

Abstract
The Key Events Dose-Response Framework (KEDRF) is an analytical approach that facilitates the use of currently available data to gain insight regarding dose-response relationships. The use of the KEDRF also helps identify critical knowledge gaps that once filled, will reduce reliance on assumptions. The present study considers how the KEDRF might be applied to pathogenic microorganisms, using fetal listeriosis resulting from maternal ingestion of food contaminated with L. monocytogenes as an initial example. Major biological events along the pathway between food ingestion and the endpoint of concern are systematically considered with regard to dose (i.e., number of organisms), pathogen factors (e.g., virulence), and protective host mechanisms (e.g., immune response or other homeostatic mechanisms). It is concluded that the KEDRF provides a useful structure for systematically evaluating the complex array of host and pathogen factors that influence the dose-response relationship. In particular, the KEDRF supports efforts to specify and quantify the sources of variability, a prerequisite to strengthening the scientific basis for food safety decision making.
Keywords: dose-response, uncertainty factors, homeostatic mechanisms, thresholds, Listeria monocytogenes
INTRODUCTION
Major challenges in risk analysis for foodborne infectious diseases include determining relationships between low level exposure to pathogenic microorganisms and likely public health outcomes, and translating those relationships into practical controls that protect public health. This paper considers these challenges as part of a broader ILSI Research Foundation (ILSI RF) effort to examine approaches to dose-response assessment for a variety of bioactive agents including allergens, nutrients, toxic chemicals, and pathogenic microorganisms. For each category of agent, the ILSI RF effort applied a mode-of-action based analytical framework, referred to as the Key Events Dose-Response Framework (KEDRF) (Julien et al., 2009). After an overview of current approaches to dose-response assessment for pathogenic microorganisms, this paper discusses how KEDRF might be applied to pathogens, using Listeria monocytogenes and fetal listeriosis as an initial example.
OVERVIEW OF CURRENT APPROACHES AND PRACTICE
“Infectious Dose” or “Minimum Infectious Dose” is a concept traditionally used to describe in quantitative terms the ability of a pathogenic microorganism to cause illness and disease (FAO/WHO, 2003). This concept presumes that all infectious agents have a particular dose level, a threshold dose, below which the organism is not expected to cause disease. This presumption is based on data from various experimental studies, with human subjects and animal models, which have identified such dose levels. These studies are the microbiological equivalent of safety assessments conducted for acutely toxic chemicals. Thus, the “minimum infectious dose” is conceptually similar to the “no observed adverse effect level” (NOAEL) in toxicology. Typically, however, studies used to identify a minimum infectious dose have been limited in sample size and thus constrained in their sensitivity to detect infrequent effects.
After a series of outbreaks of foodborne disease associated with the consumption of low levels of pathogenic bacteria, the minimum infectious dose or “threshold” presumption has been increasingly replaced with a risk-based approach that takes into account mechanistic considerations and, in particular, the probabilistic nature of the infectious disease process (Haas, 1983; FAO/WHO, 2003). This risk-based approach recognizes the three major categories of foodborne pathogens—toxigenic, toxicoinfectious, and invasive—which differ in their general underlying mechanisms of pathogenesis. Table A-1 in the Appendix describes these three categories, which are outlined very briefly here.
For toxigenic pathogens, which release a toxin into food products prior to food ingestion, the dose-response relationship is generally assumed to have a threshold or minimum toxic dose. Thus, dose-response assessments for toxigenic pathogens adopt assumptions similar to those for noncarcinogenic toxicants. On the other hand, for toxicoinfectious and invasive pathogens, the widely accepted underlying assumption is that there is no biological threshold. In these cases, the ingestion of a single bacterium is assumed to have some potential, albeit generally very small, to cause infection1 and illness.
Accordingly, widely accepted mathematical dose-response models for toxicoinfectious and invasive pathogens are based on two fundamental assumptions (FAO/WHO 2003):
Single hit assumption, i.e., the probability that any given microorganism will survive a number of barriers to establish infection is non-zero.
Independent action assumption, i.e., the probability that any given organism will cause infection is independent of the number ingested. Thus, the chance that a given bacterium will cause an infection is not affected by dose, but the chance that infection will occur increases directly with the number of bacteria i.e., increases with dose.
There is substantial indirect evidence to support these two assumptions (Meynell and Stocker, 1957; Moxon and Murphy, 1978; Rubin, 1987). There is limited knowledge, however, regarding the underlying biology and the variability of interaction between pathogens and their hosts; this limits the possibility to generalize from experimental datasets. For example, a recent study on infection of insect larvae with baculoviruses, while confirming these hypotheses for several pathogen-host combinations on the simple assumption of constant interaction, also showed the need to incorporate variability into the predictive models for other pathogen-host pairs (Zwart et al., 2009). An examination of specific sources and the extent of variability across these host-pathogen combinations would be needed in order to refine the predictive model.
Thus, at a minimum, current risk assessment approaches require the following information to estimate the probability of infection and illness for an individual host or for a population:
Mechanistic category of the pathogen
Specific nature of the pathogen (i.e., virulence factors, gene expression, stress response system, etc.)
Dose (i.e., number of organisms ingested) over the defined period of time.
In addition, several other factors may substantially influence the dose-response relationship and thus may need to be considered. These include the susceptibility of the exposed person or population, matrix effects associated with the food in which the pathogen is suspended, and the particular strain of the pathogenic microorganism. There can be substantial strain-to-strain variability within a species or serotype, making it difficult to collect the basic data needed to generate a single dose-response curve for that pathogen (FDA/FSIS, 2003).
Data used to assess dose-response for pathogenic microorganisms generally come from one of three types of studies—outbreak, human volunteer, or experimental animal model studies. Each type of study has strengths and limitations. All three approaches have substantial uncertainty due to the inherent variability in the pathogen, the host, and the food vehicle. In general, it is difficult to collect data on the concentration of pathogen to which a person was exposed and also difficult to collect relevant data on response.
For example, in most outbreak investigations the ingested dose itself must be modeled because the pathogen may multiply or die in the remaining contaminated food between the time of ingestion and the collection of samples following onset of symptoms (FAO/WHO, 2002; FAO/WHO, 2004; Teunis et al., 2004, 2008). Typically, dose information is simply not available, and the variability among doses consumed by individuals is almost never known. An exception may be the limited cases where the food involved neither supports pathogen growth nor causes rapid die-off. In such cases an analysis of food samples and epidemiological investigations can provide reasonably accurate estimates of pathogen levels and the amount of food consumed.2
Another limitation with outbreak studies is the fact that the specific nature/genetic makeup of the organism can change between the time of ingestion and the collection of food samples, making the evaluation of the initial causative organism problematic. Also, except in cases where the infection is associated with a very specific illness (e.g., hemolytic uremic syndrome associated with E. coli O157:H7), an outbreak caused by a very low dose (e.g., where less than 5% of those ingesting the pathogen become ill) is unlikely to be detected due to the statistical difficulty in identifying cases. In the infrequent situation where extensive data exist on likely exposure of a population to a particular foodborne pathogen for a specific ready-to-eat food, annual disease statistics in combination with the single parameter exponential model have been used to generate dose-response curves (Buchanan et al., 1997).
Human volunteer studies have the advantage of a known dose level and known agent. But in this case, it is necessary to extrapolate to account for potential differences between the test population and the exposed population, which for many infectious pathogens include individuals with immune systems weakened by age, immunosuppression, cytotoxic drugs, or pre-existing disease. Volunteer studies cannot be done with individuals from such susceptible populations because of the possibility of serious or fatal infections.
Enhanced investigation of outbreaks is one means of acquiring information that is representative of foodborne disease in the typical population, which includes individuals of varying immune-status. In an enhanced outbreak investigation, the number of people who consumed a given food, the number who became ill, the health status of those who became ill, the number of pathogens consumed (if available), and the properties of the relevant pathogen strain are determined. Typically, information from one outbreak investigation provides a single data point on a population dose-response curve (the probability of illness given a particular dose). An accumulation of outbreak data sets is needed to establish the position of the dose response curve, and a range of doses is needed to determine the shape of the curve. A risk assessment conducted for Salmonella spp. (FAO/WHO, 2002) is a good example of the types of data needed; however, such data sets are rarely available.
