

Abstract
The goal of this study was to design paclitaxel (PTX)-monoclonal antibody (MAb) prodrug conjugates (PTXMAbs) with the ability to deliver therapeutically significant doses of the drug to the tumor while avoiding the previously observed solubility limitations of conjugates with PTX : MAb molar ratios of >3. New PTX conjugates were synthesized using the discrete poly(ethylene glycol) (dPEG) as linkers. These compounds, PTX-L-Lys[(dPEG12)3-dPEG4]-dPEG6-NHS (9a and 9b, for L=GL or SX, respectively), were then conjugated to the anti-epidermal growth factor receptor MAb, C225 at increasing PTX : C225 ratios, producing completely soluble conjugates. Unlike the earlier PTXMAbs, buffered solutions of these conjugates remained homogeneous for extended periods of time. Fluorescence-activated cell sorting (FACS) analysis indicated preserved immunogenicity of the conjugates at all 4 substitution ratios, while cytotoxicity studies in MDA-MB-468 breast cancer cells indicated preservation of drug cytotoxicity. These conjugates may have potential in the development of high-drug-load tumor-targeting taxanes.
Introduction
Research on targeted therapy of malignancies has attracted a great deal of interest during the past several years based on its well-justified rationale, the efficient and specific delivery of cytotoxic agents to the tumor tissue. The immediate benefit of this tumor-specific delivery is two-fold: Increasing the effective dose of the antitumor agent, and decreasing the extent of its related side effects and toxicity. Receptor-based targeted treatment of cancer through the application of tumor-recognizing molecules (TRMs) has advanced considerably during the past several years with the development of monoclonal antibodies,1-4 and later, small molecule peptides capable of binding to tumor cell surface receptors.5, 6 A number of drug-,7-10 toxin-,11, 12 and radioisotope4, 13, 14 conjugates of TRMs have been developed with some currently in clinical use. A rationally designed conjugate capable of recognizing and localizing on its target, and delivering the cytotoxic payload in an efficient and timely fashion, will be an invaluable armament against cancer.
Since its discovery, paclitaxel (PTX, taxol, 1, Figure 1) has been the focus of much research towards the design and synthesis of analogues with improved physical, chemical, and pharmacological properties.15, 16 The first clinical trial of PTX for metastatic breast cancer was reported by Holmes et al. in 1991 and involved 25 patients treated with 250 mg/m2 over 24 h. Three complete and 11 partial responses for an overall response rate of 57% were observed;17 ever since, the drug has generated impressive responses from patients with metastatic disease such as the anthracycline-resistant breast cancer patients.18 Reichman et al. reported an overall response rate of 62% for 26 patients, with metastatic breast cancer and with prior adjuvant therapy, including 3 complete responses.19 Seidman et al. reported a 32.8% overall response in patients, 66% of whom had failed two or more prior treatments for metastatic disease.20 In a study by Chang et al., a 62% overall response was reported in patients treated with taxol.21
Figure 1.

Structure of paclitaxel
Despite these positive therapeutic features, taxanes suffer from such drawbacks as dose-limiting toxicities at the clinically administered quantities and lack of aqueous solubility. The negligible solubility of PTX (0.25 μg/mL in water) has led to the currently used formulations of this drug which have been reported to cause medium to severe hypersensitivity reactions.22, 23 A peripheral neuropathy is induced by PTX which becomes more severe by cumulative dosing and an arthralgia-myalgia syndrome results 2-5 days after administration of taxol.24 Other systemic toxicities include cardiac arrhythmias, alopecia, mucositis, and fatigue.25
Accordingly, design and development of water-soluble tumor-targeting taxane derivatives for excipient-free formulations, and with no or ameliorated human toxicity, is of great interest.
To this end, our laboratory previously reported the syntheses of bombesin receptor-directed PTX-peptide7, 26 and epidermal growth factor receptor-directed PTXMAb conjugates.27, 28 All conjugates showed an enhancement in the drug cytotoxicity presumably due to receptor-specific delivery, with small but significant antitumor activity as compared to the unconjugated drug.28 However, although the peptide conjugates, in which the drug and peptide moieties were connected by a PEG linker, showed high aqueous solubility, the solubility of the antibody conjugates were limited to a PTX : MAb molar ratio of 3.27 Conjugates with drug-to-MAb ratios (D : MAb) higher than 3 precipitated from the solution and were lost. At the same time, antitumor activity studies in animals showed that conjugates with higher drug loads were required for a significant therapeutic activity. This could be expected since with a large molecular weight (>155,000 Da), the antibody conjugate could deliver only a small dose of the drug (MW 853.9 Da) at a D : MAb of 3. Furthermore, the fractional tumor uptake of MAbs would be an added hurdle to the delivery of therapeutically significant doses of MAb-conjugated drugs.
In an attempt to overcome these shortcomings, we hypothesized that conjugation of a highly water-soluble PTX, designed with the appropriate functionality for MAb attachment, might yield high-drug-load (HDL) conjugates which would retain the native aqueous solubility of the antibody moiety. Furthermore, in contrast to the previously prepared low-drug-load compounds,27 the HDL conjugates would be expected to have enhanced antitumor efficacy as well. This article reports the design and synthesis of new and highly water-soluble PTX derivatives and their corresponding HDL antibody conjugates with sustained solubility, immunoreactivity, and cytotoxic activity.
Results
Two basic requirements for a successful tumor-targeted drug delivery are the preservation of the targeting element integrity and the delivery of a therapeutically significant dose of the drug to the intended site. In our earlier work, we reported the synthesis and evaluation of paclitaxel conjugates to the chimeric anti-epidermal growth factor receptor (EGFR) antibody C225 3, 29-32 for the targeted chemotherapy of EGFR-positive tumors.27, 28 In that work, PTX was linked to the antibody by the short and lipophilic linkers, succinic (SX) or glutaric (GL) acids. EGF is a transmembrane protein of the tyrosine kinase growth factor family and its receptor33, 34 is overexpressed in a number of human malignancies including breast, colorectal, ovarian, lung, and head and neck cancers.35-39
Despite the development of a successful and reproducible conjugation chemistry, the results indicated a solubility barrier which was consistently encountered beyond a PTX : MAb of 3. To circumvent this problem, the present approach employed a branched and highly water-soluble derivative of PTX containing an amine-directed conjugation site for MAb attachment.
