

Abstract
[PSI+] is a genetic element in yeast for which a heritable change in phenotype appears to be caused by a heritable change in the conformational state of the Sup35 protein. The inheritance of [PSI+] and the physical state of Sup35 in vivo depend on the protein chaperone Hsp104 (heat shock protein 104). Although these observations provide a strong genetic argument in support of the “protein-only” or “prion” hypothesis for [PSI+], there is, as yet, no direct evidence of an interaction between the two proteins. We report that when purified Sup35 and Hsp104 are mixed, the circular dichroism (CD) spectrum differs from that predicted by the addition of the proteins’ individual spectra, and the ATPase activity of Hsp104 is inhibited. Similar results are obtained with two other amyloidogenic substrates, mammalian PrP and β-amyloid 1-42 peptide, but not with several control proteins. With a group of peptides that span the PrP protein sequence, those that produced the largest changes in CD spectra also caused the strongest inhibition of ATPase activity in Hsp104. Our observations suggest that (i) previously described genetic interactions between Hsp104 and [PSI+] are caused by direct interaction between Hsp104 and Sup35; (ii) Sup35 and PrP, the determinants of the yeast and mammalian prions, respectively, share structural features that lead to a specific interaction with Hsp104; and (iii) these interactions couple a change in structure to the ATPase activity of Hsp104.
Keywords: prion, protein structure, aggregation, chaperone protein
Recently, a mode of inheritance has been discovered in Saccharomyces cerevisiae (1, 2). Phenotypes transmitted by two dominant, cytoplasmically inherited genetic elements, [PSI+] and [URE3], seem to depend on the inheritance of altered protein structures, rather than altered nucleic acids. The “protein-only” hypothesis for their inheritance led these elements to be called “yeast prions” (1). The term “prion” was first coined to describe the infectious agent hypothesized to cause mammalian transmissible spongiform encephalopathies (TSEs) by a “protein-only” mechanism: a normal cellular protein (PrPC) adopts an altered conformation (PrPSc) and interacts with other PrPC proteins to change their conformation as well (3) .
The yeast [PSI+] element, the subject of our work, does not generally kill cells. It reduces the fidelity of ribosome translation termination and thereby suppresses nonsense codons (2). This phenotype is thought to result from a change in the state of the translation-termination factor, Sup35, that interferes with its normal function. In [psi−] cells, Sup35 is protease sensitive and is mostly soluble; in [PSI+] cells, Sup35 has increased protease resistance and is mostly aggregated (4–6). (“Aggregate” is used in a general sense; Sup35 may be polymerized into an amyloid-like structure, or coalesced in a less ordered state.) When pre-existing Sup35 is in the aggregated state, newly made Sup35 also aggregates, causing a self-perpetuating loss of function in the termination factor and a heritable change in translational fidelity (5, 6) .
[PSI+] depends on the chaperone protein Hsp104 (heat shock protein 104). The first known function of Hsp104 was in thermotolerance in yeast, where it increases survival after exposure to extreme temperatures up to 1,000-fold (7). It does so by promoting the reactivation of proteins that have been damaged by heat and have begun to aggregate (8). At normal temperatures, Hsp104 overexpression cures cells of [PSI+]. Sup35 becomes soluble and the fidelity of translation termination is restored. This state is heritable, even when overexpression of Hsp104 ceases (9). Because the only known function of Hsp104 is to alter the conformational state of other proteins, these observations provide a strong argument that [PSI+] is indeed based on a heritable (self-perpetuating) change in the conformational state of Sup35.
Surprisingly, deletions of HSP104 also cure cells of [PSI+], and Sup35 is soluble in such cells as well (5, 9). This is very different from heat-induced aggregates, which remain insoluble in hsp104 deletion strains. Clearly, the relationship between Hsp104 and [PSI+] is more complex than the relationship between Hsp104 and thermotolerance.
It is possible to construct coherent models to explain these, and other, perplexing interactions between Hsp104 and [PSI+] (4–6, 9). However, a critical missing link in the protein-only hypothesis for [PSI+] inheritance is any evidence that Hsp104 actually interacts directly with Sup35. Indeed, little is known about the interaction of Hsp104 with any substrate, as the heat-denatured aggregates that constitute its other likely in vivo substrates are inherently difficult to study. Here we provide evidence for a highly specific interaction in vitro between Hsp104 and Sup35. This interaction produces a change in protein structure and inhibits the ATPase activity of Hsp104. We also report that Hsp104 interacts in a remarkably similar way with mammalian PrP, the protein determinant of the neurodegenerative prion diseases (3, 10), and with β-amyloid peptide (11). Our data add to previous reports identifying biochemical similarities between these otherwise unrelated amyloidogenic proteins.
MATERIALS AND METHODS
Protein and Peptide Preparation.
