

Abstract
Glutaredoxins are small, heat-stable proteins that exhibit a characteristic thioredoxin fold and a CXXC/S active-site motif. A variety of glutathione (GSH)-dependent catalytic activities have been attributed to the glutaredoxins, including reduction of ribonucleotide reductase, arsenate, and dehydroascorbate; assembly of iron sulfur cluster complexes; and protein glutathionylation and deglutathionylation. Catalysis of reversible protein glutathionylation by glutaredoxins has been implicated in regulation of redox signal transduction and sulfhydryl homeostasis in numerous contexts in health and disease. This forum review is presented in two parts. Part I is focused primarily on the mechanism of the deglutathionylation reaction catalyzed by prototypical dithiol glutaredoxins, especially human Grx1 and Grx2. Grx-catalyzed protein deglutathionylation proceeds by a nucleophilic, double-displacement mechanism in which rate enhancement is attributed to special reactivity of the low pKa cysteine at its active site, and to increased nucleophilicity of the second substrate, GSH. Glutaredoxins (and Grx domains) have been identified in most organisms, and many exhibit deglutathionylation or other activities or both. Further characterization according to glutathionyl selectivity, physiological substrates, and intracellular roles may lead to subclassification of this family of enzymes. Part II presents potential mechanisms for in vivo regulation of Grx activity, providing avenues for future studies. Antioxid. Redox Signal. 11, 1059–1081.
Part I: Glutaredoxins and Catalysis of Thiol-Disulfide Exchange
Glutaredoxins are GSH-disulfide oxidoreductases reported to catalyze a variety of GSH-dependent thiol-disulfide exchange reactions including protein glutathionylation and deglutathionylation, turnover of ribonucleotide reductase, and reduction of dehydroascorbate and arsenate; and some glutaredoxins are also implicated in FeS cluster homeostasis (reviewed in refs. 68, 80, 81). Among the reported catalytic activities of the glutaredoxins, protein deglutathionylation (reduction of protein-glutathione mixed disulfides, protein-SSG) has received much attention because of its regulatory roles in redox signal transduction and sulfhydryl homeostasis (reviewed in refs. 23, 80). Glutathionylation is an oxidative posttranslational modification that occurs on some protein cysteines under basal conditions [e.g., β-actin (137), mitochondrial complex II (19)]; for others, it is a transient modification that occurs during oxidative stresses such as ischemia/reperfusion [e.g., α-actin (18), GAPDH (26), mitochondrial complex I (56)]. For many proteins, glutathionylation affects function, and thus the reversible glutathionylation of specific proteins has been implicated in regulation of cellular homeostasis in health and disease (reviewed in refs. 23, 80). Grx is the primary intracellular deglutathionylating enzyme in mammalian cells (21, 52), and manipulation of Grx levels has been shown to affect protein glutathionylation status and, subsequently, downstream signaling events (1, 2, 22, 98, 137). Thus, understanding mechanisms of deglutathionylation by glutaredoxin enzymes, as well as the ways in which the deglutathionylation activity is regulated in vivo, is of great interest to the field of redox homeostasis.
“Glutaredoxin” and “thioltransferase” enzymes were first described independently. The name “glutaredoxin” was given to an enzyme discovered by Holmgren (53) in Escherichia coli mutants lacking thioredoxin. This E. coli Grx, coupled to GSH, glutathione reductase (GR), and NADPH, was characterized as a hydrogen-donor system for turnover of ribonucleotide reductase and production of deoxyribonucleotides for DNA synthesis. Somewhat earlier, an enzyme purified from rat liver was named “thioltransferase” (3) to reflect its characteristic, nucleophilic thiol-disulfide exchange activity, and to distinguish its activity from “transhydrogenase” reactions in which electrons are transferred from cofactors (103). Since then, “thioltransferase” and “glutaredoxin” enzymes from a variety of organisms and mammalian tissues have been isolated and characterized, and a high degree of structure–function congruence supports the conclusion that they simply represent alternative names for the same family of enzymes. The name “glutaredoxin” has become the more widely accepted name internationally, although it does not reflect the chemical nature of the deglutathionylation reaction (i.e., sequential thiol-disulfide exchange reactions).