Synthesis
The syntheses of the soluble pre-conjugation PTX (compounds 9a and 9b) are shown in Scheme 1. As the central branching point, Nα-Boc-protected lysine was used to provide three orthogonal coupling functionalities. The free ε-amine group of Boc-Lys was attached to the branched dPEG by an amide bond through the NHS-activated polymer 2. This reaction did not require any C-terminal protection of the amino acid which simplified the overall process. In order to further increase the water solubility of the conjugate, the free carboxylic acid of the intermediate compound 5 was then ligated to the dPEG hexamer 3, leading to compound 6. The MALDI mass spectra of compounds 5 and 6 showed MWs with 100 mass units less than calculated, indicating the cleavage of the Boc group (MW 101 Da), and regeneration of the corresponding amine, during the course of the spectroscopy. This was presumably due to the limited stability of this protecting group under high-energy conditions such as heating and laser irradiation.
Scheme 1.

Synthesis of water-soluble PTX-dPEG conjugates
(i) DCM/MT, RT; (ii) NHS, DCC, DCM, 3; (iii) TIPS/TFA; (iv) PTX-L-NHS, DIEA, DCM; (v) NHS, DCC, DCM
Acid deprotection of 6, followed by the coupling of compound 7 to PTXGLNHS or PTXSXNHS yielded products 8a or 8b, respectively. Activation of the carboxyl functions of 8a and 8b afforded the conjugation-ready activated esters 9a and 9b, respectively. The overall yields of these syntheses were 27% for 9a and 30% for 9b. The calculated and measured molecular weights of compounds 5 – 9, as well as the corresponding deviations, are shown in Table 1. Attempts to produce well-resolved and informative NMR spectra for these compounds were not successful due to high signal interference, large peak size differences, and crowded patterns of the corresponding spectra (see sample 1H-NMR of PTX, and dPEGs 2 and 3 in the Supporting Information). However, mass spectrometry and analytical HPLC clearly established the compounds identities and degrees of purity.
Table 1.
Calculated and measured molecular weights (MWs) of the dPEG linkers and PTX-dPEG conjugates.
| Compound | MWca | MWeb | %ΔMWc |
|---|---|---|---|
| 5 | 2552 | 2551 | 0.04 |
| 6 | 2887 | 2886 | 0.03 |
| 7 | 2787 | 2788 | 0.04 |
| 8a | 3737 | 3740 | 0.08 |
| 8b | 3723 | 3759 | 0.97 |
| 9a | 3834 | 3830 | 0.1 |
| 9b | 3820 | 3857 | 0.97 |
calculated molecular weight (MW)
experimental MW by MALDI-TOF MS
absolute value calculated from MWc-MWe/MWc • 100
The MALDI mass spectroscopic data for the intermediate and final products of Scheme 1 are shown in Table 1. Although the large size of the dPEG linkers used in this study necessitated the use of MALDI TOF MS, we found that some molecular fragmentation of the dPEGs could occur under the experimental conditions of this analytical modality. This fragmentation resulted in the formation of multi-peak spectral patterns of the intermediate and final products of Scheme 2 and was true even of the commercially supplied branched dPEG. This compound (2 of Figure 2, MW 2420) did not show the exact MW when analyzed by MALDI. When analyzed by positive-mode electrospray (ES) MS, however, the doubly charged MW was readily observed (see Supporting Information). This fragmentation, and not presence of impurities, explains the multi-peak patterns in MALDI spectra, although the observed MWs were in complete agreement with the calculated ones (Table 1). It should be emphasized that the ES MS could not be used for MWs larger than 1800 Da due to the inherent limitations of the instrument.
Scheme 2.

(i) 50 mM PBS, pH 8.1
Figure 2.

Structures of the single-MW dPEG branched (2) and linear (3) linkers used in the synthesis of the soluble PTX, 9.
Antibody conjugation
Targeting the lysine ε-amine groups of the antibody (10, Scheme 2), the activated esters 9a and 9b were readily conjugated to C225, at observed (MALDI) molar ratios which corresponded to the stoichiometrically calculated ratios, (PTX : C225)c. The product conjugates were purified by dialysis and the batch purity was evaluated by analytical SE-HPLC which indicated very high purities. This high degree of purity was due to the exhaustive dialysis in a large (50,000 Da) MW-cut-off membrane, which was expected to exclude any and all small-molecule starting material and reagents, and retain only the large protein molecules. The purified products were then analyzed by MALDI-TOF mass spectrometry which clearly showed the formation of the target conjugates in real time. Using the MALDI MWs of the whole conjugate, the unconjugated C225, and the substituting drug-linker conjugate, the PTX : C225 molar ratios were calculated through Equation 1.
| (eq. 1) |
where:
MWmconj, MWC225, and MWdconj are the MWs of the final MAb conjugate, C225, and drug conjugates (compounds 9a or 9b), respectively, and 115 is the MW of the departing NHS after MAb coupling.
The results of conjugation reactions are shown in Figure 3 and Table 2. As shown in this table, the measured PTX : C225 ratios, (PTX : C225)m, increased from 2 to 12 (11a-11d), and from 2 to 10 (11e-11h) when the (PTX : C225)c was raised from 5 to 50. The increase in the number of substituted drugs is also reflected in the SEC-HPLC retention times (tR) as indicated by an inverse correlation, shown in Figure 3 for the representative conjugates 11a-11d. A tR shortening of about 0.35 min was noticed for the conjugates with the highest PTX : C225 (compounds 11d and 11h, Scheme 2) as compared to the unconjugated antibody and due to a MW increase of about 37,000 to 47,000 Da. At the same time, conjugation efficiency (%CE, Table 2) was decreased, presumably due to the decrease in the availability of the lysine ε-amine binding sites of the antibody.
Figure 3.