Hsp104 (prepared as in ref. 12), Hsp70 (gift of J. Glover, University of Chicago), aldolase (Pharmacia), and IgM (Rockland, Gilbertsville, PA) were stored in 20 mM Tris (pH 8.0), 2 mM EDTA, 1.4 mM 2-mercaptoethanol, 5% glycerol, and 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF). Sup35 and the fragment MN (purified as in ref. 13) were dialyzed against HSB [high salt buffer; 20 mM Hepes (pH 7.5), 10 mM MgCl2, 140 mM KCl, and 15 mM NaCl freshly supplemented with 5 mM 2-mercaptoethanol and 1 mM AEBSF] to remove imidazole, and then dialyzed against either HSB or LSB [low salt buffer; 10 mM Mes (pH 6.5) and 10 mM MgSO4]. Concentrations were determined by the Bradford assay with BSA as a standard. Concentrations of PrP (prepared and folded into either β-sheet or α-helical forms; refs. 14 and 15), PrP peptides (as in ref. 16), and β-amyloid (Sigma) were determined spectroscopically by using calculated extinction coefficients.
ATPase Assays.
PrP peptides (1 mM resuspended in H2O) were assayed in 40 mM Tris (pH 7.5), 175 mM NaCl, 5 mM MgCl2, and 5 mM ATP in a 25 μl reaction volume containing 1 μg of Hsp104. Peptides A, G, H, and K were resuspended in dimethyl sulfoxide (DMSO) that was also added to controls containing Hsp104 alone. Effects of other proteins on the ATPase activity of Hsp104 were measured in LSB or HSB. Phosphate released (mean and standard deviation of at least three independent reactions) after 8 min at 37°C was measured with Malachite Green (17).
Spectropolarimetry.
Hsp104, Hsp70, aldolase, IgM, or storage buffer were added to Sup35 or recombinant PrP (rPrP) in the buffers indicated. When aldolase and IgM were tested as substrates of Hsp104, they were first dialyzed against HSB and subsequently LSB, so that their treatment matched that of Sup35. In LSB Sup35 solutions were somewhat cloudy, suggesting some aggregation, but little or no protein precipitated to the bottom of cuvettes during analysis. Chaperones and control proteins were added to Sup35 at a ≈1:2.5 gram/weight ratio (e.g., Sup35 at ≈0.4 mg/ml and Hsp104 at 0.15 mg/ml). Reactions were incubated for 1 hr with 1 mM ATP at 37°C and transferred to a 0.1-mm path-length cuvette. Spectra were recorded at 25°C in a Jasco 715 spectropolarimeter (bandwidth 1.0 nm, response time 16 sec, speed 20 nm/min, step resolution 0.2 nm, accumulations 4).
Proteins and rPrP were mixed with each other (each at 0.5 mg/ml) or with the appropriate storage buffer in LSB2 (20 mM phosphate buffer, pH 6.5/10 mM MgSO4/1.25 mM ATP). PrP peptides were mixed with Hsp104 in 20 mM Tris buffer containing 10 mM MgSO4, 50 mM KCl, and 1.25 mM ATP at pH 7.5 to approximate ATPase assay conditions. After 1 hr at 37°C, reactions were diluted 5-fold with cold water, and spectra were measured at 12°C as above. (A larger spectral shift was observed with these conditions for peptide F, presumably because the structural changes obtained with this peptide are unstable at higher temperatures. Although temperature had little effect on the spectra obtained with other peptides or rPrP, for consistency, 12°C was used for all experiments.)
Congo Red Dye Binding Assays.
Reaction conditions were as for circular dichroism (CD) experiments with the addition of Congo red to a final concentration of 10 μM. After 30 min at 25°C, absorbances at 320, 477, and 540 nm were determined. Congo red dye binding was measured by using the equation [(OD540/25,295) − (OD477/46,306)] (18).
RESULTS
CD of Hsp104 and Sup35 Mixtures.
Attempts to detect an interaction between Sup35 and Hsp104 by co-immunoprecipitation or by affinity chromatography with immobilized Hsp104 were unsuccessful (J. R. Glover, M. M. Patino, E.C.S., and S.L., unpublished observations), suggesting that if Hsp104 interacts with Sup35, this interaction is weak, transient, or depends on unique conditions, conformations, or cofactors. Because changes in the expression of Hsp104 lead to changes in the physical state of Sup35 in vivo, as an alternative mechanism for probing interactions between these proteins, we asked whether changes in state could be detected by CD when purified Hsp104 and Sup35 were mixed in vitro. If two proteins do not interact, or if they interact without a substantial change in secondary structure, the CD spectrum of their mixture should equal that predicted from the simple addition of their individual spectra. Indeed, when either Sup35 or Hsp104 was mixed with any of several control proteins—aldolase, Igs (IgG and IgM), α2-macroglobulin, apoferritin, and α-lactalbumin—observed spectra matched the predicted spectra (Fig. 1 A and B, and data not shown). These control proteins encompass a wide variety of structural features, including proteins that are largely α-helical or β-sheet, monomeric or oligomeric, large or small. Furthermore, spectral shifts observed when another chaperone, Hsp70, was mixed with Sup35 were small (Fig. 1A).
