conditions, RFC1 is still susceptible to proteolysis in the absence of DNA (panel 5) and is protected in the presence of primer-template DNA (panel 6), with similar kinetics as in panels 1 and 2 at high NaCl, respectively. However, under these conditions, ssDNA (panel 7) and dsDNA (panel 8) protect RFC1 from trypsin as well. Thus, it appears that all three DNAs confer similar protease resistance on RFC1, presumably by binding at the same site on RFC and possibly even inducing similar changes in conformation. It is clear, however, that the RFC·primed DNA complex is more stable to NaCl concentration and therefore different from the other two complexes. Relatively high resistance of RFC·primed DNA to NaCl was observed also in nitrocellulose binding experiments, although Kd values could not be determined accurately at high NaCl, since the binding did not reach saturation.
Is the ability of RFC to load PCNA clamps specifically on primed DNA related to the observed resistance of RFC·primed DNA complex to NaCl? We searched for a possible connection by assaying the effect of different DNA substrates on RFC ATPase activity, which is coupled to its clamp loading activity. Steady-state ATPase assays were performed with RFC (0.2 μM), PCNA (1 μM), and [α-32P]ATP (1 mM) in the presence of ssDNA, dsDNA, or primer-template DNA (1 μM) at varying NaCl concentrations. As shown in Fig. 5B, the RFC ATPase rate is stimulated by the presence of any DNA substrate by at least 2-fold over no DNA (at low NaCl, kcat = 0.1 s−1 without DNA and 0.25 s−1 with DNA). As NaCl concentration increases, RFC ATPase activity in the presence of primed DNA increases to kcat = 0.45 s−1 (peak at 150 mM NaCl), but the activity in the presence of ssDNA or dsDNA remains constant or declines (kcat = 0.2 s−1 at 150 mM NaCl). Thus, primed DNA binding has a distinctive effect on RFC activity, which corresponds to the NaCl-stable nature of the RFC·primed DNA complex.
Release of Primed DNA from RFC Is Slow Compared with Other DNA Structures
Results from competitive DNA binding and partial proteolysis assays indicate that RFC·primed DNA complex is more stable than other RFC·DNA complexes. The next experiment tests this stability directly, by measuring the rate of dissociation of the three DNAs from RFC (Fig. 6). RFC (0.016 μM) was preincubated with 32P-labeled DNA (0.02 μM) and then chased with 1 μM unlabeled self over time (1 μM DNA is a sufficient chase according to the data in Fig. 4C). At time 0, ~80% of RFC in the reaction is bound by labeled DNA, which dissociates over time. The experiment performed with 32P-labeled ssDNA substrate and unlabeled ssDNA chase shows that the DNA dissociated completely from RFC at the first measurable time point, which yields an off rate equal to or faster than 0.2 s−1. Double-stranded DNA exhibits a similar, rapid rate of dissociation from RFC (Fig. 6A). In contrast, primer-template DNA dissociates from RFC at a rate of 0.025 s−1, at least 10-fold slower than single- or double-stranded DNA. The RFC·primed DNA complex is highly stable compared with the other RFC·DNA complexes, and this property probably contributes to specificity of the interaction. It should be noted, however, that the half-life of the RFC·primed DNA complex is ~28 s; therefore, the 10-min incubation in the competition experiments of Fig. 4 should be more than adequate time for single- or double-stranded DNA in the reaction to gain access to free RFC. However, as shown in Fig. 6B, when 1 μM ssDNA or dsDNA is used as chase for 32P-labeled primer-template, there is almost no loss of the primer-template from RFC over time (tested up to 30 min). Thus, even with a high excess of ssDNA or dsDNA in the reaction, RFC preferentially binds and maintains its interaction with primed DNA.
Fig. 6. Interaction of RFC with primed DNA is more stable than with ssDNA or dsDNA.