Glutaredoxins are small, heat-stable proteins that conform to a “thioredoxin fold,” that is, four mixed β-sheets surrounded by α-helices (27, 77). Most glutaredoxins contain a CXXC active-site motif that is exposed to solvent, although in some recently described glutaredoxins (and glutaredoxin domains of multidomain proteins), the C-terminal active-site Cys is replaced by Ser (CXXS). Many glutaredoxins contain residues that have been implicated in the stabilization of the covalently bound glutathionyl moiety (i.e., the catalytic intermediate), and it has been proposed that some Grxs may exhibit affinity for reduced GSH. The majority of glutaredoxins tested to date exhibit deglutathionylase activity, although some subforms do not, suggesting alternative functions in vivo. The “glutaredoxin” family of enzymes is growing increasingly complex, with forms containing dithiol or monothiol active sites; forms that exist exclusively as monomers and those that form dimers; forms that catalyze thiol-disulfide oxidoreductase reactions and those that do not; forms that bind iron–sulfur clusters and those that do not; and forms that represent domains of multidomain proteins. As more is learned about the reactions catalyzed by each of these enzymes, we expect that a subclassification of glutaredoxins will be described according to their structural and functional characteristics.
This forum review is presented in two parts. The first section is focused on the details of the catalytic mechanism of the most well-studied dithiol glutaredoxins, and emphasis is placed on the mammalian enzymes in the context of physiologic function. Next, we discuss evidence for deglutathionylation activity or glutathionyl selectivity of glutaredoxins or both (and Grx domains on multidomain proteins) from model organisms. The catalytic activities and physiologic functions of many of these glutaredoxins are still being explored; thus, this section is provided primarily to direct readers to other forms and potential functions of glutaredoxins not featured in depth in this review.
The second portion of the review is focused on the localization and regulation of the deglutathionylase activity of particular glutaredoxins that have been implicated in physiologic functions.
Mechanistic and kinetic details of the catalysis of thiol-disulfide exchange by glutaredoxins
Uncatalyzed thiol-disulfide exchange reaction
The uncatalyzed reaction of thiol-disulfide exchange is deceptively simple:
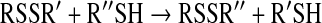 |
In this reaction, a reduced thiol is exchanged with a disulfide, resulting in formation of a new disulfide and a new thiol. Although this reaction is an oxidation–reduction reaction, it occurs as a nucleophilic displacement, the thiol nucleophile attacking the electrophilic disulfide. The rate of this reaction is dependent on the nucleophilicity of the thiol reactant (R′′SH), the reactivity of the central atom being attacked (the one sulfur of the RSSR′), and the stability of the leaving group thiol product (R′SH) (132). Because protonated thiols (-SH) are not good nucleophiles, this uncatalyzed reaction is slow at physiologic pH.
The thiol-disulfide exchange reaction is reversible and reaches an equilibrium based on the initial concentrations of reactants and products, governed by the redox potentials of the thiol disulfide couples involved. A slightly more complicated reaction of two sequential thiol-disulfide exchange steps is shown in Fig. 1, in which hydroxyethyl disulfide (HEDS) is reduced to two molecules of β-mercaptoethanol by reaction with two molecules of GSH. In previous studies, HEDS reduction by GSH was observed to reach equilibrium within 60 min (81, 83). Starting with either 0.9 mM HEDS and 1.8 mM GSH (forward reaction) or 1.8 mM β-mecaptoethanol and 0.9 mM GSSG (reverse reaction), the same equilibrium point was reached, as expected. This point is predicted by the relative redox potentials of the reactants.
FIG. 1.

Sequential thiol-disulfide exchange between hydroxyethyl disulfide (HEDS) and GSH. In the first step, the GSH thiolate attacks one sulfur of HEDS, forming hydroxyethyl-SSG (βME-SSG) and β-mercaptoethanol (βME). In the second step, a second GSH attacks the βME-SSG mixed disulfide, forming GSSG and a second molecule of β-mercaptoethanol. When HEDS is used as a pro-substrate for Grx, the first step of this reaction creates the glutathionyl mixed-disulfide substrate that can be deglutathionylated by the enzyme (81).