Figure 1.
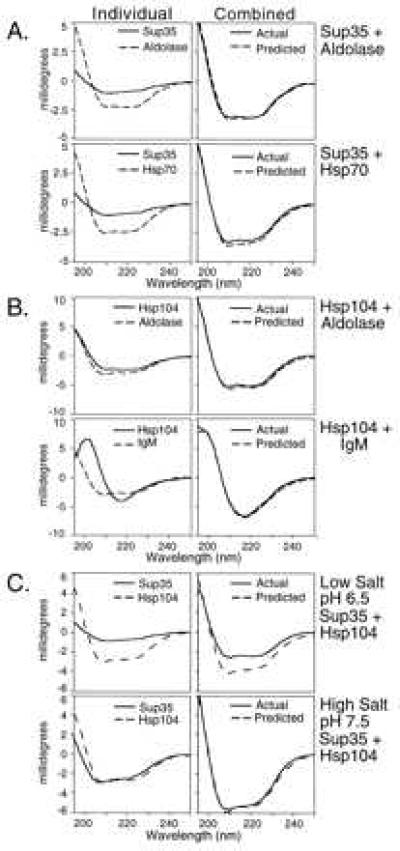
Specificity of CD spectral shifts with Hsp104 and Sup35. (Right) Predicted (−−) and actual (—) spectra of mixed proteins. (Left) Individual spectra used to generate predicted spectra. (A) Sup35 in LSB1 (low salt buffer) with aldolase or Hsp70. (B) Hsp104 and aldolase or IgM in LSB1. (C) Hsp104 and Sup35 in LSB1 (Upper) and Hsp104 and Sup35 in HSB (Lower). Data, buffer spectra subtracted, are presented in millidegrees because the possibility of proteins partitioning out of solution invalidates molar ellipticity calculations. Hsp104, Hsp70, aldolase, or the buffer in which they were prepared was directly added to Sup35 at an ≈1:2.5 gram/weight ratio. Reactions were incubated for 1 hr with 1 mM ATP at 37°C, and spectra were then recorded at 25°C.
In contrast, when Hsp104 and Sup35 were mixed, the observed spectrum differed dramatically from the predicted spectrum (Fig. 1C, Upper Right). Thus, these two proteins interacted in a highly specific manner to produce a change in the physical state of one or both proteins. ATP is required for some Hsp104 functions (12, 19), but was not required for the change in CD spectrum with Sup35 and Hsp104 (data not shown). However, ATP markedly increased the rate at which this change occurred (Table 1).
Table 1.
ATP affects rate of CD change
| Time, min | − ATP | + ATP |
|---|---|---|
| 0 | 2.9 | 2.2 |
| 3 | 7.9 | 15.2 |
| 6 | 9.9 | 17.2 |
| 10 | 10.9 | 18.2 |
| 15 | 11.8 | 18.8 |
| 20 | 12.4 | 19.2 |
| 30 | 13.3 | 19.8 |
| 72 | 16.2 | 21.9 |
The difference between the actual spectrum and the predicted spectrum at 225 nm for each time point is presented in millidegrees. In each of three separate experiments, the spectral change in mixtures of Sup35 and Hsp104 proceeded more rapidly with ATP than without ATP, although the absolute rates varied, most likely due to differences in the Sup35 preparations.
The interaction between Hsp104 and Sup35 apparently depended on the structural state of Sup35. When Sup35 was dialyzed against low salt buffer at pH 6.5 (LSB, Fig. 1C Upper Left, solid line) or a higher salt buffer at pH 7.5 (HSB, Fig. 1C Lower Left, solid line) a difference in the CD spectra indicated that the protein was in a different structural state. When Hsp104 was added, the actual CD spectrum deviated from the predicted spectrum only when Sup35 had been dialyzed in LSB (Fig. 1C, compare Right panels). Mixtures of Sup35 and several control proteins showed no deviation from predicted spectra in LSB or HSB (data not shown). Similarly, control proteins mixed with Hsp104 showed no spectral shifts in either buffer (data not shown). Moreover, the CD spectrum of Hsp104 itself did not change with the buffer (Fig. 1C Left, dashed line).
Sup35 Aggregation.