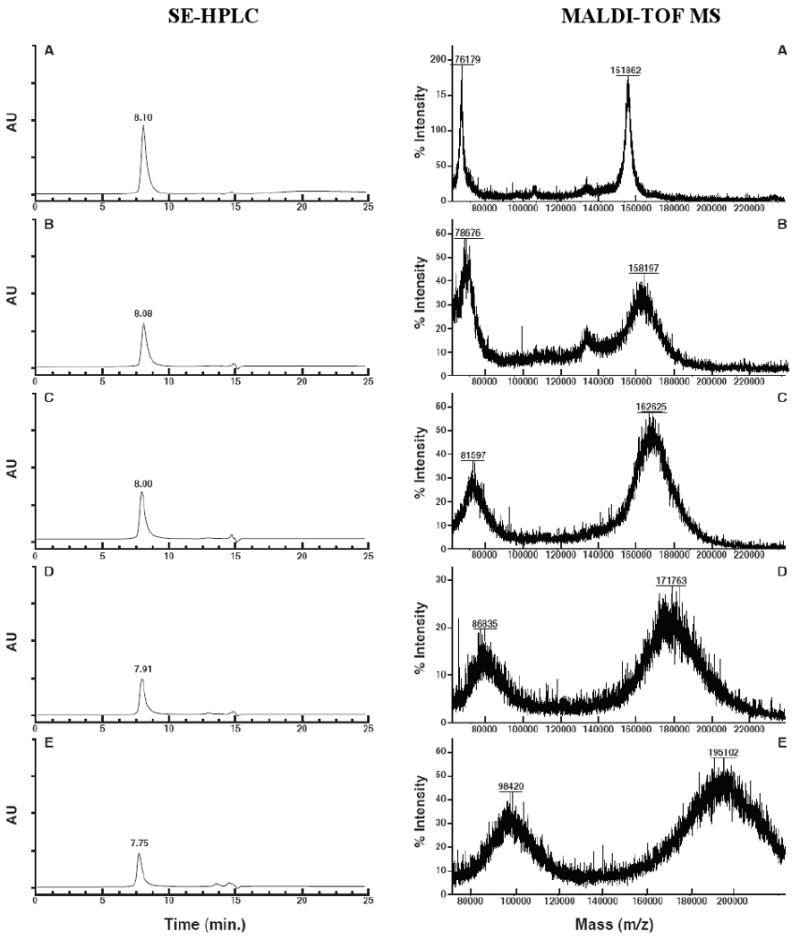
Size-exclusion chromatograms (SE-HPLC) and MALDI-TOF mass spectra of C225 (A) and conjugates 11a-11d (B-E) illustrating the degrees of purity and MWs for these compounds, respectively. SEC retention times decrease while the MALDI peaks broaden as the MW increases. The smaller left peaks in the mass spectra are the doubly charged half-MW peaks. All MALDI analyses were made with external calibration against bovine serum albumin at 66,430
Table 2.
Physicochemical properties of the 9a/9b-C225 conjugation reactions and their corresponding conjugate products. Values for the 9b-C225 conjugates are shown in parentheses.
| Conjugate | MWea | (PTX : C225)cb | (PTX : C225)mc | %yieldpd | %CEe | tR (min)f |
|---|---|---|---|---|---|---|
| 225 | 151,800 | 0 | 0 | NAg | NA | 8.1 |
| 11a (11e) | 158,298 (158,050) | 5 | 2 (2) | 77 (72) | 40 (60) | 8.08 (8.02) |
| 11b (11f) | 162,625 (169,414) | 10 | 3 (5) | 90 (86) | 30 (50) | 8.00 (8.00) |
| 11c (11g) | 171,763 (173,674) | 20 | 5 (6) | 88 (86) | 25 (35) | 7.91 (7.94) |
| 11d (11h) | 198,575 (188,800) | 50 | 12 (10) | 96 (92) | 24 (20) | 7.75 (7.77) |
measured by MALDI-TOF MS
calculated stoichiometric ratios
measured ratios from MALDI-TOF MWs and Equation 1
%yield of protein recovery
conjugation efficiency, calculated from (PTX : C225)m/(PTX : C225)c • 100
SEC-HPLC retention times at 280 nm
NA: not applicable
Furthermore, it should be noted that MALDI MS always yields broadened signals for MAbs. The reason is the existence of a combination of antibodies with different MWs but with the same antigen-binding activity. In fact, the term “monoclonal” refers to this latter property of MAbs and not to a MW homogeneity. This variation in molecular size, in turn, results in a variation in the response of individual molecules to conjugation reactions, thus leading to further peak broadening in the spectra of the product conjugates. This broadening usually increases with the conjugation number as is evident here in the spectra shown in Figure 3.
Whereas the conjugates with PTX : C225 of >3 invariably precipitated and were lost in the previously reported conjugations,27 no precipitation and loss of antibody was observed in this work: The yields of protein recovery ranged from 77% to 96%, and interestingly, with the latter yield belonging to the conjugates with the highest PTX : C225.
Aqueous solubility
During the course of this work, a number of potential water-soluble linkers were tested (Safavy, A., unpublished results). Among these, were the dendrimer polyamine PAMAM and linear amino PEG-carboxylic acids, with different molecular sizes and all suitably heterofunctionalized for MAb conjugation. Although the search for the right linker was not exhaustive, it was observed that even with these inherently hydrophilic linkers, the PTX loading of >3-5 would result in the precipitation of the resulting conjugates. In general, when the linker was placed in a “middle” position, that is, flanked by PTX on one side and another relatively large molecule such as PAMAM or a MAb on the other, the overall solubility was lost beyond the 3-PTX substitution number. The other alternative, the use of a solubilizing linker as a pendant group with only one end attached to the rest of the molecule and possessing a branched structure was, therefore, considered. An expected advantage of a branched design was to provide a larger “hydrophilic sphere” in contrast to a long straight-chain solubilizer of the same molecular weight. These considerations led to the use of the branched and single-MW (discrete) PEG (dPEG) 2, shown in Figure 2. The rationale for using a dPEG, as opposed to the conventional polydispersed PEGs, was the possibility of exact stoichiometric calculations. A single-MW preconjugation PTX would make possible the execution of controlled conjugation reactions with known D : MAb molar ratios. As it turned out, this design of the PTX derivatives 9a and 9b did indeed produce completely water-soluble, high-drug-load antibody conjugates. Refrigerated buffered solutions of the conjugates (Scheme 2, compounds 11a-11h) remained indefinitely clear and homogeneous.
Cell binding activity
An important consideration in the practice of MAb conjugation is the preservation of the immunoreactivity (IR) of the antibody as a direct factor in target recognition. An impaired IR would lead to reduced tumor-delivered doses of the drug, and therefore, to reduced therapeutic efficacies. As the preservation of the IR is expected to be inversely proportional to the conjugation degree, the cell binding activity of these high-load conjugates were of primary interest. FACS analysis was performed on each conjugate to compare their binding ability to MDA-MB-468 human breast carcinoma cells as compared to the unconjugated antibody C225. This experiment was carried out with the conjugates of the glutarate derivative 9a as the main candidate for in vivo studies (see Cytotoxicity section, below). As shown in Figure 4, in all compounds, conjugation of compound 9a at different D : MAb ratios, had no detectable effect on the IR of the antibody. All conjugates did bind to the cell surface EGFRs, to the same extent as did the parent antibody. Any reduction in the antigen binding ability of the conjugate would have lead to a shift in the location of the corresponding fluorescence peak with respect to that of the unconjugated C225 peak (panel B of Figure 4).