When human data are not available, animal models may be used to establish the shape and position of the dose response curve. With animal model studies, the mechanism of infection and resulting illness should be shown to be the same as in humans; otherwise, extrapolation is needed to account for the inter-species differences. Such inter-species differences can be substantial. For example, at the time of a recent FDA-USDA risk assessment for L. monocytogenes, the only studies with sufficient data to obtain a dose-response curve for risk of stillbirth from maternal exposure during gestation were mouse studies (FDA/FSIS, 2003). The mouse fetus is susceptible to L. monocytogenes only under certain conditions (Abram et al., 2003; Sahaghian et al., 2009, Lammerding et al., 1992; Klink and Runkiki, 1995; Ito et al., 2004), and the mouse dose-response curve differs from humans due to differences in the E-cadherin receptor, the enterocyte cell surface receptor required for uptake of the pathogen from the intestine (Lecuit, 1999). More recently, oral route dose-response data for L. monocytogenes have been collected in the pregnant guinea pig and pregnant non-human primate. In these species the E-cadherin receptor is the same as in the human and oral exposure results in stillbirth (Smith et al., 2003; Williams et al., 2007; Smith et al., 2008).
Regardless of the data source—outbreak data, volunteer studies, or animal models—current approaches to dose-response assessment for pathogenic microorganisms generally require a number of extrapolations and assumptions. In addition to the major extrapolations for species differences and the differences in susceptibility among humans discussed above, other assumptions are typically adopted, including i) each episode of illness is acute though the associated exposure event may be cumulative for 1–2 days; ii) each exposure/dose is a separate independent event; and iii) no immunity exists. Also, assumptions regarding the distribution of pathogens in food products are usually made.
Another very critical extrapolation, which in large part prompted the present study, involves extrapolation from high dose to the low doses typically relevant for public health. In volunteer and in animal model studies, the number of subjects is almost always too small to measure probabilities of infection at very low doses. Thus, administered doses are typically much higher than the dose levels of practical concern. And as already noted, it is very difficult to detect outbreaks from ingestion of low-dose levels. Because regulations are generally based on areas of the dose-response curve below actual data points, assumptions and extrapolations must be made in order to generate a full dose-response curve that includes relevant dose levels. For toxicoinfectious and invasive pathogens, the single hit and independent action assumptions underlie the widely accepted mathematical modeling approaches for predicting the low end of the dose-response curve (Buchanan et al., 2000; Haas, 2002; Zwietering and Havelaar, 2006), and these assumptions have a major impact on the shape of the low end of the curve.
The significant obstacles to collecting robust data for relevant and typically low doses will likely remain for some time. Thus, the best strategy for refining the dose-response assessment for foodborne pathogens is to advance the understanding of the underlying biology, and by doing so, refine the assumptions that underlie predictive models. In particular, it is necessary to improve the understanding of the sources of variability in dose-response, and to quantify these various sources. The remainder of this paper will discuss the Key Events Dose-Response Framework, an analytical approach that is intended to shed light on the critical factors that determine response to dose, including variability in such response.
KEY EVENTS DOSE-RESPONSE FRAMEWORK: POTENTIAL FOR USE WITH L. MONOCYTOGENES
Background
As described in the first paper of this series (Julien et al., 2009), the KEDRF involves the examination of the major biological steps that occur between the intake of a bioactive agent and the onset of a specified adverse effect. At each step the range of factors that may influence response are systematically examined, including dose (i.e., number of organisms), pathogen factors (e.g., virulence), and protective host mechanisms (e.g., immune response or other adaptive or homeostatic mechanisms). It should be noted that, for pathogens, as opposed to other types of agents considered in this ILSI RF project, the interactions between the bioactive agent and the host can be especially complex. As living organisms, pathogens have the potential to develop highly dynamic interactions with hosts. For example, pathogens often evolve systems that commandeer host resources, which are then used to help the pathogen survive and grow.
Host characteristics (e.g., age, health status, etc.) are also essential to consider. They may modify the effectiveness of the homeostatic or immune response mechanisms of the host, or otherwise contribute to the variability in response.
The impact of host and pathogen factors has been recognized for some time. The potential value of the KEDRF is in gaining insight regarding how these factors influence dose-response, i.e., how specific factors influence the probability of progression from one step to the next toward the ultimate effect of concern, and how they may contribute to variability in the ultimate dose-response relationship. A deeper understanding of these factors and their relative importance can be expected to improve accuracy and reliability of the predictive models used in public health. It will also help focus research on the types of data needed to reduce reliance on the assumptions routinely used when estimating risk from foodborne pathogenic bacteria. Thus, the KEDRF approach complements, integrates, and builds upon in vivo and in vitro approaches. The KEDRF helps identify research questions that may be answered by those experimental approaches. In turn, the data they generate will refine the understanding of specific events in the KEDRF pathway.
As a means of illustrating the potential of this approach, we consider how it might be applied to study dose-response relations for fetal listeriosis resulting from maternal ingestion of L. monocytogenes. After a brief overview of L. monocytogenes and the pathogenesis of fetal listeriosis, this paper will discuss individual Key Events and issues that could be examined further in a full Key Events Dose-Response Analysis.
L. monocytogenes is typically ingested via ready-to-eat foods such as soft cheeses and deli meats (FDA/FSIS, 2003; FAO/WHO, 2004). The pathogen is invasive; it crosses the intestinal epithelium and is transported to distal sites in the host (e.g., spleen, liver, bone marrow, brain) (Swaminathan et al., 2007). Illness is relatively rare, but is associated with a high fatality rate. A 2003 FDA/FSIS risk assessment for L. monocytogenes estimated that each U.S. consumer has an 80% probability each year of consuming a serving of food containing more than 1 million L. monocytogenes; however, only an estimated 2500 illnesses occur (Mead et al., 1999). These illnesses lead to about 500 deaths, resulting in a case fatality rate of about 20% (Mead et al., 1999). Deaths can occur from septicemia or meningitis in the elderly and immunocompromised individuals such as transplant patients, chemotherapy patients, or AIDS patients (FDA/FSIS, 2003; FAO/WHO, 2004). A significant public health concern is the ingestion of contaminated food by pregnant women. The listeriosis rate among pregnant women is similar to other adults, but the fetus is 20 times more likely to develop listeriosis compared to healthy individuals between the ages of 30 days and 60 years (FDA/FSIS, 2003; FAO/WHO, 2004). Fetal listeriosis can potentially lead to spontaneous abortion, stillbirths, or neonatal meningitis.
Overview of Pathogenesis for Fetal Listeriosis
The pathogenesis of L. monocytogenes for fetal listeriosis has been reviewed (Goldfine and Shen, 2007; Doyle and Beuchat, 2007; Ryser and Marth, 2007) and is only briefly summarized here. Following the intake of a food product containing L. monocytogenes, the pathogen is exposed to the acidic environment of the upper GI tract. Bacteria that survive that environment enter the intestines where they may bind to enterocytes, the epithelial cells that line the intestinal mucosa. The binding and uptake into enterocytes is facilitated by interaction of a protein (internalin A) on the bacterial surface with a host receptor (E-cadherin) on the surface of the enterocytes. Once taken into enterocytes by endocytosis, the bacteria escape from the phagosomes and proliferate intracellularly (Gaillard et al., 1987; Conte et al., 2000). The pathogen can then be transferred to phagocytes, again escaping and proliferating (Conte et al., 2000). Because of its ability to survive and multiply within circulating phagocytic cells, L. monocytogenes can be disseminated throughout the body via blood and lymphatic circulation into various tissues including liver, spleen, gall bladder, CNS and, during pregnancy, the placenta and the fetus. Ultimately, the pathogen is transferred from phagocytes to endothelial cells at the ultimate site of the infection (i.e., the fetus) (Drevets et al., 1995).