In vivo, the inheritance of [PSI+] is associated with the partitioning of Sup35 into aggregates, a change in state that requires Hsp104 (4, 5, 9). In vitro, Sup35 forms highly ordered, amyloid-like fibers after prolonged incubations in the absence of Hsp104 (13). In CD experiments the proteins did not precipitate to the bottom of the cuvette or exhibit significant binding to the walls of the tube. However, the upward shift in the spectrum might be caused by, at least in part, a partitioning of protein from solution while it remains in suspension (20). To determine whether the interaction between Hsp104 and Sup35 detected by CD analysis in vitro is related to the biological interaction between the two proteins in vivo, we asked whether it was associated with a change in protein aggregation. Indeed, solutions containing mixtures of Sup35 and Hsp104 invariably scattered more light at 320 nm (typically ≈30% more) than the simple sum of light scattering by each protein alone. An increase in Congo red dye binding was also detected by the characteristic spectral shift that occurs when this dye binds amyloid proteins (18). (Absolute values for dye binding were not calculated because light scattering makes background subtraction difficult to compute.)
Effects of Sup35 on the ATPase Activity of Hsp104.
When other members of the HSP100 (clp) family are incubated with substrates, the rate at which they hydrolyze ATP is increased (21–23). Thus, changes in the ATPase activity of Hsp104 provide another method for detecting an interaction with Sup35. When assayed in HSB, in which no CD changes were observed, Sup35 weakly stimulated the ATPase activity of Hsp104 (Table 2). Surprisingly, in LSB, in which CD changes were observed, Sup35 strongly inhibited the ATPase activity of Hsp104.
Table 2.
Effects of proteins and peptides on the ATPase activity of Hsp104
| HSB | LSB | |
|---|---|---|
| Hsp104 alone | 1.0 ± 0.05 | 1.0 ± 0.1 |
| Sup35 | 1.2 ± 0.1 | 0.6 ± 0.1 |
| N-term Sup35 | 1.2 ± 0.1 | 0.7 ± 0.1 |
| PrPβ | 1.2 ± 0.1 | 0.6 ± 0.05 |
| β-Amyloid 1-42 | 0.8 ± 0.05 | 0.3 ± 0.05 |
| β-Amyloid 1-40 | 1.1 ± 0.05 | 0.5 ± 0.1 |
| Reverse amyloid 40-1 | 1.1 ± 0.1 | 0.8 ± 0.1 |
| Aldolase | 1.1 ± 0.05 | 1.0 ± 0.1 |
| BSA | 1.0 ± 0.05 | 1.0 ± 0.05 |
| Apoferritin | 1.0 ± 0.1 | 1.0 ± 0.05 |
| IgM | 1.1 ± 0.05 | 1.1 ± 0.05 |
Hsp104 ATPase activity was measured in HSB or LSB1 and is presented as the activity of Hsp104 with protein divided by the activity of Hsp104 in buffer alone. Within individual experiments very little variance was observed; however, even with the results from three different preparations of Sup35 averaged here, only ≈10% variability was observed.
Previous studies identified the N-terminal domain of Sup35 as the essential “prion-determining” region (24). This domain is also responsible for the formation of self-seeded amyloid fibrils by Sup35 in vitro. (13, 25). The ATPase activity of Hsp104 was inhibited by this domain to an extent similar to that observed with Sup35 itself (Table 2).
Effects of Other Amyloids on the ATPase Activity of Hsp104.
The expansion of the mammalian prion hypothesis to the yeast [PSI+] element was initially based on genetic arguments. PrP and Sup35 are unrelated in sequence and in biological function (1, 2, 9). Nonetheless, the capacity for both proteins to form amyloid-like aggregates (13, 26, 27) suggests an underlying biochemical similarity between them. We asked whether this similarity would extend to shared molecular features in the two proteins that allow recognition by Hsp104. The change in state of mammalian PrP associated with TSEs is characterized by increased β-sheet content and protease resistance in amino acid segment 90–231 (28, 29). A recombinant hamster protein corresponding to this segment, in a β-sheet-rich conformation, rPrPβ (14, 15), produced the same unexpected effect on the ATPase activity of Hsp104 as did Sup35 (Table 2).
We also tested another amyloidogenic peptide, β-amyloid 1-42, a fragment often found in the neural plaques associated with Alzheimer’s disease (11). Again, the ATPase activity of Hsp104 was inhibited (Table 2). Less inhibition was observed with a less amyloidogenic derivative, β-amyloid 1-40, still less with a peptide containing the same amino acids in the reverse order, and no inhibition was observed with a wide variety of control proteins (Table 2, and data not shown). Thus, the unexpected inhibitory effects of these three amyloidogenic polypeptides on the ATPase activity of Hsp104 are specific and strongly suggest an underlying biochemical similarity between them.
CD of Hsp104 and PrP Mixtures.