Release of 32P-labeled DNA (0.02 μM) from RFC (0.016 μM) was measured over time in the presence of 1 μM unlabeled DNA chase by nitrocellulose membrane filtration assays, as described under “Experimental Procedures.” A, 32P-labeled primer-template (●), 32P-labeled ssDNA (□), and 32P-labeled dsDNA (△) chased with excess unlabeled self as competitor. The dissociation rates are 0.025 ± 0.003 s−1, ≥0.2 s−1, and ≥0.2 s−1, respectively. B shows that if 32P-labeled primer-template is chased with excess unlabeled ssDNA (□) or dsDNA (△), the RFC·primed DNA complex persists over time, in contrast to the chase with unlabeled primer-template DNA (●).
Primer-Template DNAs with Either 5′ or 3′ Single-stranded Overhang Can Mimic Full Primed DNA to Varying Extents
The assays described thus far have been performed with primer-template containing a 31-nt duplex region flanked by 25-nt 5′ and 3′ single-stranded DNA overhangs. What features of this DNA are important for specific recognition by RFC: the 3′ primer-template junction, the 5′ junction, or perhaps arrangement of double- and single-stranded regions adjacent to each other? We addressed this question by measuring binding of primer-template DNAs with either a 25-nt 5′ ssDNA overhang (3′ primer junction) or 25-nt 3′ ssDNA overhang (5′ primer junction) to RFC. Both DNAs bind RFC with high affinity (apparent Kd = 10 nM; data not shown), and ssDNA cannot compete effectively against either primer-template for RFC in competition experiments with 0.016 μM RFC, 0.02 μM 32P-labeled primer-template, and 0–1 μM unlabeled ssDNA (Fig. 7A). In complementary experiments (Fig. 7B), when 32P-labeled ssDNA (0.02 μM) is competed with unlabeled 5′primer junction or 3′primer junction DNA (0–1 μM), only 0.004 and 0.003 μM of the competitor, respectively, is required to reduce ssDNA binding to RFC by half (as observed with the full primer-template DNA competitor; Fig. 4E). In contrast, the same experiment performed with only the 56-nt template strands of the 5′ or 3′ primer junction DNAs yields a K1/2 of 0.035 μM as expected from simple competition between the 31- and 56-mer single-stranded DNAs. Thus, it appears that a single-stranded plus double-stranded DNA structure, with a 5′ or 3′ ssDNA overhang, is sufficient for specific interaction with RFC.
Fig. 7. A 5′ or 3′ primer-template junction supports specific interaction between RFC and primed DNA.

A, binding of 0.02 μM 32P-ssDNA (□), 32P-31/56 5′ primer junction DNA (●), or 32P-31/56 3′ primer junction DNA (○) to RFC (0.016 μM) in the presence of 0–1 μM unlabeled ssDNA competitor. B, a complementary experiment in which 32P-ssDNA 0.02 μM binding to RFC (0.016 μM) is competed with 0–0.4 μM unlabeled 31/56 5′ primer junction DNA (●), 56-mer template ssDNA (■), 31/56 3′ primer junction DNA (○), and the corresponding 56-mer template ssDNA (□). For both 56-mer single-stranded DNAs, the K1/2 is 0.035 μM, indicating simple competition between the 31- and 56-mer ssDNAs. In contrast, both 3′ and 5′ primer junction DNA competitors yield a K1/2 of 0.004 μM. C, the ATPase activity of RFC (0.2 μM) measured in the presence of 5′ primer junction DNA (●) or 3′ primer junction DNA (○), as described under “Experimental Procedures.” The peak steady-state ATPase rate is 0.45 s−1 for the 5′ primer junction (similar to full primer-template DNA) and 0.25 s−1 for 3′ primer junction (similar to ssDNA).
Next, we tested the effect of the two different primer-template junctions on the ATPase activity of RFC. Fig. 7C shows that the 5′ primer junction DNA (3′ ssDNA overhang) stimulates RFC ATPase activity similar to full primer-template DNA (peak ATPase rate is 0.45 s−1 at 150 mM NaCl). In contrast, in the presence of 3′ primer junction DNA (5′ ssDNA overhang), the ATPase rate remains at levels observed with single-stranded DNA substrate. Thus, RFC can distinguish the two half-primer-template DNA structures from single- or double-stranded DNA and also appears capable of distinguishing between the two. Since it is not clear yet exactly how DNA binding is linked to individual steps in the ATPase pathway, the implication of this difference between the 3′ and 5′ primer-template junction DNAs awaits investigation.