Figure 4.

FACS analyses of the unconjugated C225 (panel B) and PTX conjugates 11a-11d (panels C through F, respectively) against MDA-MB-468 human breast carcinoma cells. All conjugates showed a significant shift in the light intensity compared to a no-MAb run (panel A) indicating the preserved immunoreactivity of the antibody.
Cytotoxicity
Glutaric acid (GL) and succinic acid (SX) are the most frequently used linkers for the attachment of PTX to antibodies, peptides, and polymers. We have shown previously that under physiologic conditions, the GL linkage was about 17 times more stable against hydrolysis, and thus PTX release, than was the SX linkage.28 In addition to the direct kinetics experiments, this difference in stability was shown by the negligible in vitro cell killing activity of the PTXGLC225 when the drug exposure time was 24 h (Safavy, unpublished results). On the other hand, the in vivo activity observed for PTXGLC225 was absent in the case of PTXSXC225, presumably due to a rapid release and excretion of the drug which occurred prior to its peak tumor uptake.28 Accordingly, we proposed the use of GL linkers for PTX conjugates intended for systemic (in vivo) applications while SX was suggested for in vitro studies.
The linker effect was confirmed also in the present study when only negligible cytotoxicity was observed for the GL-linked conjugates 11a-11d (data not shown), although the FACS studies had demonstrated the preservation of the immunoreactivity of the C225 moiety. To extend the validity of the previous findings on linker-cytotoxicity relationships to the conjugates of the present study, we also synthesized the SX-linked versions of PTXdPEGC225 conjugates solely for cytotoxicity evaluation. The synthesis was carried out through the same method as for the GL conjugates and as shown in Schemes 1 and 2, leading to the preparation of conjugates 11e-11h. As expected, all SX conjugates showed cytotoxic activity against MDA-MB-468 human breast carcinoma cell line in a cell proliferation assay. In this assay, the cells were treated with either the blank medium, free PTX, or each of the 11e-11h conjugates for 24 h, followed by removal of the treatments and continuation of the incubation for an additional 72 h. An equimolar dose of PTX was used in all treated groups. Viable cells were then counted, and the cell numbers in each group was normalized against the untreated controls. The conjugates showed the same cytotoxic activity as the unconjugated drug, indicating complete preservation of PTX activity. These results are shown in Figure 5.
Figure 5.
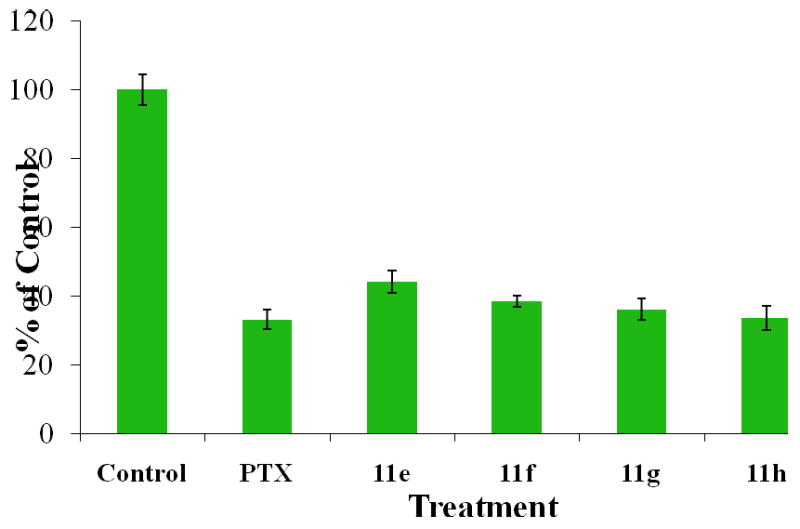
Cell proliferation inhibition in MDA-MB-468 human breast carcinoma cell line treated with either free PTX or each of the 11e-11h conjugates at 10 nM drug equivalent concentration and with a 24-h exposure time. Surviving cell numbers were normalized against the untreated controls.
Conclusions
In an effort to produce high-drug-load, yet water-soluble, paclitaxel conjugates for tumor-targeted chemotherapy of cancer, novel derivatives of the antitumor drug paclitaxel (PTX, taxol) were synthesized and were conjugated to the chimeric anti-EGFR MAb, C225. The conjugation number increased in direct proportion to the increasing drug stoichiometry, reaching a D : MAb ratio of 12 and 10 for the two highest load compounds. Unlike the previously reported low-drug-load conjugates with limited aqueous solubility at D : MAb of >3, these conjugates were highly soluble in physiologic buffers at ambient and refrigerated temperatures, and thus, may be suitable for excipient-free clinical formulations. As evidenced by FACS analysis, conjugation of these compounds did not alter the immunoreactivity of the parent antibody at up to a PTX : MAb molar ratios of 12. The retention of the drug cytotoxicity was also demonstrated through cell proliferation assays against a human breast cancer cell line. To our knowledge, no other PTXMAb conjugates with these high D : MAb ratios have been previously reported. With the ability to carry, target, and deliver a therapeutically significant load of the drug, these compounds may have potential in the development of tumor-targeting PTX conjugates for clinical applications.