When Hsp104 was mixed with rPrPβ (14, 15), the CD spectrum of the solution differed dramatically from the spectrum predicted by the addition of individual spectra (Fig. 2A Right). This result was very reproducible in both degree and effect, with two different preparations of rPrP and two of Hsp104. When rPrPβ was mixed with several other chaperones, only GroEL (Hsp60) yielded a substantial spectral shift (Fig. 2B Right, and data not shown). Other chaperones (Cdc37, Hsp90, Hsp70), as well as some nonchaperone proteins (apoferritin, β-galactosidase, α2-macroglobulin, and α-lactalbumin) yielded spectral shifts with PrP, but they were much smaller than those observed with Hsp104 and GroEL. Finally, when rPrPβ was mixed with BSA or aldolase (Fig. 2C Right, and data not shown), predicted and actual spectra were virtually identical.
Figure 2.
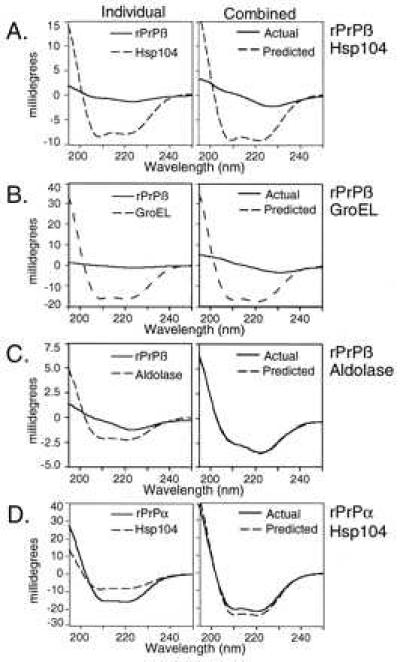
Specificity of CD spectral shifts with Hsp104 and rPrP. Predicted (−−), actual (—), and individual spectra as in Fig. 1. rPrP was prepared and folded into either β-sheet or α-helical forms. (A) Hsp104 and rPrPβ. (B) GroEL and rPrPβ. (C) Aldolase and rPrPβ. (D) Hsp104 and rPrPα. Proteins and rPrP were mixed (each at 0.5 mg/ml) with each other in LSB2. Reactions were incubated at 37°C for 1 hr, diluted 5-fold with cold water, and spectra were measured at 12°C.
As with Sup35, the interaction between Hsp104 and rPrP depended on the structural state of rPrP. When rPrP was pre-incubated under conditions (14, 15) that promote an α-helical conformation (rPrPα) rather than a β-sheet-rich conformation (rPrPβ), and mixed with Hsp104, the actual spectrum matched the predicted spectrum (Fig. 2D Right). The α-helical and β-sheet-rich forms of rPrP, once acquired, were stable after transfer to the same buffer. Because they were in the same buffer when mixed with Hsp104, the different results obtained with rPrPα and rPrPβ can be attributed to an effect of substrate structure on interaction with Hsp104, rather than to an effect of buffer.
Correlation Between Structural Transitions and ATPase Inhibition with PrP Peptides.
Because for both Sup35 and PrP the inhibition of the ATPase activity of Hsp104 occurred under the same conditions where a spectral shift occurred, we postulated that these amyloidogenic proteins might inhibit the ATPase activity of Hsp104 by coupling it to a major change in structure. To investigate this possibility further, we took advantage of various PrP peptide derivatives (Fig. 3A) and of previous work characterizing the structural transitions of both PrP and these derivatives (14–16, 27, 30). Several peptides from the N-terminal region had little or no effect on the ATPase activity of Hsp104 (Fig. 3B, peptides A and B); a peptide corresponding to amino acids 90–145 strongly stimulated ATP hydrolysis by Hsp104 (Fig. 3B, peptide F); several peptides derived from the C terminus inhibited it (Fig. 3B, peptides G–K).
Figure 3.
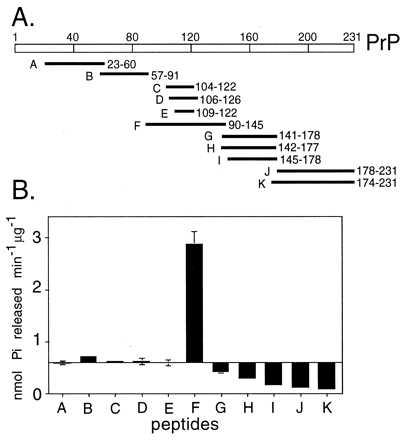
Effects of mixing PrP peptides with Hsp104. (A) Location of peptides used in this study. Peptides prepared as in ref. 16 were derived from the hamster PrP sequence except for peptide K, derived from mouse PrP. (B) Effects of PrP peptides on the ATPase activity of Hsp104. Bars extend from the value obtained for Hsp104 without added peptide.