PCNA Suppresses RFC Binding to ssDNA and dsDNA
It is not yet completely clear whether RFC binds PCNA or DNA first in the clamp-loading pathway. If PCNA is bound first to RFC, it may also influence the ability of RFC to recognize primed DNA as a target for clamp assembly. We examined this possibility by performing DNA binding experiments with PCNA (1 μM) and ATPγS (1 mM) in the reaction with 0.2 μM RFC and 0–1 μM 32P-labeled DNA (30 mM NaCl); the experiments were performed at high RFC concentration to ensure stable RFC·PCNA complex formation. Fig. 8A shows that primer-template DNA binding to RFC is unaffected by the presence of PCNA, but binding of ssDNA (Fig. 8B) and dsDNA (Fig. 8C) is significantly lower (note that inclusion of PCNA does not increase NaCl concentration in the reaction). Thus, the RFC clamp loader can discriminate against single- and double-stranded DNA when free or in complex with the PCNA clamp.
Fig. 8. PCNA increases selectivity of RFC for primed DNA.

A–C, respectively, show the effect of PCNA on interaction between RFC and DNA in assays measuring 32P-primer-template, 32P-ssDNA, and 32P-dsDNA binding to RFC (0.2 μM) in the absence (■) and in the presence (□) of PCNA (1 μM) plus ATPγS (1 mM).
DISCUSSION
The RFC clamp loader is a critical component of the protein machinery responsible for DNA replication and repair/recombination. It catalyzes assembly of circular PCNA clamps onto primed DNA sites, where they are bound by replicative DNA polymerases and used as sliding tethers for rapid, processive DNA synthesis (reviewed in Ref. 1). Initiation of processive DNA synthesis is influenced by the efficiency of clamp assembly on DNA, and rapid, specific recognition of primer-template DNA is important for efficient RFC function. Rapid clamp loader action is especially important during synthesis of lagging strand DNA, since a clamp must be loaded about every second for the initiation of each new Okazaki fragment. RFC is known to bind primer-template DNA with high affinity, but it binds other DNA structures, such as single- and double-stranded DNA, as well (reviewed in Ref. 15). This raises the question of whether (and how) RFC can distinguish a primed DNA site from the considerable background of single- and double-stranded DNA during replication. In this study, we examine RFC binding to different DNA substrates and demonstrate that RFC selectively binds and forms a stable complex with primer-template DNA even in the presence of a large excess of single- and double-stranded DNA.
The affinity of RFC for primer-template DNA as well as ssDNA and dsDNA was measured by nitrocellulose membrane filtration assays that provide a quantitative measure of protein-DNA interactions. The apparent Kd values are close to 10 nM for all three DNA structures, indicating that RFC binds ssDNA and dsDNA with as high affinity as primer-template DNA. The RFC1 subunit of RFC contains an amino-terminal domain (~275 amino acids) that reportedly binds both dsDNA and ssDNA but appears to be dispensable for its PCNA-loading activity (43). In fact, many in vitro studies have been performed with RFC deleted for the RFC1 amino-terminal domain, since it appears unnecessary and its removal seems to improve protein stability and activity (38, 44–47). Thus, it was possible that the high affinity DNA binding we observed with the full-length, wild-type RFC complex is unrelated to its clamp loading function. It should be noted, however, that even if it were unrelated to clamp loading, tight binding of RFC to DNA structures other than primer-template could titrate out the clamp loader and negatively impact DNA replication efficiency in vivo. We did, however, assay a truncated version of RFC (trRFC = RFC1:Δ2–283 and RFC2, -3, -4, and -5) for DNA binding and observed that trRFC also binds ssDNA and dsDNA with similar affinity as primer-template DNA. Additionally, nucleotide cofactors of RFC (ATPγS (for ATP) and ADP) do not appear to significantly affect the interaction (as measured by nitrocellulose membrane filtration assays).