An Initial Key Events Framework for Fetal Listeriosis
When applying the analytical approach of KEDRF, one of the first tasks is to describe the relevant pathway of Key Events. The term “Key Event” has been defined in the field of chemical risk assessment as an empirically observable precursor step that is itself a necessary element of a chemical's mode-of-action, or a biologically based marker for such an element (Sonich-Mullin et al., 2001). Thus, for purposes of examining the KEDRF as a potentially useful analytical approach, we identified five major and necessary steps as Key Events that lead to fetal listeriosis.
Future work on this subject will likely refine this initial description, and may describe additional, more specific steps within the five discussed here. But, for purposes of this initial study, the following five steps, i.e., the Key Events, will be discussed with regard to factors that may influence the probability of progression from one step to the next, and also factors that may contribute to the variability in the ultimate dose-response relationship:
Survival of the pathogen in the upper GI tract
Establishment in the intestine; attachment to and uptake into epithelial cells
Survival and escape from phagosomes in enterocytes, and transfer of pathogen to phagocytes
Transfer of pathogen across placenta
Pathogen growth leading to fetal morbidity and mortality
It should be noted that the specific steps associated with fetal listeriosis may differ from those leading to listeriosis in other susceptible populations (e.g., elderly, patients with chronic disease, transplant patients). Also, the steps discussed in this L. monocytogenes example are likely to differ from steps for other mechanistic categories (toxicoinfectious, toxigenic) and even for other pathogens in the invasive category.
Figure 1 outlines the five Key Events that will be discussed. Note that while each event between the initial intake and effect of concern is part of a conditional chain of events, certain events may also be considered “control points” in that they engage host mechanisms that influence whether the cascade of events progresses. That is, these control points engage mechanisms, such as immune or homeostatic mechanisms, that serve to protect the normal environment. If these mechanisms fail (i.e., if control is lost), the pathogen has an opportunity to continue along the Key Events pathway toward the effect of concern. Thus, at each event, there are several factors—dose level, pathogen characteristics, and host mechanisms—that in combination determine the probability (P) that progression occurs.
Figure 1.
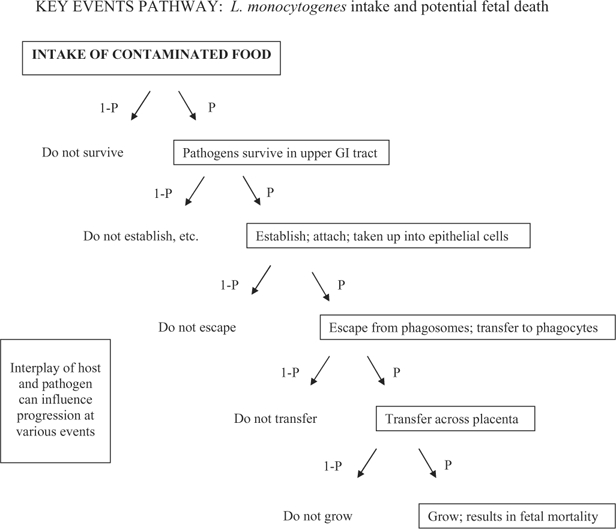
Key biological events occurring between intake of pathogen and the specific effect of concern (fetal listeriosis). At each event, both host and pathogen factors can be examined with regard to: i) how they may influence probability of progression toward the effect of concern (i.e., how they affect the number of organisms exiting a given event, given the number of organisms entering the event), and ii) how they influence inter- and intra- host response variability.
Note that Fig. 1 could be used to represent events within a given individual, or events within a given population. If Fig. 1 represents an individual, the outcome of each event can be characterized by a binary yes or no (i.e., the outcome either progresses toward the effect of concern, or it does not). If Fig. 1 refers to a population the probabilities of progression past each event will reflect variability across the population.
Observations Regarding Key Events for Fetal Listeriosis
Key Event 1: Survival of the Pathogen in the Upper GI Tract
Several factors affect how many, if any, L. monocytogenes survive the acidic environment of the upper GI tract, including the food matrix, the quantity and composition/acidity of foods consumed, and the general level of acidity in the GI tract. Certain host characteristics or behaviors (e.g., antacids consumption, achlorhydria) may reduce acid production and thus increase the probability of pathogen survival. Pathogen adaptability can also affect the outcome of this event. L. monocytogenes is known to have mechanisms that increase acid resistance, and these mechanisms are induced by prior exposure to moderately acid conditions (Kroll and Patchett, 1992; Buchanan et al., 1994; Wiedmann et al., 1998; Formato et al., 2007; Adriao et al., 2008; Ryan et al., 2009). These adaptive mechanisms have also been associated with increased expression of virulence determinants (O'Driscoll et al., 1996; Cotter et al., 1999; Gahan et al., 1999; Hill et al., 2000). Despite the various host and pathogen factors that are available to influence L. monocytogenes survival, the numbers that survive the GI tract are generally assumed to be proportional to the numbers ingested, and the dose response curves in animal models support this assumption (Williams et al., 2007; Smith et al., 2008).
Key Event 2: Establishment in the Intestine, Attachment to and Uptake into Epithelial Cells
Little is known regarding specific pathogen factors that influence the extent of the L. monocytogenes’ growth in the intestinal lumen or after it has attached to the intestinal mucosa. But the growth of the microorganism in the lumen would be expected to increase the probability that the bacteria find and bind to receptor sites on the intestinal epithelium. Similarly, growth on the surface of the enterocyte would be expected to increase the number of L. monocytogenes cells internalized. Host factors that affect the local GI response are not well characterized in quantitative terms, but the host innate immune response (i.e., proinflammatory cytokines macrophages, monocytes, polymorphonuclear leukocytes, and natural killer cells) will reduce the numbers of microbes that colonize the intestine.
L. monocytogenes are taken into enterocytes via receptor mediated endocytosis. Host E-cadherin receptors bind with internalin (InlA), a bacterial surface protein that is controlled by the prfA regulatory gene. Differences in InlA are believed to be one of the factors that differ in the three genetic lineages recognized for L. monocytogenes (Jia et al., 2007). Factors potentially affecting the probability of uptake via endocytosis into enterocytes include strain differences, growth stage, prior growth conditions, induction of bacterial stress response, and various host differences such as age or immune status (Dramsi et al., 1993; Domann et al., 1997; Lecuit et al., 1997, 2001; McGann et al., 2007a, 2007b; Linden et al., 2008; Milillo et al., 2009).
Key Event 3: Survival and Escape from Phagosomes and Transfer to Phagocytes
Once taken up into the enterocyte by endocytosis, pathogens would generally be subject to degradation in phagosome vacuoles. However, most L. monocytogenes strains have the ability to survive, and ultimately escape, from phagosomes due to their ability to synthesize listeriolysin O (LLO) (Gaillard et al., 1987; Del Corral et al., 1990; Lam and Brumell, 2008). In response to acidification of the phagosome, LLO undergoes structural reconfiguration that renders the protein stable and active. It then forms small pores in the phagosome, blocking the acidification of this compartment and thereby preventing its fusion with lysosomes (Singh et al., 2008). This provides a “window of opportunity” for L. monocytogenes to escape the phagosome and initiate growth in the cytoplasm, and involves the combined actions of LLO and two phospholipase enzymes (Mengaud et al., 1991; Camilli et al., 1991; Notermans et al., 1991; Marquis et al., 1995, 2000; Smith et al., 1995; Goldfine et al., 1998; Das et al., 2001; Wei et al., 2005). LLO activity is promoted by γ -interferon inducible lysosomal thiol reductase (GILT) in the host, and is further augmented by the induction of an acid tolerance system within L. monocytogenes (Conte et al., 2000). GILT appears to be a critical host factor, and mice that lack GILT are resistant to L. monocytogenes infection (Singh et al., 2008). Once released into the cytoplasm, the actively growing pathogen can spread from cell to cell via ActA polymerization of the host cell actin, which supports movement within the cytoplasm to the membrane-membrane interface with adjacent cells. The bacteria are then spread to other enterocytes and ultimately to phagocytes, which disseminate the pathogen through the maternal circulatory and lymph system to various organs including the placenta.