Adding Grx at the starting point of either reaction increases the rate of the reaction, but consistent with its role as a catalyst, it does not change the position of the equilibrium (Fig. 2). For most proteins, Kox (i.e., the GSH/GSSG ratio at which PSH/PSSG = 1) is ∼1 (42). Practically this means, for reactions involving protein thiols and GSSG in the presence of much larger concentrations of reduced GSH (e.g., intracellular conditions, GSH/GSSG ∼100), protein glutathionylation is unlikely to proceed by thiol-disulfide exchange spontaneously [see (38)]. Thus, except for protein thiols with unusual Kox values, such as c-Jun [Kox ∼13; i.e., 50% c-Jun-SSG when GSH/GSSG = 13 (66)], experiments in which proteins are glutathionylated in vitro by GSSG in the absence of reduced GSH probably do not reflect what occurs in vivo.
FIG. 2.

Grx catalysis of reversible thiol-disulfide interchange—approach to equilibrium. Reaction mixtures were 2.2 ml at 30°C, containing 0.091 M K phosphate, pH 7.5. Lower lines: initial reaction mixtures also contained 0.91 mM HEDS and 1.82 mM GSH ± Grx1 from human red blood cells (0.01 units). Upper lines: initial reaction mixtures contained 0.91 mM GSSG and 1.82 mM β-mercaptoethanol (β-ME) ± Grx1 (0.01 units). Solid symbols, +Grx; open symbols, -Grx. Separate stock solutions of the reactants were prewarmed to 30°C, and each reaction was initiated by adding the reduced substrate (GSH or β-ME). At time points indicated, 0.2 ml of each reaction mixture was withdrawn and added to 0.8 ml of 0.25 mM NADPH in 0.125 mM K phosphate, pH 7.5; after mixing and reading an initial A340nm value, 2 units of GSSG reductase was added, and the decrease in A340nm was monitored until it reached a plateau (ca. 2 min). Total GSSG was calculated from the values of A340nm (1 nmol/0.005 ΔA340nm), determined from a separate standard curve for authentic GSSG). All data points represent the mean ± standard error of two separate experiments. Reprinted with permission from (83).
Overall catalytic scheme for glutaredoxin
The steps of Grx-catalyzed protein deglutathionylation are presented in Fig. 3A. The overall reaction is a series of thiol-disulfide exchange reactions and involves a glutaredoxin-glutathione mixed-disulfide intermediate (Grx-SSG). Kinetically, this corresponds to a ping-pong mechanism, and two-substrate kinetic analyses have documented this mechanism (38, 47, 122). The KM for a particular substrate, under these conditions (that is, at a fixed concentration of the second substrate) is not a reflection of affinity, but a composite of the kinetic constants of the individual reactions. Accordingly, when the second substrate is varied, the KM and Vmax values both change in a coordinated way so that parallel lines result when plotted in a Lineweaver-Burk double-reciprocal format after nonlinear regression analysis of the primary data (38, 47, 122). Evolution of intrinsic kinetic constants is approached by replotting the reciprocals of the Vmax or KM values from varied first substrate at discrete second-substrate concentrations. If such plots have positive y-intercepts, then the corresponding x-intercepts yield intrinsic KM values. This kind of analysis for Grx1 and Grx2 instead gave a surprising result: the x and y intercepts (related to the reciprocal KMint and Vmaxint, respectively) projected to the origin (38, 122). This result can be explained in one of two ways. The first is the relation of k2 ≫ k-1 (Fig. 4A), in which k2 is the rate constant of the chemical step (the thiol exchange reaction), and k-1 is the dissociation of the enzyme–substrate complex. This is termed complete commitment to catalysis, meaning that each substrate-binding event leads to a bond-transformation reaction. The second explanation refers to the absence of a reversible complex. This is called an encounter reaction, meaning that no binding occurs, that the substrate and enzyme react without any binding modes playing a role in the reaction (Fig. 4B).
FIG. 3.