Experimental Section
General information
Paclitaxel hemi-glutarate and hemi-succinate, and their corresponding N-hydroxysuccinimide esters (PTXGLNHS and PTXSXNHS) were prepared by the methods of Deutsch et al.40 and Luo and Prestwich,41 respectively. C225 (Erbitux®) was generously provided by ImClone Systems (New York, NY). Discrete PEG (dPEG) linkers (Figure 2, compounds 2 and 3) were purchased from Quanta BioDesign, Ltd. (Powell, OH). Buffers and aqueous solvents were prepared with metal-free purified water (18.2 MΩ) obtained from a Milli-QF system (Millipore, Bedford, MA). Dry solvents were of Sigma-Aldrich (Milwaukee, WI) anhydrous grade which were kept over 4 Å molecular sieves before use. All compounds used in this work were of ≥95% purity. Measures of purities were provided by the manufacturers for the commercially supplied compounds. The purities of the intermediate and final product organic compounds were determined by thin-layer chromatography (TLC) (for non-uv-active compounds) or by reversed-phase (RP) HPLC and TLC (for uv-active compounds). The purities of the drug-antibody conjugates were determined by size-exclusion (SE) HPLC. TLC analyses were performed on aluminum backed silica gel 60 F254 plates (Merck, Darmstadt, Germany), eluted with 15 : 1 : 84, v/v, methanol : HOAc : chloroform. TLC results are reported for each compound in the form of the corresponding Rf values with the corresponding purities shown in parentheses. Products 5 and 6, and 7, were visualized by iodine (I2) and ninhydrine staining, respectively. Products 8a, 8b, 9a, and 9b were detected by both uv light and I2 staining. RP-HPLC was performed with a Beckman System Gold instrument operated by Beckman 32 Karat Version 5.0 software and equipped with a System Gold 166 uv/vis detector (Beckman Coulter, Fullerton, CA) at 254 nm. Analytical and preparative RP-HPLC was performed in 4.6 mm × 250 mm, C18, and 22 mm × 250 mm, C8 columns with 5 μm particle size (GraceVydac, Deerfield, IL), respectively. Elution solvents were 0.1% TFA : H2O (solvent A) and 0.1 : 60 : 40, TFA : CH3CN : H2O, v/v, (solvent B). A solvent B gradient of 10% - 90% was used in each run and within 20 min (analytical runs) or 90 min (preparative runs). SEC-HPLC was performed with a BioRad model 5000 Titanium System (BioRad, Richmond, CA) equipped with a model 1806 UV/Vis detector at 280 nm. For analytical SEC-HPLC, a Biosil 7.8 mm × 300 mm column (BioRad) equipped with a Biosil 7.8 mm × 80 mm guard column was used. A 10 mM, phosphate buffer containing 300 mM NaCl, 10 mM Na2SO4, and 10% (v/v) DMSO, was used as solvent. Protein concentrations were determined using a BCA protein assay kit (Thermo Scientific, Rockford, IL) according to the manufacturer protocol. FACS analyses were performed on a Becton Dickinson FACScan (BD Biosciences, San Jose, CA) using CellQuest v.3.1 software. Fluorescein-labeled mouse anti-human secondary antibody (Invitrogen, Frederick, MD) was used as the secondary antibody and was excited by 488 nm blue laser followed by detection in FL1 channel on a log scale of mean fluorescence intensity.
MALDI-TOF mass spectrometry
For compounds 5-9 the MALDI MS analyses were performed with a Voyager Elite mass spectrometer in positive mode and with delayed extraction technology (PerSeptive Biosystems, Framingham, MA). Sinapinic acid was used as matrix, and samples were prepared in a 50 : 50 (v/v) mixture of 0.1% TFA/acetonitrile. One μL of the prepared sample was then spotted onto MALDI stainless steel target and air dried before analysis. A 1-pmol/L solution of bovine serum albumin was used as internal standard.
For C225 and all antibody conjugates MALDI MS was performed with a Bruker Reflex™ III MALDI-TOF with a linear detector and in positive mode. The protein solution was mixed with sinapinic acid matrix solution (6 mg/mL, 50 : 50 CAN : H2O, v/v, containing 0.1% TFA) at proper ratios depending on the sample concentration. One μL of the prepared sample was then spotted onto MALDI stainless steel target and air dried before analysis. External calibration was performed with bovine serum albumin with [M+H] average mass (m/z) of 66430.
Boc-Lys[(dPEG12)3-dPEG4]-OH, 5
To the solution of dPEG 2 (450 mg, 186 μmol) in DCM was added a solution of Boc-lysine (4, 54.5 mg, 221.3 μmol) in three parts of 43.5 mg, 5 mg, and 6 mg, within 24 h, under argon and with stirring. The complete disappearance of the starting material 2, as monitored by TLC, was observed after an additional 48 h. The solvents were distilled in vacuum and the residual oil was re-dissolved in 10 mL of purified water. The turbid mixture was transferred into a dialysis membrane and was dialyzed first against 2 L of 150 mM chloride buffer for 2 h, and then in water (3 × 2 L) for 5 h, both at 4 °C. Lyophilization of the clear solution afforded the product 5 as a light yellow oil. Yield: 366 mg, 74%. Rf: 0.62 (I2, one spot). MALDI MS: (M+H)+-Boc, 2551-100 = 2451 (calc. 2552).
Boc-Lys[(dPEG12)3-dPEG4]-dPEG6-OH, 6
Conjugate 5 (351 mg, 132.5 μmol) was dissolved in DCM and a solution of NHS (61 mg, 530 μmol) in THF was added under argon. After cooling the mixture in ice, a solution of DCC (34.25 mg, 166 μmol) in DCM was added in several portions. The ice bath was removed and the reaction mixture was stirred at ambient temperature for 48 h. The solid precipitate was filtered off and the solvents were distilled in vacuum to a viscous clear oil.
The oily activated ester was re-dissolved in dry DMF (1 mL) and a solution of the dPEG6 (3, 51.5 mg, 145.7 μmol) in MTL (800 μL), containing 38.2 μL (219 μmol) DIEA, was slowly added. A second portion of 3 (5.5 mg, 15.6 μmol) in 500 μL of MTL was added after 18 h. After another 3 hours, the solvents were removed in vacuum and the residual oil was re-dissolved in 2 mL of 50 mM, acetate buffer at pH 4.1. The solution was dialyzed against 3 × 2 L of water, at 4 °C and for 20 h. The slight quantity of particulate mass was removed by centrifugation and the clear solution was lyophilized to afford 6 as a highly viscous clear oil. Yield: 244 mg (61%). Rf: 0.6 (I2, product + one minor spot). MALDI MS: (M+H)+-Boc, 2886-100 = 2786 (calc. 2887).
H-Lys[(dPEG12)3-dPEG4]-dPEG6-OH•TFA, 7
To an argon-purged ice-cold round-bottomed flask containing conjugate 6 (230 mg, 77 μmol) was added 5 mL of a mixture of TIPS and TFA (5 : 95, v/v) and the solution was incubated at ambient temperature for 30 min. The solvents were removed in vacuum, followed by addition and distillation of 10 mL of CHL. The oily product 7 was kept under high vacuum for 12 h. Yield: 224 mg (100%). Rf: 0.3 (ninhydrin, product + one minor spot). MS: (M+H)+ 2788 (calc. 2787).