Two attributes that are central to the ability of L. monocytogenes to cause disease are its ability to synthesize LLO and thus escape from the phagosomes, and its ability to transfer among enterocytes, phagocytes, and endothelial cells while remaining in an intracellular environment. This movement of L. monocytogenes in cells via phagocytosis/endocytosis evades the humoral immune system (Tilney and Portnoy, 1989; Del Corral et al., 1990; Dabiri et al., 1990; Mounier, et al., 1990; Kocks et al., 1992; Sanger et al., 1992; Tilney et al., 1992; Pistor et al., 1994; Grenklo et al., 2003). Listeriolysin synthesis is a distinguishing characteristic of L. monocytogenes. If a strain does not have a functional listeriolysin gene or cannot synthesize LLO, it will not escape the phagosome. The extent of LLO production varies among L. monocytogenes strains (Parrisius et al., 1986; Datta and Kothary, 1993) and may be a determinant in their relative virulence.
Key Event 4: Transfer of Pathogen across Placenta
The specific mechanism by which L. monocytogenes crosses the placenta is under study, and two different mechanisms have been hypothesized—the invasion of the endothelial cells via InlA-E-cadherin interaction (Lecuit et al., 1999, 2004), and actin-mediated cell-to-cell transfer from infected phagocytes to placental endothelial cells (Drevets et al., 1995). Cells with E-cadherin receptors (e.g., liver cells, intracerebral microvascular endothelial cells, chorionic villi of the placenta) are the sites associated with invasive listeriosis (Lecuit et al., 1999). This suggests a direct interaction between bloodborne L. monocytogenes cells and the placental endothelial cells (Lecuit et al., 2004). Other investigations suggest that transfer across the placenta involves stimulation of polymorphonuclear leukocytes adhesion and cell-to-cell transfer (Drevets, 1998). This hypothesis is reinforced by evidence of a decreased ability of the pathogen to invade placental tissue in ActA− mutants (Le Monnier et al., 2007), and also by the lack of a maternal humoral immune response prior to stillbirth in studies using rhesus monkeys (Smith et al., 2003). In these studies, L. monocytogenes could not be isolated from the serum, nor was there any increase in antisera titers at 4 days or 14 days after oral exposure to L. monocytogenes. There was a significant increase in antisera titers at the time of stillbirth, however, which occurred concurrently with tissue breakdown in the placenta and fetus.
The difference in these proposed mechanisms has implications with regard to the host's ability to prevent progression toward fetal listeriosis. A mechanism that utilizes actin-mediated cell-to-cell transmission would protect the pathogen from the humoral immune response, whereas a receptor-mediated bacterial-endothelial interaction would offer the possibility of humoral immune protection if the maternal immune system can respond rapidly enough.
Key Event 5: Pathogen Growth Leading to Fetal Morbidity and Mortality
If L. monocytogenes successfully transfer across the placenta, they gain entry to fetal circulation and may spread cell-to-cell ultimately colonizing the fetal liver and brain (Bakardjiev et al., 2005; Williams et al., 2007; Jensen et al., 2008). Fetuses lack a fully competent immune system, and thus are at great risk of infection if the pathogen crosses the placenta. While fetal colonization results in an asymptomatic maternal infection, it may also result in spontaneous abortion, delivery of a stillborn infant, or delivery of an infant infected with L. monocytogenes. The outcome may be affected by the timing of the infection (early or late in the gestational period). The details of maternal and placental immunity (Irvin et al., 2008a; Irvin et al., 2008b) are not fully known but specialized immune functions during pregnancy and in the placenta may be associated with Th1 cytokines (Barber et al., 2005) and changes in the Th1/Th2 cytokine ratios (Lin et al., 2003). It is not known whether death of the fetus occurs by reaction of the maternal system to fetal infection, loss of placental integrity, infection of the fetus directly, or some combination of these processes.
DISCUSSION
This brief overview of Key Events in the L. monocytogenes pathway illustrates how a systematic analytical approach would help to “tease out” the complex factors that determine response to a given initial dose. A more in–depth analysis of current knowledge at individual events should be conducted, and would be a basis for generating specific hypotheses and for identifying research priorities. Even this initial examination generated some new insights.
For example, this analysis highlighted some potential differences across the various Key Events. Several events do not appear to engage to any significant extent, specific host mechanisms in response to the pathogen (e.g., survival through the upper intestinal tract, attachment to the intestinal epithelium). Thus, the outcomes of these Key Events appear to be fundamentally probabilistic in nature. As such, the dose-response relationship for the individual event would most appropriately be described as nonthresholded or log-linear. Other Key Events in the pathway, however, engage more specific host mechanisms that work to preserve homeostasis and prevent progression toward fetal listeriosis. But these host mechanisms may have a finite capacity that can be overcome with “high enough” numbers of pathogens. At such a point, an increase in pathogen dose would not simply increase the probability of progression; rather, more complex (predator-prey) interactions would need to be considered. If host mechanisms have been exhausted, an increase in pathogen dose might ensure progression along the pathway. This possibility that certain Key Events might be best characterized by a linear dose-response curve and others by a nonlinear curve could be examined in a comprehensive analysis of Key Events for L. monocytogenes.
The above discussion of Key Events also noted some places where specific host attributes might explain some of the overall variability in dose-response. Clearly, certain host attributes, in particular deficiencies in the immune response, underlie and define the susceptible population for many pathogens. But even within susceptible populations, the attack rate for some pathogens, such as L. monocytogenes, is low (FDA/FSIS, 2003). Thus, other host factors (i.e., genetic differences, exposure patterns, etc.) likely contribute. Again, a full analysis of individual Key Events would likely generate hypotheses regarding the contribution of specific host factors to overall variability. Once identified and quantified, these factors could be used to more precisely characterize susceptible individuals within a susceptible population.
Similarly, pathogen variability at particular Key Events may be equally important in determining whether a given infection results in illness. Various genetic lineages and polymorphisms associated with determinants of virulence result in substantial strain-to-strain variability, affecting how quickly the pathogen can grow and overcome the barriers and response of the host. The stress responses of a pathogen (e.g., pH tolerance, resistance to oxidative stress) may also affect the dose-response curve, shifting the curve left along the x-axis. Also, quorum sensing processes may become engaged and regulate genes that influence the virulence of a pathogen (e.g., toxins, proteases, hemolysins, adhesins), and stress responses, thereby affecting the outcome of later key events (Anand and Griffiths, 2003; Joelsson et al., 2007). These multiple sources of variability are already known, but the Key Events approach helps to pinpoint the specific steps in the overall pathway where these pathogen factors may exert their influence and where a more comprehensive examination would be especially fruitful.
An example of a pathogen attribute that strongly affects the ultimate outcome in the fetal listeriosis pathway is the ability of L. monocytogenes to escape from the phagosome. This ability to escape requires the production of sufficient LLO and phospholipases before the pathogen becomes inactivated. Thus, high numbers of L. monocytogenes in phagosomes, or the presence of strains that produce elevated levels of LLO, would seem more likely to lead to fetal listeriosis. As noted previously, this attribute varies across strains and may contribute to differences in virulence. Likewise, the ability to spread among enterocytes, phagocytes, and endothelial cells (via actin mediated transfer) while remaining in an intracellular environment is an essential attribute if infection is to lead to fetal listeriosis. It may be useful to examine whether variability in this particular attribute contributes to overall variability in virulence.
CONCLUSION
Current approaches to dose-response assessment for low-dose exposures to microorganisms in foods rely on multiple assumptions and extrapolations to cope with limitations in knowledge and also limitations in our ability to effectively measure variability (e.g., variability among humans, strain differences among pathogens). The KEDRF offers an analytical framework for better utilizing current and future data to gain insight regarding dose-response based on understanding the critical biological events that occur between exposure and disease. The present study provides an initial example of the potential of this approach for evaluating pathogenic microorganisms.