Peptides with different effects on the ATPase activity of Hsp104 were then tested for spectral shifts in the presence of Hsp104. (Because CD analysis requires large quantities of peptide, we did not have sufficient material to test them all.) When peptide B was mixed with Hsp104, the CD spectrum was equivalent to that predicted from the addition of the individual spectra (Fig. 4A.) The actual and predicted spectra of Hsp104 and peptide F were not identical, but the deviation was small (Fig. 4B). In contrast, the spectra obtained from mixing Hsp104 with the C-terminal peptides G and J were very different from the predicted spectra (Fig. 4 C and D). Thus, the PrP peptides that inhibited the ATPase activity of Hsp104 yielded the strongest spectral shift.
Figure 4.
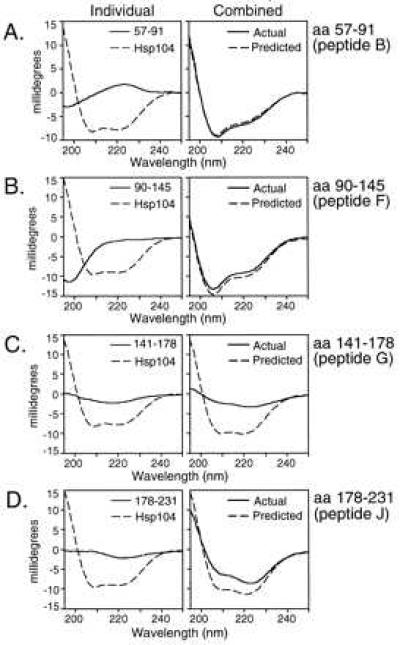
Specificity of CD spectral shifts with different PrP peptides. Predicted (−−), actual (—), and individual spectra as in Fig. 1. Reactions were performed as in Fig. 2, except that the buffer used was 20 mM Tris, 10 mM MgSO4, 50 mM KCl, and 1.25 mM ATP at pH 7.5 to approximate ATPase assay conditions.
DISCUSSION
The dependence of [PSI+] on the protein chaperone Hsp104 provides one of the strongest genetic arguments that the inheritance of a phenotypic trait can be caused by the inheritance of a change in protein conformation, in this case, the conformation of Sup35 (4, 5, 9). The validity of this argument rests on two assumptions: (i) that Hsp104 and Sup35 interact directly, and (ii) that this interaction influences the physical state of Sup35. Here we provide evidence in support of both. Remarkably, very similar results were obtained with PrP, the mammalian protein whose altered conformation is thought to propagate the TSEs (3, 10). β-Amyloid, the peptide whose deposition in amyloids is thought to contribute to Alzheimer’s disease (11), was characterized less extensively, but it too interacted with Hsp104 in a similar manner. These findings reveal an underlying biochemical similarity between these otherwise unrelated proteins.
CD experiments provided one line of evidence for the direct interaction of Hsp104 with Sup35 and with PrP. The actual spectra observed when Hsp104 is mixed with either of these proteins is different from the spectra predicted by the simple addition of their individual spectra. These spectral shifts are highly specific. When control proteins, encompassing a wide variety of structural features, are mixed with Hsp104, actual spectra match the predicted spectra. Further, when Sup35 or rPrPβ are mixed with control proteins (including other chaperones), spectral deviations are relatively small or undetectable (except in the case of rPrPβ and GroEL). Finally, the interactions of Hsp104 with Sup35 and PrP themselves appear to depend on the initial structural states of Sup35 and PrP. Currently, producing different structural states of Sup35 depends on using different buffers and, although these buffers did not influence the CD spectum of Hsp104, they might influence the interaction of Hsp104 with Sup35. However, in the case of rPrP distinct conformational states, once established, are stable on transfer to the same buffer (14, 15). A large spectral shift occurs with rPrPβ, a β-sheet-rich, multimeric conformation (15) thought to be associated with TSE diseases, but not with rPrPα, an α-helix-rich, monomeric conformation thought to mimic the normal cellular form.
The changes in spectra we observe are large, but their nature is unclear. One issue is that the interpretation of CD spectra is based on comparisons with defined structures, and no structures of amyloid fibers have yet been solved. No protein precipitated to the bottom of the cuvette, nor was much protein lost to the walls of the cuvette. However, proteins may leave solution without visible precipitation, resulting in a weaker (upwardly shifted) CD signal (20). The changes we observe are therefore consistent with either a reduction in α-helix and the production of other (perhaps novel) structures, and/or a partitioning of proteins from the solvent phase while they remain in suspension. Another issue is that the contributions made by individual proteins to the CD signal cannot be distinguished in mixed solutions. In [PSI+] and [psi−] cells, it is the aggregation state of Sup35 that changes whereas Hsp104 remains soluble (4, 5). Because Sup35 has a known capacity to form amyloid-like fibers in vitro (13), the increase in light scattering and Congo red binding in our experiments suggest that Hsp104 is facilitating a change in the structure of Sup35. However, the states of these proteins are too poorly characterized, both in vivo and in vitro, to determine whether the structural transitions we report mimic those that occur in vivo. Additional methods and materials will be required to resolve this question. However, the CD data do establish that direct interactions occur between the chaperone and these substrates and that these interactions result in a change in the state of the interacting protein(s).