Competitive DNA binding experiments were performed next, in order to assay directly if the presence of ssDNA or dsDNA in the reaction reduces formation of the RFC·primer-template DNA complex. Surprisingly, ssDNA and dsDNA were unable to compete effectively with the primer-template even at 50-fold higher concentrations (100-fold higher than the apparent Kd). RFC appears to interact selectively with primed DNA among excess ssDNA and dsDNA, although it binds these DNAs with high affinity in the absence of primed DNA. This selectivity could result from a highly stable interaction between RFC and primed DNA that renders RFC unavailable for interaction with other DNA or because primer-template binding to RFC lowers its affinity for other DNA or because primer-template binds RFC with high affinity whether RFC is free or in complex with single- or double-stranded DNA (but not vice versa).
The first hypothesis predicts a slow rate of dissociation of primed DNA from RFC, the second hypothesis implies a primed DNA-specific change in RFC conformation, and both the second and third hypotheses predict tight binding of primer-template to RFC in the absence and presence of ssDNA or dsDNA. These predictions were tested by measuring the rate of dissociation of 32P-labeled primer-template from RFC in chase experiments, by partial proteolysis of RFC to detect DNA-dependent conformational changes, and by several competitive DNA binding experiments. The chase experiments revealed that dissociation of primer-template DNA from RFC is at least 10-fold slower than that of single- or double-stranded DNA. Thus, the RFC·primed DNA complex is much more stable than RFC·ssDNA or RFC·dsDNA complexes, which probably contributes to the selectivity of interaction between RFC and primed DNA. It should be noted that differences in the dissociation rates (koff) imply that the nearly identical Kd values determined for the three DNAs from membrane filtration assays may not be absolutely correct; these assays may not necessarily measure binding under true equilibrium conditions, because the reactants could undergo significant changes in concentration as the solution passes through the membrane. It is also quite possible that the DNAs bind RFC with different association rates (kon); there is evidence that DNA-binding proteins (RPA, for example) can bind different DNA structures with different bimolecular association rates (48). It will be interesting to measure the “on” and “off ” rates of RFC-DNA interaction in solution to determine whether RFC does bind primed DNA with a different “on” rate and whether this plays a role in its selection as the site for clamp assembly.
The second possibility, that primed DNA binding to RFC induces loss of affinity for single- and double-stranded DNA, is still likely, because the half-life of RFC·primed DNA complex is only about 30 s (koff = 0.025 s−1), and this does not explain complete resistance of the complex to competition by excess ssDNA and dsDNA even on prolonged exposure (10 min; Fig. 6). A partial tryptic digest indicates a change in RFC conformation on binding DNA; the RFC1 subunit is predominantly affected, consistent with earlier reports that RFC1 is the primary DNA-binding subunit in RFC. There are no obvious differences in the profile and extent of RFC proteolysis in the presence of ssDNA, dsDNA, or primed DNA at low NaCl concentration, indicating that all DNAs bind the same site and have the same effect on RFC. However, the RFC·primed DNA complex is notably more stable to NaCl concentration. Furthermore, all three DNAs stimulate the steady-state ATPase activity of RFC, but unlike ssDNA and dsDNA, primed DNA-stimulated activity increases with salt concentration and peaks at a 2-fold higher rate than observed with other DNAs. Taken together, these results hint at a primed DNA-specific effect on RFC conformation/activity, although they could also simply reflect higher stability of the RFC-DNA interaction.
Evidence from competitive DNA binding experiments is consistent with primed DNA lowering RFC affinity for other DNAs or with the ability of primed DNA to bind RFC, whether free or in complex with other DNA, with high affinity. We observe identical tight binding of primer-template to RFC in the absence or presence of excess ssDNA. Also, a very low amount of primer-template DNA is required to disrupt the interaction between ssDNA and RFC. Similar results are obtained in experiments with double-stranded DNA. These results could indicate that primed DNA binds RFC·ssDNA or RFC·dsDNA complexes with high affinity, displaces the DNA from RFC, and prevents rebinding. Alternatively, these results can be explained by a decrease in RFC affinity for ssDNA and dsDNA on binding primer-template DNA, which persists even after the primer-template dissociates from RFC. Thus, primed DNA functions as a catalyst and substoichiometric amounts in the reaction can prevent RFC binding to other DNA. The end result is preferential interaction between RFC and primed DNA that is resistant to competition by other DNA structures. Experiments measuring changes in intrinsic RFC fluorescence that might reflect primed DNA-induced conformational change are under way, to further explore the basis for this selectivity.