The initial Key Events analysis developed for fetal listeriosis provided a structure for systematically considering the complex array of host and pathogen factors that influence dose-response. These factors were considered at the level of individual Key Events, where they interact; but they could also be further considered in the context of the overall pathway of events. The present exercise also highlighted some key research needs. Future work should refine the description of the pathway and conduct a more comprehensive analysis of currently available data. It will be especially useful to examine information on host homeostatic mechanisms, and the conditions under which they may reach their capacity. In summary, the use of this approach can be expected to generate new hypotheses and focused research, which will provide new data and subsequently refine the understanding of the overall pathway. Thus, the KEDRF approach provides a basis for continuous iterative improvement in microbial dose-response assessment.
Note that the KEDRF, in combination with data on interactions at specific events, should be particularly helpful for informing the development of predictive models. The framework provides a connection between dose-response relations as measured at individual events and the dose-response relationship that is ultimately of concern. Data and knowledge for specific Key Events will provide more direct evidence to inform and refine assumptions that underlie dose-response models. Also, the framework helps connect data on variability at individual Key Events to the variability observed at the population level. These connections will lead to more informed dose-response characterizations for foodborne pathogenic bacteria and ultimately strengthen the scientific basis of food safety regulation. A clear understanding of the underlying biology is critical to making scientifically-based, risk-based decisions about the level of control needed in order to meet public health goals (CAC, 2007).
The type of analysis described above for L. monocytogenes could be applied to other foodborne pathogens as well, each of which has key events associated with the expression of specific virulence factors. For example, a substantial portion of Escherichia coli strains have the ability to produce shiga toxin; however, only a small percentage of the strains are associated with hemorrhagic colitis and hemolytic uremic syndrome, for example, E. coli O157:H7. The likely key events associated with enterohemorrhagic strains are survival through the upper gastrointestinal tract, attachment to the intestinal epithelium, the colonization of the attachment site, the production of shigatoxin, and the dissemination of the toxin throughout the body. In this instance, eae gene-mediated attachment to enterocytes may be the critical attribute that could be used to effectively differentiate enterohemorrhagic E. coli from other shiga toxin-producing strains. In turn, such knowledge could be used to establish appropriate biologically based regulatory criteria for the various classes of potentially pathogenic E. coli strains.
ACKNOWLEDGMENTS
This paper is one of the work products of an international group of experts convened by the International Life Sciences Institute (ILSI) Research Foundation. ILSI is a nonprofit, worldwide organization established in 1978 to advance the understanding of scientific issues relating to nutrition, food safety, toxicology, risk assessment, and the environment. The ILSI Research Foundation was established in 1984 to create a vehicle for ILSI to support research. Its risk assessment program sponsors, organizes, and participates in a wide range of activities to develop and disseminate new scientific knowledge, encourage exchange of ideas, and build consensus among scientists from academia, industry, government, and public interest groups in its efforts to improve the scientific basis of risk assessment. Financial support for this project from the following sources is gratefully acknowledged: ILSI Research Foundation, Health Canada, Ajinomoto, Coca-Cola Company, Groupe Danone, Kellogg Company, Kraft Foods, Inc., Mars, Inc., Mead Johnson Nutritionals, Monsanto Company, Nestlé, PepsiCo, Inc., The Procter & Gamble Company, Syngenta Ltd., Flavor Extract Manufacturers Association, and Grocery Manufacturers Association. Assistance from Ms. Julie Fitzpatrick with coordination of the final preparation of these papers is also gratefully acknowledged.
Declaration of Interests: Dr. Buchanan has served as a consultant to the Dairy Marketing Institute and Science Applications International Corporation (SAIC) and serves on the scientific advisory committees of ConAgra, Inc. and Mars Snackfood USA. All other authors declare that they have no relevant interests to disclose.
APPENDIX
Table A-1.
Overview of Mechanistic Catagories for Pathogenic Bacteria
| Toxigenic bacteria: Produce preformed toxins in foods |
| Staphylococcus aureus produces a highly heat stable enterotoxin. This bacterium is traditionally associated with products that have been temperature abused sufficiently to allow S. aureus to attain high levels and produce its enterotoxins. Even if food is subsequently cooked, the extreme heat stability of the enterotoxin would result in illness. |
| Clostridium botulinum neurotoxin (BoTN) is produced during the growth of C. botulinum –an obligate anaerobe and spore former –in canned foods, various fermented, dried or salted meat and fish that are not properly produced and/or refrigerated, and various other foods where the oxygen content is limited. The toxin appears to be associated with the outer bacterial cell coat and is sloughed off into the food product as the culture grows. |
| Toxicoinfectious bacteria: Colonize surface of the GI tract |
| One subgroup (e.g., Vibrio cholerae and enterotoxigenic Escherichia coli (ETEC)) delivers toxin directly into epithelial cells to which the bacterium has attached. Once inside the epithelial cell, the enterotoxins typically affect cyclic AMP- or cyclic GMP-mediated net Cl1 secretion, often resulting in severe diarrhea. |
| A second subgroup (e.g., enterohemorrhagic E. coli [EHEC]), colonize the surface of endothelial cells and elicit toxin (Shiga toxin (Stx)), which locally disrupts the integrity of the intestinal tract, most likely as a means of acquiring limiting nutrients such as iron. Focal necrosis of epithelial cells leads to the primary adverse effect - hemorrhagic colitis (HC). Stx is also taken up by the host and transported to distant internal tissue where it can lead to primary sequelae -hemolytic uremic syndrome (HUS). |
| A third subgroup (e.g., Clostridium perfringens) produces an enterotoxin associated with sporulation. Microorganism typically sporulates poorly in foods so high levels of enterotoxin are not observed. When microorganisms are ingested, cells sporulate in the intestinal tract, undergo autolysis, and enterotoxin is released into the lumen. |
| Invasive bacteria: Invade across intestinal epithelium and disseminate within the host |
| One subgroup (e.g., Salmonella enterica, Shigella spp. and enteroinvasive E. coli) invades the epithelium, spreads intercellularly, and causes focal microulcerations of the mucosa. |
| A second subgroup (e.g., Yersinia enterocolitica and Yersinia pseudotuberculosis) crosses the epithelium and proceeds to regional lymph nodes, causing severe abdominal pain and, occasionally, mesenteric lymphadenitis. Campylobacter jejuni may also translocate across the mucosa and survive submucosally, but does not typically reach the bloodstream. |
| A third subgroup (e.g., Salmonella Typhi and Paratyphi, Listeria monocytogenes) translocates across the epithelium, and may then be transported to distal sites in the host (e.g., spleen, liver, and bone marrow, brain). |
Footnotes
1Infection is usually defined as a condition in which a pathogen can actively multiply in the body of the host. Infection may or may not be accompanied by clinical signs of illness in the host (i.e., there may be symptomatic or asymptomatic infection).
2For example, the 1994 outbreak of Escherichia coli O157:H7 associated with dry, fermented pork and beef salami (a product in which this pathogen can survive, but not grow, for extended storage periods) provided clear evidence of the ability of low levels of this organism to cause a significant rate of disease (Getty et al., 2007).