The ability of both Sup35 and PrP to inhibit the ATPase activity of Hsp104 provides independent evidence for an interaction between these proteins. The same specificity was observed as with CD: (i) control proteins do not inhibit the ATPase activity of Hsp104; (ii) Sup35 inhibits it under the conditions that lead to a change in CD spectrum, but not under the conditions where no change in CD spectrum occurred; and (iii) rPrPβ also inhibited it under the conditions that lead to a change in the CD spectrum. Studies with a series of peptides spanning the PrP sequence provide another link between the Hsp104–substrate interactions that lead to structural transitions and those that inhibit ATPase activity. The strongest inhibition in the ATPase activity of Hsp104 occurred with the peptides that produced the strongest CD shifts.
The inhibition of the ATPase activity of Hsp104 was itself surprising. Interactions between other HSP100 proteins and their substrates generally stimulate the chaperone’s ATPase activity (21–23). At least some of these interactions, however, seem to involve less dramatic structural transitions (19). For example, ClpA (an Escherichia coli relative of Hsp104) converts the RepA protein from dimers to monomers (31). Both ClpA and Hsp104 are hexameric proteins with multiple ATP binding sites and, presumably, multiple substrate binding sites. Perhaps the structural transitions of more complex, amyloidogenic substrates involve more coupled or “concerted” work from the chaperone and this inhibits its free-running ATPase activity.
We did not monitor CD spectral shifts with β-amyloid peptide and Hsp104, but it is striking that this peptide too inhibited the ATPase activity of Hsp104. β-Amyloid, Sup35, and PrP differ in size and biological function and have unrelated sequences (except for weak homology in a few oligopeptide repeats of Sup35 and PrP). Yet, all share the capacity to assemble into amyloid-like aggregates (11, 13, 26). The [PSI+] genetic trait is linked to the aggregation of Sup35; the pathologies of TSEs and Alzheimer’s disease are generally associated with the aggregation of PrP and β-amyloid, respectively (3, 10, 11). Presumably, it is the shared capacity for such conformational transitions that leads to recognition by Hsp104.
Strong genetic evidence supports the biological significance of the interaction between Hsp104 and Sup35 (4, 5, 9). The biological relevance of the interactions between Hsp104 and PrP is supported by a separate study from our laboratory. DebBurman et al. (32) have found that in the presence of the infectious form of PrP (PrPSc) Hsp104 accelerates the rate at which full-length cellular form, PrPC, assumes the protease resistance pattern that is the hallmark of PrPSc. Of the many chaperones tested in that study, only Hsp104 and GroEL accelerated conversion. It is notable therefore that of the several chaperones tested here (Hsp70, Hsp90, Ydj1, Cdc37, GroEL, and Hsp104), only Hsp104 and GroEL produced a strong spectral shift with PrP. We do not mean to suggest that Hsp104 (or GroEL) homologs regulate the structural transitions of PrP or β-amyloid in vivo. Rather, we suggest that protein chaperones provide common mechanisms for controlling certain types of conformational switches and, thus, might provide potential avenues for therapeutic intervention. In any case, the strikingly similar and highly specific interactions we observe between Hsp104 and these three very different proteins provides another link between the so called yeast and mammalian prions and between these prions and β-amyloid.
Acknowledgments
We are grateful to D. Ware for technical assistance, M. Patino for Sup35 expression constructs, T. Sosnick and P. Speros for invaluable CD advice, T. Serio and S. K. DebBurman for comments on the manuscript, T. Sosnick and S. B. Prusiner for use of their spectropolarimeters, and most particularly to M. A. Baldwin and S. B. Prusiner for PrP peptides and proteins, technical help, and advice. This work was supported by the Howard Hughes Medical Institute and the National Institutes of Health Grant GM25874.