Structural features peculiar to primer-template DNA guide assembly of clamp·DNA polymerase complexes at primed sites for initiation of processive DNA synthesis. The PCNA ring is a trimer of identical subunits arranged in a head-to-tail fashion (4). This arrangement imparts directionality to the PCNA ring and to its interactions with other proteins, including DNA polymerase. The carboxyl-terminal face and the interdomain connector loops of PCNA have been implicated in binding polymerase δ and ε, as well as RFC (49–51); therefore, PCNA is probably loaded on DNA with its C-terminal face toward the 3′ primer-template junction. Presumably, the clamp loader binds primed DNA in a particular orientation in order to load the clamp correctly.
Does RFC recognize the 3′ or the 5′ primer-template junction or simply the presence of single- and double-stranded DNA adjacent to each other? In vivo, the 3′ primer-template junction may be blocked by Pol α primase after it synthesizes a 35–50-nucleotide RNA-DNA hybrid primer, as it maintains stable contact with the primed site and RPA on template DNA (RFC displaces Pol α from the primed site to initiate clamp assembly) (52, 53). Thus, it is possible that the 5′ primer-template junction is more exposed than the 3′ junction (at least initially when Pol α is present at the site), and RFC recognizes this particular feature of primed DNA. Consistent with this hypothesis, the human RFC p140 subunit is found to cross-link primed DNA at the 5′ junction, indicating that the clamp loader is bound in its vicinity (54). We tested DNAs containing either a 3′ or a 5′ primer-template junction and found that both structures support specific and stable interaction with RFC when challenged with single-stranded DNA. However, differences in RFC ATPase activity in the presence of a 3′ or 5′ primer-template junction DNA suggest that RFC distinguishes between these two DNA structures. The 5′ primer-template junction effects the high, NaCl-resistant RFC ATPase rate observed also with full primer-template DNA, while the 3′ primer-template junction effects the low, NaCl-sensitive ATPase rate observed with single- or double-stranded DNA. Since the ATPase activity of RFC is coupled to its PCNA loading activity, one can speculate that changes in the ATPase rate reflect how RFC reacts to DNA structure during the clamp assembly process. More detailed information on the RFC ATPase and DNA binding mechanisms is necessary for better insight into the mechanism of primer-template recognition and clamp assembly.
A Model for Efficient Recognition of Primed DNA Sites for PCNA Clamp Assembly by RFC
Interaction of RFC with single- and double-stranded DNA has been reported by several laboratories, but since RFC loads PCNA predominantly on primed DNA, this interaction is generally considered nonspecific and has not been attributed any particular function. Furthermore, since RFC does not appear to bind RPA-coated single-stranded DNA (33, 38), it is assumed that in vivo RPA blocks RFC binding to ssDNA, thereby increasing its selectivity for the primer-template. Our finding that RFC·ssDNA and RFC·dsDNA complexes are much more labile than RFC·primer-template DNA reveals a mechanism by which RFC itself can find and selectively bind primed DNA. The half-life of RFC on single- or double-stranded DNA is at least 10-fold lower than on the primer-template, and these DNAs are unable to compete effectively with primed DNA even at a 50-fold higher concentration. Therefore, as depicted in Fig. 9, this rapid ssDNA or dsDNA binding and release could constitute a scanning mechanism for speedy recognition of a primed DNA site at the replication fork against a background of excess single- and double-stranded DNA.
Fig. 9. A model for rapid recognition of primed DNA sites by RFC at the DNA replication fork.

During DNA replication, the RFC clamp loader can transiently interact with single- and double-stranded DNA at the replication fork until it recognizes a primed DNA site, where it pauses to assemble a clamp. The ability to scan DNA rapidly as well as bind a primer-template structure with high specificity and stability among excess single- and double-stranded DNA may improve the efficiency with which RFC catalyzes clamp assembly at primed DNA sites.