REFERENCES
- Abram M., Schluter D., Vuckovic D., Wraber B., Doric M., Deckert M. Murine model of pregnancy-associated Listeria monocytogenes infection. FEMS Immunol. Med. Microbiol. 2003;35:177–182. doi: 10.1016/S0928-8244(02)00449-2. [DOI] [PubMed] [Google Scholar]
- Adriao A., Vieira M., Fernandes I., Barbosa M., Sol M., Tenreiro R. P. Marked intra-strain variation in response of Listeria monocytogenes dairy isolates to acid and salt stress and the effect of acid or salt adaptation on adherence to abiotic surfaces. Int. J. Food Microbiol. 2008;123:142–150. doi: 10.1016/j.ijfoodmicro.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Anand S. K., Griffiths M. W. Quorum sensing and expression of virulence in Escherichia coli O157:H7. Int. J. Food Microbiol. 2003;85:1–9. doi: 10.1016/s0168-1605(02)00482-8. [DOI] [PubMed] [Google Scholar]
- Bakardjiev A.I., Stacy B. A., Portnoy D. A. Growth of Listeria monocytogenes in the guinea pig placenta and role of cell-to-cell spread in fetal infection. J. Infect. Dis. 2005;191:1889–1897. doi: 10.1086/430090. [DOI] [PubMed] [Google Scholar]
- Barber E. M., Fazzari M., Pollard J. W. Th1 cytokines are essential for placental immunity for Listeria monocytogenes. Infect. Immun. 2005;73:6322–6331. doi: 10.1128/IAI.73.10.6322-6331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan R. L., Golden M. H., Whiting R. C., Phillips J. G., Smith J. L. Model for the non-thermal inactivation of Listeria monocytogenes. J. Food Sci. 1994;59:179–188. [Google Scholar]
- Buchanan R. L., Damert W. G., Whiting R. C., van Schothorst M. An approach for using epidemiologic and microbial food survey data to develop a “purposefully conservative” estimate of the dose-response relationship between Listeria monocytogenes levels and the incidence of foodborne listeriosis. J. Food Prot. 1997;60:918–922. doi: 10.4315/0362-028X-60.8.918. [DOI] [PubMed] [Google Scholar]
- Buchanan R. L., Smith J. L., Long W. Microbial risk assessment: Dose-response relations and risk characterization. Int. J. Food Microbiol. 2000;58:159–172. doi: 10.1016/s0168-1605(00)00270-1. [DOI] [PubMed] [Google Scholar]
- CAC (Codex Alimentarius Commission) Principles and Guidelines for the Conduct of Microbial Risk Management (MRM) 2007. CAC/GL 63-2007.
- Camilli A., Goldfine H., Portnoy D. A. Listeria monocytogenes mutants lacking phosphatidylinositol-specific phospholipase C are avirulent. J. Exp. Med. 1991;173:751–754. doi: 10.1084/jem.173.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte M. P., Petrone G., Di Biase A. M., Ammendolia M. G., Superti F., Seganti L. Acid tolerance in Listeria monocytogenes influences invasiveness of enterocyte-like cells and macrophage-like cells. Microbial Pathog. 2000;29:137–144. doi: 10.1006/mpat.2000.0379. [DOI] [PubMed] [Google Scholar]
- Cotter P. D., Emerson N., Gahan C. G. M., Hill C. Identification of genes involved in acid resistance in Listeria monocytogenes with a view to examining their importance in virulence and survival in foods. 1999. Food Microbiology and Food Safety into the Next Millenium: Proceedings of the 7th International Conference of the International Committee on Food Microbiology and Hygiene (ICFMH), Veldhoven, The Netherlands, 13-17 Sept. 1999. Ziest: Foundation Food Micro’99.
- Dabiri G. A., Sanger J. M., Portnoy D. A., Southwick F. S. Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proceedings of the National Academy of Science USA. 1990;87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A., Asatryan L., Reddy M. A., Wass C. A., Stins M. F., Joshi S., et al. Differential role of cytosolic phospholipase A2 in the invasion of brain microvascular endothelial cells by Escherichia coli and Listeria monocytogenes. J. Infect Dis. 2001;184:732–737. doi: 10.1086/322986. [DOI] [PubMed] [Google Scholar]
- Datta A. H., Kothary M. R. Effects of glucose, growth temperature, and pH on listerolysin production by Listeria monocytogenes. Appl. Environ. Microbiol. 1993;59:3495–3497. doi: 10.1128/aem.59.10.3495-3497.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Corral F., Buchanan R. L., Bencivengo M. M., Cooke P. H. Quantitative comparison of selected virulence associated characteristics in food and clinical isolates of Listeria. J. Food Prot. 1990;53:1003–1009. doi: 10.4315/0362-028X-53.12.1003. [DOI] [PubMed] [Google Scholar]
- Domann E., Zechel S., Lingnau A., Hain T., Darji A., Nichterlein T., et al. Identification and characterization of a novel PrfA-regulated gene in Listeria monocytogenes whose product, IrpA, is highly homologous to internalin proteins, which contan leucine. Infect. Immun. 1997;65:101–109. doi: 10.1128/iai.65.1.101-109.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle M. P., Beuchat L. R., editors. Food Microbiology: Fundamentals and Frontiers. Washington DC: ASM Press; 2007. [Google Scholar]
- Drevets D. A. Listeria monocytogenes virulence factors that stimulate endothelial cells. Infect. Immun. 1998;66:232–238. doi: 10.1128/iai.66.1.232-238.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets D. A., Sawyer R. T., Potter T. A., Campbell P. A. Listeria monocytogenes infects human endothelial cells by two distinct mechanisms. Infect. Immun. 1995;63:4268–4276. doi: 10.1128/iai.63.11.4268-4276.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S., Kocks C., Forestier C., Cossart P. Internalin-mediated invasion of epithelial cells by Listeria monocytogenes is regulated by the bacterial growth state, temperature and the pleiotropic activator Prf A. Mol. Microbiol. 1993;9:931–941. doi: 10.1111/j.1365-2958.1993.tb01223.x. [DOI] [PubMed] [Google Scholar]
- FAO/WHO (Food and Agriculture Organization/World Health Organization) “Microbiological Risk Assessment Series 2: Risk Assessments of Salmonella in Eggs and Broiler Chickens.”. Geneva, Rome: World Health Organization, Food and Agricultural Organization of the United Nations; 2002. [Google Scholar]
- FAO/WHO (Food and Agriculture Organization/World Health Organization) “Microbiological Risk Assessment Series 3: Hazard Characterization for Pathogens in Food and Water”. Geneva, Rome: World Health Organization, Food and Agricultural Organization of the United Nations; 2003. [Google Scholar]
- FAO/WHO (Food and Agriculture Organization World Health Organization) “Microbiological Risk Assessment Series 5: Risk assessment of Listeria monocytogenes in Ready to Eat Foods: Technical report”. Geneva, Rome: World Health Organization, Food and Agricultural Organization of the United Nations; 2004. [Google Scholar]
- FDA/FSIS (HHS Food and Drug Administration/USDA Food Safety and Inspection Service) “Quantitative Assessment of the Relative Risk to Public Health from Foodborne Listeria monocytogenes among Selected Categories of Ready-to-Eat Foods”. US Food and Drug Administration; 2003. [Google Scholar]
- Formato G., Geornaras I., Barmpalia I. M., Skandamis P. N., Belk K. E., Scanga J. A., Kendall P. A., Smith G. C., Sofos J. N. Effect of acid adaptation on growth during storage at 10 degrees C and resistance to simulated gastric fluid of Listeria monocytogenes inoculated onto bologna formulated with or without antimicrobials. J. Food Prot. 2007;70:65–69. doi: 10.4315/0362-028x-70.1.65. [DOI] [PubMed] [Google Scholar]
- Gahan C. G. M., Hill C. The relationship between acid stress responses and virulence in Salmonella Typhimurium and Listeria monocytogenes. Int. J. Food Microbiol. 1999;50:93–100. doi: 10.1016/s0168-1605(99)00079-3. [DOI] [PubMed] [Google Scholar]
- Gaillard J.-L., Berche P., Mounier J., Richard S., Sansonetti P. In vitro model ofpenetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect. Immun. 1987;55:2822–2829. doi: 10.1128/iai.55.11.2822-2829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getty J. K. J., Phebus R. K., Marsden J. L., Fung D. Y. C., Kastner C. L. Escherichia coli O157:H7 and fermented sausages: A review. J. Rapid Methods and Automation in Microbiol. 2007;8:141–170. [Google Scholar]