PTXGL-Lys[(dPEG12)3-dPEG4]-dPEG6-OH, 8a
The amine salt 7 (223.4 mg, 77 μmol) was dissolved in DCM (5 mL) and solution of PTXGLNHS or PTXSXNHS (93.2 mg, 87.5 μmol) in 2 mL dry DCM was added under argon. DIEA (200 μmol) was added in 30-μL portions to raise the pH of the reaction to ∼pH 8, as tested on a wet pH paper. The clear solution was stirred at ambient temperature for 12 h. Solvent was removed in vacuum and the crude product was purified by column chromatography in a 2.3 Cm × 12 Cm column eluted with 0.1% HOAc in chloroform and a 0% - 7% MTL gradient. The isolated product was re-dissolved in ACN : water (70 : 30, v/v) followed by lyophilization, to remove traces of HOAc and to afford a semi-solid product. Yield: 297 mg (62%). Rf: 0.79 (uv and I2, one spot). tR (RP-HPLC) 23.5 min (>98%). MALDI MS: (M+H)+ 3740 (calc. 3737).
PTXSX-Lys[(dPEG12)3-dPEG4]-dPEG6-OH, 8b
The same procedure as for 8a was used except for the use of PTXSXNHS (92 mg, 87.5 μmol). Yield: 215 mg (73%). Rf: 0.71 (uv and I2, one spot). tR (RP-HPLC) 23.4 min (98.7%). MALDI MS: (M+H)+ 3759 (calc. 3723).
PTXGL-Lys[(dPEG12)3-dPEG4]-dPEG6-NHS, 9a
A solution of NHS (19,94 mg, 173.3 μmol) in 500 μL THF was added to that of 8a (297 mg, 75.2 μmol) in 5 mL DCM under argon, followed by the addition of DCC (24.5 mg, 118.4 μmol) in DCM (1 mL). The solution was stirred at room temperature overnight producing a white solid precipitate. A second portion of DCC (10 mg, 48.5 μmol) in 300 μL DCM was added and the reaction was stirred at ambient temperature for an additional 48 h. The solvents were distilled in vacuum, the semi-solid residue was treated with a mixture of ACN : WTR (60 : 40, v/v), and the solid precipitate was removed by centrifugation. The product 9a was purified by preparative RP-HPLC. Yield: 140 mg (47%). Rf: 0.65 (uv and I2, one spot). tR (RP-HPLC) 24.16 min (>98%). MALDI MS: (M+H)+ 3830 (calc. 3834).
PTXSX-Lys[(dPEG12)3-dPEG4]-dPEG6-NHS, 9b
The same procedure as for 9a was used except for the use of 8b (287.5 mg, 75.2 μmol). Yield: 268.3 mg (91%). Rf: 0.54 (uv and I2, one spot). tR (RP-HPLC) 23.18 min (96.4%). MALDI MS: (M+H)+ 3857 (calc. 3820).
General procedure for antibody conjugation
The antibody solution was buffer-exchanged by filtration in Centricon-50 centrifuge funnels (Millipore, Bedford, MA) at 4,000 rpm, into an 8.1 mM PBS at pH 8.1, and the protein concentration was measured. For each conjugation, 600 μL of the antibody solution containing 2 mg (1.3 × 10-5 mmol) was chilled in an ice bath and a measured amount of 9a or 9b in the same buffer was added with gentle stirring at 0 °C. The calculated 9a/9b : C225 molar ratios were 5, 10, 20, and 50. After 1 h, the solution was dialyzed in a 50-K MWCO membrane against 4 × 2 L of DPBS for 12 h at 4 °C. The product was analyzed for purity by SEC-HPLC. Formation of the product conjugates and the number of substituted drugs per C225 were determined by MALDI-TOF MS and using Equation 1.
Cell proliferation assays
MDA-MB-468 human breast carcinoma cells (American Type Culture Collection, Manassas, VA) were maintained as monolayers in 75-cm tissue culture flasks using their respective cell culture medium containing 10% fetal bovine serum and 2 mM L-glutamine. Incubation was at 37 °C under a humidified 5% CO2 : air atmosphere (standard conditions) for five days. The cells were harvested when in mid-log growth and their concentration was determined using a particle counter (Beckman Coulter, Inc, Fullerton, CA). An aliquot of the cell suspension was diluted in culture medium for delivery to a 24-well tissue culture plate at a range of 10,000 to 30,000 per mL per well. After 24 h, quadruplicate wells were inoculated with either the medium as untreated controls, or a test compound at a 10-nM drug equivalent concentration. After 24 h of incubation, the wells were aspirated, washed once with 1 mL PBS, and were refilled with 1 mL treatment-free medium. Following a 96-h post-treatment incubation under standard conditions, the viable cells were counted and the numbers were normalized to the number of untreated controls. The extent of cytotoxicity in treated wells as compared to the controls was calculated using Microsoft Excel software program (Microsoft Corp., Redmond, WA).
Supplementary Material
Acknowledgments
This work was supported in part through grants from the Susan G. Komen for the Cure (AS, Award Number BCTR0707430), Department of Defense (DOD) Breast Cancer Research Program (AS, Award Number DAMD17-02-1-0268), and DOD Prostate Cancer Research Program (AS, Award Number DAMD17-02-1-0004). The UAB Flow Cytometry Core Facility was funded by NIH P30 grant Number AR48311. The authors acknowledge the assistance of D. Ray Moore and Landon Wilson (UAB Mass Spectroscopy Shared Facility) in the molecular weight determinations, and of Enid Keyser (FACS Core Facility) in the antibody binding activity analyses. Contribution of Dr. Qiaoli Liang (University of Alabama, Chemistry Mass Spectrometry Facility) in the determination of antibody conjugates molecular weights and formatting of the related data is greatly acknowledged. Sally Lagan and David Fisher are thanked for preparing the manuscript and preparation of figures, respectively.