Once RFC recognizes a primed-DNA site, it forms a relatively stable complex that could bind PCNA, load it on the DNA, and release PCNA·DNA to complete clamp assembly. It is also possible that RFC has to release the DNA to form an RFC·PCNA complex first. At present, evidence from studies of E. coli γ complex (26) as well as truncated S. cerevisiae RFC (44) suggests that these clamp loaders bind their clamps prior to binding primed DNA in the clamp loading pathway. For example, presteady-state analysis of the γ complex mechanism reveals a lag in β assembly if γ complex is preincubated with primed DNA before the addition of β. In the proposed model, the lag is ascribed to dissociation of DNA that must occur before γ complex can start clamp assembly correctly by binding β first (26). In the case of S. cerevisiae RFC, analysis of the interaction between RFC and PCNA by surface plasmon resonance indicates that PCNA does not bind an RFC·primed DNA complex with high affinity. Thus, it has been proposed that RFC binds PCNA before it binds primed DNA in the clamp assembly pathway (44). The ATP-bound RFC·PCNA complex has low affinity for both single- and double-stranded DNA (Fig. 8), indicating that RFC can select for primed DNA even when bound to PCNA. However, it appears unlikely that in vivo RFC exists predominantly in complex with PCNA, since several other proteins including replication proteins (Pol δ/ε, Fen1, and DNA ligase I) and repair proteins (XPG endonuclease, Msh2–6, and Msh2–3 complexes) among many others are known to bind PCNA (reviewed in Ref. 55). Thus, the inherent ability of RFC to rapidly scan single- and double-stranded DNA and form a stable complex with primer-template DNA allows the free clamp loader to efficiently recognize primed DNA sites for clamp assembly.
Acknowledgments
We appreciate the gift of trRFC and PCNA over-expression clones as well as SSB protein from Dr. Mike O’Donnell and bacteriophage T7 DNA polymerase from Dr. Smita Patel.
Footnotes
The abbreviations used are: PCNA, proliferating cell nuclear antigen; RFC, replication factor C; Pol, polymerase; ssDNA, single-stranded DNA; dsDNA, double-stranded DNA; ATPγS, adenosine 5′-O-(thiotri-phosphate); DTT, dithiothreitol; SSB, single-stranded binding protein; nt, nucleotide.
This work was supported by National Institutes of Health Grant GM64514-01.
References
- 1.Waga S, Stillman B. Annu Rev Biochem. 1998;67:721–751. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- 2.Kelman Z, O’Donnell M. Annu Rev Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- 3.Jeruzalmi D, O’Donnell M, Kuriyan J. Curr Opin Struct Biol. 2002;12:217–224. doi: 10.1016/s0959-440x(02)00313-5. [DOI] [PubMed] [Google Scholar]
- 4.Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 5.Stukenberg PT, Studwell-Vaughan PS, O’Donnell M. J Biol Chem. 1991;266:11328–11334. [PubMed] [Google Scholar]
- 6.Bauer GA, Burgers PM. Proc Natl Acad Sci U S A. 1988;85:7506–7510. doi: 10.1073/pnas.85.20.7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgers PM. J Biol Chem. 1991;266:22698–22706. [PubMed] [Google Scholar]
- 8.O’Donnell ME. J Biol Chem. 1987;262:16558–16565. [PubMed] [Google Scholar]
- 9.Li X, Burgers PM. J Biol Chem. 1994;269:21880–21884. [PubMed] [Google Scholar]