- Goldfine H., Bannam T., Johnston N. C., Zukert W. R. Bacterial phospholipases and intracellular growth: The two distinct phospholipases of Listeria monocytogenes. J. Appl. Microbiol. 1998;84:7S–14S. doi: 10.1046/j.1365-2672.1998.0840s107s.x. [DOI] [PubMed] [Google Scholar]
- Goldfine H., Shen H., editors. Listeria Monocytogenes Pathogenesis and Host Response. New York: Springer; 2007. [Google Scholar]
- Grenklo S., Geese M, Lindbery U., Wehland J., Karlsson R., Sechi A.S. A crucial role for profiling-actin in the intracellular motility of Listeria monocytogenes. EMBO Reports. 2003;4:523–528. doi: 10.1038/sj.embor.embor823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas C. N. Estimation of risk due to low doses of microorganisms: a comparison of alternative methodologies. Am. J. Epidemiol. 1983;118:573–582. doi: 10.1093/oxfordjournals.aje.a113662. [DOI] [PubMed] [Google Scholar]
- Haas C. N. Conditional dose-response relationships for microorganisms: Development and application. Risk Anal. 2002;22:455–463. doi: 10.1111/0272-4332.00035. [DOI] [PubMed] [Google Scholar]
- Hill C., Gahan C. Listeria monocytogenes: Role of stress in virulence and survival in food. Irish J. Agric. Food Res. 2000;63:721–726. [Google Scholar]
- Irvin E. A., Williams D., Voss K. A., Smith M. A. Listeria monocytogenes infection in pregnant guinea pigs is associated with maternal liver necrosis, a decrease in maternal serum TNF-? concentrations, and an increase in placental apoptosis. Reprod. Toxicol. 2008a;26:123–129. doi: 10.1016/j.reprotox.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Irvin E.A., Williams D., Hamler S. E., Smith M. A. Immunological and pathological changes in the placenta during infection with Listeria monocytogenes in pregnant guinea pigs. Reprod. Toxicol. 2008b;26((2)):151–155. doi: 10.1016/j.reprotox.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Ito S. O., Ishii K. J., Shirota H., Klinman D. M. CPG oligodeoxynucleotides improve the survival of pregnant and fetal mice following Listeria monocytogenes infection. Infect. Immun. 2004;72:3543–3548. doi: 10.1128/IAI.72.6.3543-3548.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen A., Williams D., Irvin E. A., Gram L., Smith M. A. A processing plant persistent strain of Listeria monocytogenes crosses the fetoplacental barrier in a pregnant guinea pig model. J. Food Prot. 2008;71((5)):1028–1034. doi: 10.4315/0362-028x-71.5.1028. [DOI] [PubMed] [Google Scholar]
- Jia Y., Nightingale K. K., Boor K. J., Ho A., Wiedmann M., McGann P. Distribution of internalin gene profiles of Listeria monocytogenes isolates from different sources associated with phylogenetic lineages. Foodborne Pathog. Dis. 2007;4:222–232. doi: 10.1089/fpd.2006.0081. [DOI] [PubMed] [Google Scholar]
- Joelsson A., Kan B., Zhu J. Quorum sensing enhances the stress response in Vibrio cholerae. Appl. Environ. Microbiol. 2007;73:3742–3746. doi: 10.1128/AEM.02804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien E., Boobis A. R., Olin S. S. the ILSI Research Foundation Threshold Working Group. The Key Events Dose-Response Framework: A Cross-Disciplinary Mode-of-Action Based Approach to Examining Dose-Response and Thresholds. Crit. Rev. Food Sci. Nutr. 2009;49:682–689. doi: 10.1080/10408390903110692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink M., Rudnicka W. Listeria monocytogenes infection in pregnant mice: Abnormalities in the function of non-adherent accessory light density dendritic cells. FEMS Immunol. Med. Microbiol. 1995;12:143–152. doi: 10.1111/j.1574-695X.1995.tb00186.x. [DOI] [PubMed] [Google Scholar]
- Kocks C., Gouin E., Tabouret M., Berche P., Ohayon H., Cossart P. Listeria monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- Kroll R. G., Patchett R. A. Induced acid tolerance in Listeria monocytogenes. Lett. Appl Microbiol. 1992;14:224–227. [Google Scholar]
- Lam G. Y., Brumell J. H. A Listeria escape trick. Nature. 2008;455:1186–1187. doi: 10.1038/4551186a. [DOI] [PubMed] [Google Scholar]
- Lammerding A. M., Glass K. A., Gendron-Fitzpatrick A., Doyle M. P. Determination of virulence of different strains of Listeria monocytogenes and Listeria innocua by oral inoculation of pregnant mice. Appl. Environ. Microbiol. 1992;58:3991–4000. doi: 10.1128/aem.58.12.3991-4000.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M., Dramsi A., Gottardi C., Fedor-Chaiken M., Gumbiner B., Cossart P. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 1999;18:3953–3963. doi: 10.1093/emboj/18.14.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M., Nelson D. M., Smith S. D., Khun H., Huerre M., Vacher-Lavenu M.-C., Gordon J. I., Cossart P. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes with trophoblast E-cadherin. Proceeding of the National Academy of Science USA. 2004;101:6152–6157. doi: 10.1073/pnas.0401434101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M., Ohayon H., Braun L., Mengaud J., Cossart P. Internalin of Listeria monocytogenes with an intact leucine-rich repeat region is sufficient to promote internalization. Infect. Immun. 1997;65:5309–5319. doi: 10.1128/iai.65.12.5309-5319.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M., Vandormael-Pournin S., Lefort J., Huere M., Gounon P., Dupuy C., Babinet C., Cossart P. A transgenic model for listeriosis: Role of internalin in crossing the intestinal barrier. Science. 2001;292:1722–1725. doi: 10.1126/science.1059852. [DOI] [PubMed] [Google Scholar]
- Le Monnier A., Autret N., Join-Lambert O. F., Jaubert F., Charbit A., Berche P., Kayal S. ActA is required for crossing of the fetoplacental barrier by Listeria monocytogenes. Infect. Immun. 2007;75:950–957. doi: 10.1128/IAI.01570-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D., Smith M. A., Elter J., Champagne C., Downey C. L., Beck J., Offenbacher S. Porphyromonas gingivalis infection in pregnant mice is associated with placental dissemination, an increase in the placental Th1/Th2 cytokine ratio, and fetal growth restriction. Infect. Immun. 2003;71:5163–5168. doi: 10.1128/IAI.71.9.5163-5168.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden S. K., Bierne H., Sabet C., Png C. W., Florin T. H., McGuckin M. A., Cossart P. Listeria monocytogenes internalins bind to the human intestinal mucin MUC2. Arch. Microbiol. 2008;190:101–104. doi: 10.1007/s00203-008-0358-6. [DOI] [PubMed] [Google Scholar]
- Marquis H., Doshi V., Portnoy D. A. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect. Immun. 1995;63:4531–4534. doi: 10.1128/iai.63.11.4531-4534.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis H., Hager E. J. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol. Microbiol. 2000;35:289–298. doi: 10.1046/j.1365-2958.2000.01708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann P., Ivanek R., Wiedmann M., Boor K. J. Temperature-dependent expression of Listeria monocytogenes internalin and internalinlike genes suggests functional diversity of these proteins among the Listeriae. Appl. Environ. Microbiol. 2007a;73:2806–2814. doi: 10.1128/AEM.02923-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann P., Wiedmann M., Boor K. J. The alternative sigma factor sigma-B and the virulence gene regulatory PrfA both regulate transcription of Listeria monocytogenes internalins. Appl. Environ. Microbiol. 2007b;73:2919–2930. doi: 10.1128/AEM.02664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead P. S., Slutsker L., Dietz V., McCraig L. F., Bresee J. S., Shapiro C., Griffin P. M., Tauxe R. V. Food-related illness and death in the United States. Emerg. Infect. Diseases. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J., Braun-Breton C., Cossart P. Identification of phosphatidlyinositol-specific phospholipase C activity in Listeria monocytogenes: A novel type of virulence factor? Mol. Microbiol. 1991;5:367–372. doi: 10.1111/j.1365-2958.1991.tb02118.x. [DOI] [PubMed] [Google Scholar]