ABBREVIATIONS
- Hsp104
heat shock protein 104
- CD
circular dichroism
- TSEs
transmissible spongiform encephalopathies
- HSB
high salt buffer
- LSB
low salt buffer
- rPrP
recombinant PrP
References
- 1.Wickner R B. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 2.Lindquist S. Cell. 1997;89:495–498. doi: 10.1016/s0092-8674(00)80231-7. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner S B. In: Fields Virology. Fields B N, Knipe D M, Howley P M, editors. Philadelphia: Lippencott-Raven; 1996. pp. 2901–2950. [Google Scholar]
- 4.Paushkin S V, Kushnirov V V, Smirnov V N, Ter-Avanesyan M D. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 5.Patino M M, Liu J-J, Glover J R, Lindquist S. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 6.Paushkin S V, Kushnirov V V, Smirnov V N, Ter-Avanesyan M D. Science. 1997;277:381–383. doi: 10.1126/science.277.5324.381. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez Y, Lindquist S L. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 8.Parsell D A, Kowal A S, Singer M A, Lindquist S. Nature (London) 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 9.Chernoff Y O, Lindquist S L, Ono B-i, Inge-Vechtomov S G, Liebman S W. Science. 1995;268:880–883. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 10.Caughey B, Chesebro B. Trends Cell Biol. 1997;7:56–62. doi: 10.1016/S0962-8924(96)10054-4. [DOI] [PubMed] [Google Scholar]
- 11.Glenner G G, Wong C W. Biochem Biophys Res Commun. 1984;122:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 12.Parsell D A, Kowal A S, Lindquist S. J Biol Chem. 1994;269:4480–4487. [PubMed] [Google Scholar]
- 13.Glover J R, Kowal A S, Schirmer E C, Patino M M, Liu J-J, Lindquist S. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 14.Mehlhorn I, Groth D, Stöckel J, Moffat B, Reilly D, Yansura D, Willett W S, Baldwin M, Fletlerick R, Cohen F E, Vandlen R, Henner D, Prusiner S B. Biochemistry. 1996;35:5528–5537. doi: 10.1021/bi952965e. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Stockel J, Mehlhorn I, Groth D, Baldwin M A, Prusiner S B, James T L, Cohen F E. Biochemistry. 1997;36:3543–3553. doi: 10.1021/bi961965r. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Kaneko K, Nguyen J T, Livshits T L, Baldwin M A, Cohen F E, James T L, Prusiner S B. J Mol Biol. 1995;250:514–526. doi: 10.1006/jmbi.1995.0395. [DOI] [PubMed] [Google Scholar]
- 17.Lanzetta P A, Alvarez L J, Reinach P S, Candia O A. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 18.Klunk W E, Pettegrew J W, Abraham D J. J Histochem Cytochem. 1989;37:1273–1279. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 19.Schirmer E C, Glover J R, Singer M A, Lindquist S. Trends Biochem Sci. 1996;21:289–296. [PubMed] [Google Scholar]
- 20.Wu C-S C, Chen G C. Anal Biochem. 1989;177:178–182. doi: 10.1016/0003-2697(89)90036-5. [DOI] [PubMed] [Google Scholar]
- 21.Maurizi M R, Thompson M W, Singh S K, Kim S-H. Methods Enzymol. 1994;244:314–331. doi: 10.1016/0076-6879(94)44025-5. [DOI] [PubMed] [Google Scholar]
- 22.Hwang B J, Woo K M, Goldberg A L, Chung C H. J Biol Chem. 1988;263:8727–8734. [PubMed] [Google Scholar]
- 23.Wawrzynow A, Wojtkowiak D, Marszalek J, Banecki B, Jonsen M, Graves B, Georgopoulos C, Zylicz M. EMBO J. 1995;14:1867–1877. doi: 10.1002/j.1460-2075.1995.tb07179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ter-Avanesyan M D, Kushnirov V V, Dagkesamanskaya A R, Didichenko S A, Chernoff Y O, Inge-Vechtomov S G, Smirnov V N. Mol Microbiol. 1993;7:683–692. doi: 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 25.King C Y, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. Proc Natl Acad Sci USA. 1997;94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prusiner S B, McKinley M P, Bowman K A, Bolton D C, Bendheim P E, Groth D F, Glenner G G. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 27.Gasset M, Baldwin M A, Lloyd D H, Gabriel J-M, Holtzman D M, Cohen F, Fletterick R, Prusiner S B. Proc Natl Acad Sci USA. 1992;89:10940–10944. doi: 10.1073/pnas.89.22.10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prusiner S B, Bolton D C, Groth D F, Bowman K A, Cochran S P, McKinley M P. Biochemistry. 1982;21:6942–6950. doi: 10.1021/bi00269a050. [DOI] [PubMed] [Google Scholar]
- 29.Caughey B W, Dong A, Bhat K S, Ernst D, Hayes S F, Caughey W S. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen J, Baldwin M A, Cohen F E, Prusiner S B. Biochemistry. 1995;34:4186–4192. doi: 10.1021/bi00013a006. [DOI] [PubMed] [Google Scholar]
- 31.Wickner S, Gottesman S, Skowyra D, Hoskins J, McKenney K, Maurizi M R. Proc Natl Acad Sci USA. 1994;91:12218–12222. doi: 10.1073/pnas.91.25.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DebBurman S K, Raymond G J, Caughey B, Lindquist S. Proc Natl Acad Sci USA. 1997;94:13938–13943. doi: 10.1073/pnas.94.25.13938. [DOI] [PMC free article] [PubMed] [Google Scholar]