- 10.Li X, Burgers PM. Proc Natl Acad Sci U S A. 1994;91:868–872. doi: 10.1073/pnas.91.3.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noskov V, Maki S, Kawasaki Y, Leem SH, Ono B, Araki H, Pavlov Y, Sugino A. Nucleic Acids Res. 1994;22:1527–1535. doi: 10.1093/nar/22.9.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howell EA, McAlear MA, Rose D, Holm C. Mol Cell Biol. 1994;14:255–267. doi: 10.1128/mcb.14.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gary SL, Burgers MJ. Nucleic Acids Res. 1995;23:4986–4991. doi: 10.1093/nar/23.24.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cullmann G, Fien K, Kobayashi R, Stillman B. Mol Cell Biol. 1995;15:4661–4671. doi: 10.1128/mcb.15.9.4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mossi R, Hubscher U. Eur J Biochem. 1998;254:209–216. [PubMed] [Google Scholar]
- 16.Neuwald AF, Aravind L, Spouge JL, Koonin EV. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 17.Bunz F, Kobayashi R, Stillman B. Proc Natl Acad Sci U S A. 1993;90:11014–11018. doi: 10.1073/pnas.90.23.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeruzalmi D, O’Donnell M, Kuriyan J. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 19.Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O’Donnell M, Kuriyan J. Cell. 2001;106:417–428. [PubMed] [Google Scholar]
- 20.Oyama T, Ishino Y, Cann IK, Ishino S, Morikawa K. Mol Cell. 2001;8:455–463. doi: 10.1016/s1097-2765(01)00328-8. [DOI] [PubMed] [Google Scholar]
- 21.O’Donnell M, Jeruzalmi D, Kuriyan J. Curr Biol. 2001;11:R935–946. doi: 10.1016/s0960-9822(01)00559-0. [DOI] [PubMed] [Google Scholar]
- 22.Shiomi Y, Usukura J, Masamura Y, Takeyasu K, Nakayama Y, Obuse C, Yoshikawa H, Tsurimoto T. Proc Natl Acad Sci U S A. 2000;97:14127–14132. doi: 10.1073/pnas.97.26.14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayanagi K, Miyata T, Oyama T, Ishino Y, Morikawa K. J Struct Biol. 2001;134:35–45. doi: 10.1006/jsbi.2001.4357. [DOI] [PubMed] [Google Scholar]
- 24.Turner J, Hingorani MM, Kelman Z, O’Donnell M. EMBO J. 1999;18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hingorani MM, O’Donnell M. J Biol Chem. 1998;273:24550–24563. doi: 10.1074/jbc.273.38.24550. [DOI] [PubMed] [Google Scholar]
- 26.Ason B, Bertram JG, Hingorani MM, Beechem JM, O’Donnell M, Goodman MF, Bloom LB. J Biol Chem. 2000;275:3006–3015. doi: 10.1074/jbc.275.4.3006. [DOI] [PubMed] [Google Scholar]
- 27.Sexton DJ, Kaboord BF, Berdis AJ, Carver TE, Benkovic SJ. Biochemistry. 1998;37:7749–7756. doi: 10.1021/bi980088h. [DOI] [PubMed] [Google Scholar]
- 28.Pietroni P, Young MC, Latham GJ, von Hippel PH. J Mol Biol. 2001;309:869–891. doi: 10.1006/jmbi.2001.4687. [DOI] [PubMed] [Google Scholar]
- 29.Podust LM, Podust VN, Sogo JM, Hubscher U. Mol Cell Biol. 1995;15:3072–3081. doi: 10.1128/mcb.15.6.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burbelo PD, Utani A, Pan ZQ, Yamada Y. Proc Natl Acad Sci U S A. 1993;90:11543–11547. doi: 10.1073/pnas.90.24.11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fotedar R, Mossi R, Fitzgerald P, Rousselle T, Maga G, Brickner H, Messier H, Kasibhatla S, Hubscher U, Fotedar A. EMBO J. 1996;15:4423–4433. [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Y, Zeft AS, Riegel AT. Biochem Biophys Res Commun. 1993;193:779–786. doi: 10.1006/bbrc.1993.1693. [DOI] [PubMed] [Google Scholar]