Footnotes
Abbreviations. ACN, acetonitrile; CAN, acetonitrile; CHL, chloroform; DCC, N, N-dicyclohexyl carbodiimide; DCM, dichloromethane; DIEA, N, N-diisopropyl ethylamine; dPEG, discrete PEG; EGFR, epidermal growth factor receptor; FACS, fluorescent-activated cell sorting; FITC, fluorescein isothiocyanate; GL, glutarate or glutaric acid; MAb, monoclonal antibody; MTL, methanol; NHS, N-hydroxy succinimide; PTX, paclitaxel; RP, reversed-phase; SE-HPLC, size-exclusion HPLC; SX, succinate or succinic acid; TIPS, triisopropylsilane; WTR, water
Supporting Information Available: Comparative 1H-NMR for paclitaxel, polymers 2 and 3, and product 9a, mass spectra, and HPLC charts. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kohler GMC. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 2.Goodwin DA. Stategies for antibody targeting. Antib Immunoconj Radiopharm. 1991;4:427–434. [Google Scholar]
- 3.Mamot C, Drummond DC, Noble CO, Kallab V, Guo Z, Hong K, Kirpotin DB, Park JW. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65:11631–11638. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- 4.Richman CM, Denardo SJ, O'Donnell RT, Yuan A, Shen S, Goldstein DS, Tuscano JM, Wun T, Chew HK, Lara PN, Kukis DL, Natarajan A, Meares CF, Lamborn KR, DeNardo GL. High-dose radioimmunotherapy combined with fixed, low-dose paclitaxel in metastatic prostate and breast cancer by using a MUC-1 monoclonal antibody, m170, linked to indium-111/yttrium-90 via a cathepsin cleavable linker with cyclosporine to prevent human anti-mouse antibody. Clin Cancer Res. 2005;11:5920–5927. doi: 10.1158/1078-0432.CCR-05-0211. [DOI] [PubMed] [Google Scholar]
- 5.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 6.Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. CA Cancer J Clin. 2009;59:111–137. doi: 10.3322/caac.20003. [DOI] [PubMed] [Google Scholar]
- 7.Safavy A, Raisch KP, Khazaeli MB, Buchsbaum DJ, Bonner JA. Paclitaxel derivatives for targeted therapy of cancer: Toward the development of smart taxanes. J Med Chem. 1999;42:4919–4924. doi: 10.1021/jm990355x. [DOI] [PubMed] [Google Scholar]
- 8.Tolcher AW, Sugarman S, Gelmon KA, Cohen R, Saleh M, Isaacs C, Young L, Healey D, Onetto N, Slichenmyer W. Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J Clin Oncol. 1999;17:478–484. doi: 10.1200/JCO.1999.17.2.478. [DOI] [PubMed] [Google Scholar]
- 9.Ojima I. Guided molecular missiles for tumor-targeting chemotherapy--case studies using the second-generation taxoids as warheads. Acc Chem Res. 2008;41:108–119. doi: 10.1021/ar700093f. [DOI] [PubMed] [Google Scholar]
- 10.Singh Y, Palombo M, Sinko PJ. Recent trends in targeted anticancer prodrug and conjugate design. Curr Med Chem. 2008;15:1802–1826. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akamatsu Y, Murphy JC, Nolan KF, Thomas P, Kreitman RJ, Leung SO, Junghans RP. A single-chain immunotoxin against carcinoembryonic antigen that suppresses growth of colorectal carcinoma cells. Clin Cancer Res. 1998;4:2825–2832. [PubMed] [Google Scholar]
- 12.Kreitman RJ, Wang QC, FitzGerald DJ, Pastan I. Complete regression of human B-cell lymphoma xenografts in mice treated with recombinant anti-CD22 immunotoxin RFB4(dsFv)-PE38 at doses tolerated by cynomolgus monkeys. Int J Cancer. 1999;81:148–155. doi: 10.1002/(sici)1097-0215(19990331)81:1<148::aid-ijc24>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 13.Safavy A, Smith DC, Jr, Bazooband A, Buchsbaum DJ. De novo synthesis of a new diethylenetriaminepentaacetic acid (DTPA) bifunctional chelating agent. Bioconjug Chem. 2002;13:317–326. doi: 10.1021/bc0100861. [DOI] [PubMed] [Google Scholar]
- 14.Sharkey RM, Goldenberg DM. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J Clin. 2006;56:226–243. doi: 10.3322/canjclin.56.4.226. [DOI] [PubMed] [Google Scholar]
- 15.Geney R, Chen J, Ojima I. Recent advances in the new generation taxane anticancer agents. Med Chem. 2005;1:125–139. doi: 10.2174/1573406053175292. [DOI] [PubMed] [Google Scholar]
- 16.Huang Q, Kirikae F, Kirikae T, Pepe A, Amin A, Respicio L, Slayden RA, Tonge PJ, Ojima I. Targeting FtsZ for antituberculosis drug discovery: noncytotoxic taxanes as novel antituberculosis agents. J Med Chem. 2006;49:463–466. doi: 10.1021/jm050920y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes FA, Walters RS, Theriault RL, Forman AD, Newton LK, Raber MN, Buzdar AU, Frye DK, Hortobagyi GN. Phase II trial of taxol, an active drug in the treatment of metastatic breast cancer. J Natl Cancer Inst. 1991;83:1797–1805. doi: 10.1093/jnci/83.24.1797-a. [DOI] [PubMed] [Google Scholar]
- 18.Robert N, Leyland-Jones B, Asmar L, Belt R, Ilegbodu D, Loesch D, Raju R, Valentine E, Sayre R, Cobleigh M, Albain K, McCullough C, Fuchs L, Slamon D. Randomized phase III study of trastuzumab, paclitaxel, and carboplatin compared with trastuzumab and paclitaxel in women with HER-2-overexpressing metastatic breast cancer. J Clin Oncol. 2006;24:2786–2792. doi: 10.1200/JCO.2005.04.1764. [DOI] [PubMed] [Google Scholar]
- 19.Reichman BS, Seidman AD, Crown JP, Heelan R, Hakes TB, Lebwohl DE, Gilewski TA, Surbone A, Currie V, Hudis CA. Paclitaxel and recombinant human granulocyte colony-stimulating factor as initial chemotherapy for metastatic breast cancer. J Clin Oncol. 1993;11:1943–1951. doi: 10.1200/JCO.1993.11.10.1943. [DOI] [PubMed] [Google Scholar]