- Meynell G. G., Stocker B. A. D. Some hypotheses on the aetiology of fatal infections in partially resistant hosts and their application to mice challenged with Salmonella paratyphi-B or Salmonella typhimurium by intraperitoneal injection. J. Gen. Microbiol. 1957;16:38–58. doi: 10.1099/00221287-16-1-38. [DOI] [PubMed] [Google Scholar]
- Milillo S. R., Wiedmann M. Contributions of six lineage-specific internalin-like genes to invasion efficiency of Listeria monocytogenes. Food-borne Pathog. Dis. 2009;6:57–70. doi: 10.1089/fpd.2008.0140. [DOI] [PubMed] [Google Scholar]
- Mounier J., Ryter A., Coquis-Rondon M., Sansonetti P. J. Intracellular and cell-to-cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocyte-like cell line Caco-2. Infect. Immun. 1990;58:1048–1058. doi: 10.1128/iai.58.4.1048-1058.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxon E. R., Murphy P. A. Haemophilus influenzae bacteremia and meningitis resulting from survival of a single organism. Proceedings National Academy of Science USA. 1978;75:1534–1536. doi: 10.1073/pnas.75.3.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notermans S. H. W., Dufrenne J., Leimeister-Wachter M., Domann E., Chakraborty T. Phosphtidylinositol-specific phospholipase C activity as a marker to distinguish between pathogenic and nonpathogenic Listeria species. Appl. Environ. Microbiol. 1991;57:2666–2670. doi: 10.1128/aem.57.9.2666-2670.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll B., Gahan C. G. M., Hill C. Adaptive acid tolerance response in Listeria monocytogenes: Isolation of an acid-tolerant mutant which demonstrates increased virulence. Appl. Environ. Microbiol. 1996;62:1693–1996. doi: 10.1128/aem.62.5.1693-1698.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrisius J., Bhakdi S., Roth M., Tranum-Jensen J., Goebel W., Sseeliger H. P. R. Production of listeriolysin by beta-hemolytic strains of Listeria monocytogenes. Infect. Immun. 1986;51:314–319. doi: 10.1128/iai.51.1.314-319.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistor S., Chakraborty T., Niebuhr K., domann E., Wehland J. The ActA protein of Listeria monocytogenes acts as a nucleator inducing reorganization of the actin cytoskeleton. EMBO J. 1994;13:758–763. doi: 10.1002/j.1460-2075.1994.tb06318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin L. G. Bacterial colonization and infection resulting from multiplication of a single organism. Rev. Infect. Dis. 1987;9:488–493. doi: 10.1093/clinids/9.3.488. [DOI] [PubMed] [Google Scholar]
- Ryan S., Begley M., Gahan C. G., Hill C. Molecular characterization of the arginine deiminase system in Listeria monocytogenes: Regulation and role in acid tolerance. Environ. Microbiol. 2009;11:432–445. doi: 10.1111/j.1462-2920.2008.01782.x. [DOI] [PubMed] [Google Scholar]
- Ryser E.T., Marth E. H., editors. Listeria, listeriosis, and food safety. Boca Raton, FL: CRC Press; 2007. [Google Scholar]
- Sahaghian R., Faith N. G., Czuprynski C. Comparison of systemic Listeria monocytogenes infection in esophageally inoculated mice anesthetized with isoflurane or pentobarbital. Lab. Anim. 2009;38((4)):126–30. doi: 10.1038/laban0409-126. [DOI] [PubMed] [Google Scholar]
- Sanger J. M., Sanger J. W., Southwick F. S. Host cell actin assembly is necessary and likely to provide the propulsive force for intracellular movement of Listeria monocytogenes. Infect. Immun. 1992;60:3609–3619. doi: 10.1128/iai.60.9.3609-3619.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Jamieson A., Cresswell P. GILT is a critical host factor for Listeria monocytogenes infection. Nature. 2008;455:1244–1248. doi: 10.1038/nature07344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. A., Marquis H., Jones S., Johnston N. C., Portnoy D. A., Goldfine H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect. Immun. 1995;63:4231–4237. doi: 10.1128/iai.63.11.4231-4237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. A., Takeuchi K., Anderson G., Ware G. O., McClure H. M., Raybourne R. B., Mytle N., Doyle M. P. Dose response for Listeria monocytogenes-induced stillbirths in nonhuman primates. Infect. Immun. 2008;76:726–731. doi: 10.1128/IAI.01366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. A., Takeuchi K., Brackett R. E., McClure H. M., Raybourne R., Williams K., Babu U. S., Ware G. O., Broderson J. R., Doyle M. P. A nonhuman primate model for Listeria monocytogenes-induced stillbirths. Infect. Immun. 2003;71((3)):1574–1579. doi: 10.1128/IAI.71.3.1574-1579.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonich-Mullin C., Fielder R., Wiltse J., Baetcke K., Dempsey J., Fenner-Crisp P., Grant D., Hartley M., Knaap A., Kroese D., Mangelsdorf I., Meek M.E., Rice J. M., Younes M. International Programme on Chemical Safety, IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul. Toxicol. Pharmacol. 2001;34((2)):146–152. doi: 10.1006/rtph.2001.1493. [DOI] [PubMed] [Google Scholar]
- Swaminathan B., Cabanes D., Zhang W., Cossart P., Doyle M. P., Beuchat L. R. Food Microbiology: Fundamentals and Frontiers. 3rd Ed. Washington, DC: ASM Press; 2007. Listeria monocytogenes; pp. 457–491. [Google Scholar]
- Teunis P., Takumi K., Shinagawa K. Dose response for infection by Escherichia coli O157:H7 from outbreak data. Risk Anal. 2004;24:401–407. doi: 10.1111/j.0272-4332.2004.00441.x. [DOI] [PubMed] [Google Scholar]
- Teunis P. F., Ogden I. D., Strachan N. J. Hierarchical dose response of E. coli O157:H7 from human outbreaks incorporating heterogeneity in exposure. Epidemiol. Infect. 2008;136:761–770. doi: 10.1017/S0950268807008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney L. G., Portnoy D. A. Actin filaments aid the growth, movement and filament of the intracellular bacterial parasite, Listeria monoctogenes. J. Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney L. G., Connelly P. S., Portnoy D. A. Actin filamento nucleation by the bacterial pathogen Listeria monocytogenes. J. Cell Biol. 1992;111:2979–2988. doi: 10.1083/jcb.111.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z., Zenewicz L. A., Goldfine H. Listeria monocytogenes phosphatidylinosito-specific phospholipase C has evolved for virulence by greatly reduced activity on GPI anchors. Proceedings National Academy of Science USA. 2005;102:12927–12931. doi: 10.1073/pnas.0501725102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedmann M., Arvik T. J., Hurley R. J., Boor K. J. General stress transcription factor sigma B and its role in acid tolerance and virulence of Listeria monocytogenes. J. Bacteriol. 1998;180:3650–3656. doi: 10.1128/jb.180.14.3650-3656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D., Irvin E. A., Chmielewski R. A., Frank J. F., Smith M. A. Dose response of Listeria monocytogenes after oral exposure in pregnant guinea pigs. J. Food Prot. 2007;70:1122–1128. doi: 10.4315/0362-028x-70.5.1122. [DOI] [PubMed] [Google Scholar]
- Zwart M. P., Hemerik L., Cory J. S., de Visser J. A., Bianchi F. J., Van Oers M. M., Vlak J. M., Hoekstra R. F., Van der Werf W. An experimental test of the independent action hypothesis in virus-insect pathosystems. Proc. R. Soc B. 2009;276:2233–2242. doi: 10.1098/rspb.2009.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwietering M. H., Havelaar A. H., Potter M. Food consumption and disease risk. Cambridge: Woodhead Publishing; 2006. Dose-response relationships and foodborne disease; pp. 422–439. [Google Scholar]