- 33.Tsurimoto T, Stillman B. J Biol Chem. 1991;266:1950–1960. [PubMed] [Google Scholar]
- 34.Cai J, Uhlmann F, Gibbs E, Flores-Rozas H, Lee CG, Phillips B, Finkelstein J, Yao N, O’Donnell M, Hurwitz J. Proc Natl Acad Sci U S A. 1996;93:12896–12901. doi: 10.1073/pnas.93.23.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen BL, Uhlmann F, Gaur LK, Mulder BA, Posey KL, Jones LB, Hardin SH. Nucleic Acids Res. 1998;26:3877–3882. doi: 10.1093/nar/26.17.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keller RC, Mossi R, Maga G, Wellinger RE, Hubscher U, Sogo JM. Nucleic Acids Res. 1999;27:3433–3437. doi: 10.1093/nar/27.17.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlmann F, Cai J, Gibbs E, O’Donnell M, Hurwitz J. J Biol Chem. 1997;272:10058–10064. doi: 10.1074/jbc.272.15.10058. [DOI] [PubMed] [Google Scholar]
- 38.Gomes XV, Burgers PM. J Biol Chem. 2001;276:34768–34775. doi: 10.1074/jbc.M011631200. [DOI] [PubMed] [Google Scholar]
- 39.Tsurimoto T, Stillman B. Proc Natl Acad Sci U S A. 1990;87:1023–1027. doi: 10.1073/pnas.87.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Studwell PS, O’Donnell M. J Biol Chem. 1990;265:1171–1178. [PubMed] [Google Scholar]
- 41.Turner J, O’Donnell M. Methods Enzymol. 1995;262:442–449. doi: 10.1016/0076-6879(95)62035-4. [DOI] [PubMed] [Google Scholar]
- 42.Burgers PM. Methods. 1999;18:349–355. doi: 10.1006/meth.1999.0796. [DOI] [PubMed] [Google Scholar]
- 43.Gomes XV, Gary SL, Burgers PM. J Biol Chem. 2000;275:14541–14549. doi: 10.1074/jbc.275.19.14541. [DOI] [PubMed] [Google Scholar]
- 44.Gomes XV, Schmidt SL, Burgers PM. J Biol Chem. 2001;276:34776–34783. doi: 10.1074/jbc.M011743200. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt SL, Gomes XV, Burgers PM. J Biol Chem. 2001;276:34784–34791. doi: 10.1074/jbc.M011633200. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt SL, Pautz AL, Burgers PM. J Biol Chem. 2001;276:34792–34800. doi: 10.1074/jbc.m011671200. [DOI] [PubMed] [Google Scholar]
- 47.Podust VN, Tiwari N, Stephan S, Fanning E. J Biol Chem. 1998;273:31992–31999. doi: 10.1074/jbc.273.48.31992. [DOI] [PubMed] [Google Scholar]
- 48.Patrick SM, Turchi JJ. J Biol Chem. 2001;276:22630–22637. doi: 10.1074/jbc.M010314200. [DOI] [PubMed] [Google Scholar]
- 49.Fukuda K, Morioka H, Imajou S, Ikeda S, Ohtsuka E, Tsurimoto T. J Biol Chem. 1995;270:22527–22534. doi: 10.1074/jbc.270.38.22527. [DOI] [PubMed] [Google Scholar]
- 50.Eissenberg JC, Ayyagari R, Gomes XV, Burgers PM. Mol Cell Biol. 1997;17:6367–6378. doi: 10.1128/mcb.17.11.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mossi R, Jonsson ZO, Allen BL, Hardin SH, Hubscher U. J Biol Chem. 1997;272:1769–1776. doi: 10.1074/jbc.272.3.1769. [DOI] [PubMed] [Google Scholar]
- 52.Yuzhakov A, Kelman Z, Hurwitz J, O’Donnell M. EMBO J. 1999;18:6189–6199. doi: 10.1093/emboj/18.21.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maga G, Stucki M, Spadari S, Hubscher U. J Mol Biol. 2000;295:791–801. doi: 10.1006/jmbi.1999.3394. [DOI] [PubMed] [Google Scholar]
- 54.Kolpashchikov DM, Hughes P, Favre A, Baldacci G, Lavrik OI. J Mol Recognit. 2001;14:239–244. doi: 10.1002/jmr.538. [DOI] [PubMed] [Google Scholar]
- 55.Warbrick E. Bioessays. 2000;22:997–1006. doi: 10.1002/1521-1878(200011)22:11<997::AID-BIES6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]