- 20.Seidman AD, Reichman BS, Crown JP, Yao TJ, Currie V, Hakes TB, Hudis CA, Gilewski TA, Baselga J, Forsythe P. Paclitaxel as second and subsequent therapy for metastatic breast cancer: Activity independent of prior anthracycline response. J Clin Oncol. 1995;13:1152–1159. doi: 10.1200/JCO.1995.13.5.1152. [DOI] [PubMed] [Google Scholar]
- 21.Chang AY, Boros L, Asbury R, Hui L, Rubins J. Dose-escalation study of weekly 1-hour paclitaxel administration in patients with refractory cancer. Semin Oncol. 1997;24:S17-69–S17-71. [PubMed] [Google Scholar]
- 22.Dorr RT. Pharmacology and toxicology of Cremophor EL diluent. Ann Pharmacother. 1994;28:S11–S14. doi: 10.1177/10600280940280S503. [DOI] [PubMed] [Google Scholar]
- 23.Sharma A, Mayhew E, Bolcsak L, Cavanaugh C, Harmon P, Janoff A, Bernacki RJ. Activity of paclitaxel liposome formulations against human ovarian tumor xenografts. Int J Cancer. 1997;71:103–107. doi: 10.1002/(sici)1097-0215(19970328)71:1<103::aid-ijc17>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 24.Payne JY, Holmes F, Cohen PR, Gagel R, Buzdar A, Dhingra K. Paclitaxel: Severe mucocutaneous toxicity in a patient with hyperbilirubinemia. South Med J. 1996;89:542–545. doi: 10.1097/00007611-199605000-00022. [DOI] [PubMed] [Google Scholar]
- 25.Rowinsky EK, Donehower RC. Paclitaxel (taxol) N Engl J Med. 1995;332:1004–1014. doi: 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- 26.Safavy A, Raisch KP, Matusiak D, Bhatnagar S, Helson L. Single-drug multiligand conjugates: Synthesis and preliminary cytotoxicity evaluation of a paclitaxel-dipeptide “scorpion” molecule. Bioconjug Chem. 2006;17:565–570. doi: 10.1021/bc050224c. [DOI] [PubMed] [Google Scholar]
- 27.Safavy A, Bonner JA, Waksal H, Buchsbaum DJ, Gillespie GY, Khazaeli MB, Arani RB, Chen DT, Carpenter M, Raisch KP. Synthesis and biological evaluation of paclitaxel-C225 conjugate as a model for targeted drug delivery. Bioconjug Chem. 2003;14:302–310. doi: 10.1021/bc020033z. [DOI] [PubMed] [Google Scholar]
- 28.Safavy A, Georg GI, Vander Velde D, Raisch KP, Safavy K, Carpenter M, Wang W, Bonner JA, Khazaeli MB, Buchsbaum DJ. Site-specifically traced drug release and biodistribution of a paclitaxel-antibody conjugate toward optimization of the linker structure. Bioconjug Chem. 2004;15:1264–1274. doi: 10.1021/bc049868v. [DOI] [PubMed] [Google Scholar]
- 29.Naramura M, Gillies SD, Mendelsohn J, Reisfeld RA, Mueller BM. Therapeutic potential of chimeric and murine anti-(epidermal growth factor receptor) antibodies in a metastasis model for human melanoma. Cancer Immunol Immunother. 1993;37:343–349. doi: 10.1007/BF01518458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res. 1995;1:1311–1318. [PubMed] [Google Scholar]
- 31.Karashima T, Sweeney P, Slaton JW, Kim SJ, Kedar D, Izawa JI, Fan Z, Pettaway C, Hicklin DJ, Shuin T, Dinney CP. Inhibition of angiogenesis by the antiepidermal growth factor receptor antibody ImClone C225 in androgen-independent prostate cancer growing orthotopically in nude mice. Clin Cancer Res. 2002;8:1253–1264. [PubMed] [Google Scholar]
- 32.Reusch U, Sundaram M, Davol PA, Olson SD, Davis JB, Demel K, Nissim J, Rathore R, Liu PY, Lum LG. Anti-CD3 × anti-epidermal growth factor receptor (EGFR) bispecific antibody redirects T-cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. Clin Cancer Res. 2006;12:183–190. doi: 10.1158/1078-0432.CCR-05-1855. [DOI] [PubMed] [Google Scholar]
- 33.Thompson DM, Gill GN. The EGF receptor: Structure, regulation and potential role in malignancy. Cancer Surv. 1985;4:767–788. [PubMed] [Google Scholar]
- 34.Schlessinger J. The epidermal growth factor receptor as a multi-functional allosteric protein. Biochem. 1988;27:3119–3123. doi: 10.1021/bi00409a002. [DOI] [PubMed] [Google Scholar]
- 35.Carpenter G, Cohen S. Epidermal growth factor. Ann Rev Biochem. 1979;48:193–216. doi: 10.1146/annurev.bi.48.070179.001205. [DOI] [PubMed] [Google Scholar]
- 36.Pathak MA, Matrisian LM, Magun BE, Salmon SE. Effect of epidermal growth factor on clonogenic growth of primary human tumor cells. Int J Cancer. 1982;30:745–750. doi: 10.1002/ijc.2910300611. [DOI] [PubMed] [Google Scholar]
- 37.Singletary SE, Baker FL, Spitzer G, Tucker SL, Tomasovic B, Brock WA, Ajani JA, Kelly AM. Biological effect of epidermal growth factor on the in vitro growth of human tumors. Cancer Res. 1987;47:403–406. [PubMed] [Google Scholar]
- 38.Real PJ, Benito A, Cuevas J, Berciano MT, de Juan A, Coffer P, Gomez-Roman J, Lafarga M, Lopez-Vega JM, Fernandez-Luna JL. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res. 2005;65:8151–8157. doi: 10.1158/0008-5472.CAN-05-1134. [DOI] [PubMed] [Google Scholar]
- 39.Reilly RM, Chen P, Wang J, Scollard D, Cameron R, Vallis KA. Preclinical pharmacokinetic, biodistribution, toxicology, and dosimetry studies of 111In-DTPA-human epidermal growth factor: an auger electron-emitting radiotherapeutic agent for epidermal growth factor receptor-positive breast cancer. J Nucl Med. 2006;47:1023–1031. [PubMed] [Google Scholar]
- 40.Deutsch H, Glinski J, Hernandez M, Haugwitz R, Narayanan V, Suffness M, Zalkow L. Synthesis of congeners and prodrugs: Water soluble prodrugs of taxol with potent antitumor activity. J Med Chem. 1989;32:788–792. doi: 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]
- 41.Luo Y, Prestwich GD. Synthesis and selective cytotoxicity of a hyaluronic acid-antitumor bioconjugate. Bioconjug Chem. 1999;10:755–763. doi: 10.1021/bc9900338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.