(A) Catalytic mechanism of deglutathionylation by human glutaredoxins (38, 47, 122). In the first step, the thiolate of the Grx N-terminal active-site cysteine attacks the glutathionyl sulfur of the protein-glutathione mixed disulfide (P-SSG), forming the Grx-SSG intermediate and releasing reduced protein-SH (P-SH). In the second step, free GSH attacks the glutathionyl sulfur of the Grx-SSG intermediate, releasing reduced Grx and GSSG. GSSG is then reduced to 2GSH by GSSG reductase (GR) and NADPH. Step 3 represents a side reaction in which the Grx C-terminal active-site cysteine competes with GSH for reduction of Grx-SSG, forming a Grx active-site disulfide and releasing GSH. The Grx-S2 side product is reduced by GSH and recruited back into the catalytic cycle. (B) Grx can use GSSG as an oxidized substrate (38, 83, 124). This scheme is analogous to Scheme 1A, except that the first substrate for Grx is glutathionylated glutathione (i.e., GSSG), and the second substrate is protein-SH. This reaction occurs under oxidizing conditions (i.e., low GSH/GSSG ratio) until the protein-SH/protein-SSG ratio reaches equilibrium. (C) Proposed mechanism of glutathione thiyl radical (GS•) scavenging by Grx (124). In the first step, the N-terminal active-site cysteine of Grx attacks GS•, forming a Grx disulfide anion radical intermediate. This radical then reacts with O2 in Step 2, forming superoxide (O2•−) and the typical Grx-SSG intermediate. In Step 3, the Grx-SSG intermediate is reduced by GSH, forming GSSG and reduced enzyme. (D) Proposed mechanism of glutathionyl transfer by Grx (124). In the first step of the reaction, the Grx catalytic cysteine thiolate attacks GS•, forming the Grx-SSG•− disulfide anion radical intermediate. This intermediate can proceed to react with protein-SH (P-SH, Step 2), forming protein thiyl radical (P-S•). In Step 3, another Grx-SSG•− molecule reacts with P-S• (Step 3), quenching the radical reaction and forming protein-SSG (P-SSG). The net reaction yields protein-SSG from two GS• and two P-SH molecules.
FIG. 4.

Complete commitment to catalysis vs. encounter-type catalytic mechanisms. (A) In the case of high commitment to catalysis, a reversible binding step exists between enzyme and substrate, followed by a chemistry step (k2) that is very fast compared with the rate of enzyme–substrate dissociation (k-1). Thus, essentially every substrate molecule that binds to enzyme undergoes a nucleophilic displacement reaction. (B) In the case of an encounter-type mechanism, enzyme and substrate react on association, but without formation of a reversible complex. This latter model is supported by two-substrate kinetic analysis of Grx1 (122) and Grx2 (38), which predicts “true” KMint values approaching infinity for both substrates.
These two possibilities are easily distinguished, in that in the first, very tight binding of substrate analogues are designed so that they cannot undergo the second step of catalysis. These analogues can be used in affinity chromatography procedures to purify the enzymes; and they can also serve as effective inhibitors of catalysis. In the second case, the encounter complex is not affected by substrate analogues; therefore, they do not affect the reaction rate, nor does the enzyme bind to chromatography resins linked to such analogues. In the case of Grx, S-methyl glutathione may serve as a mimetic of either the oxidized or reduced substrate (protein-SSG or GSH). This analog efficiently inhibits glutathione-S-transferases (64), which display high affinity for their substrate GSH. Remarkably for human Grx1 and Grx2, this substrate analogue does not inhibit the enzymatic reaction, even at millimolar concentrations (38, 122), supporting an encounter-type mechanism for both enzymes. In summary, the lack of specific and potent inhibitors of Grx strongly suggests that these reactions are encounter reactions and occur without formation of enzyme–substrate complexes.
Catalysis of deglutathionylation by E. coli Grx1 also appears to operate via an analogous ping-pong mechanism (14). However, a recent report of the kinetics of yeast glutaredoxin 7 (ScGrx7, a monothiol glutaredoxin) displayed ping-pong kinetics, but nonzero intercepts were shown on secondary plots of 1/KM or 1/Vmax vs. 1/Cys-SSG at three cysteinylglutathione (Cys-SSG) concentrations (79). It is possible that a more complete analysis involving more than three Cys-SSG concentrations extending higher than the estimated KM might yield a result corresponding to those for hGrx1 and hGrx2 (above); alternatively, this yeast Grx may be dissimilar in this aspect of the double-displacement mechanism, (i.e., reversible ES complexes may precede the covalent reactions).
Characteristics of individual steps of glutaredoxin catalysis of thiol-disulfide exchange
Step 1: Reaction of the oxidized disulfide substrate with reduced Grx (Fig. 3A, Step 1)
This reaction is very fast relative to the overall rate of the catalyzed reaction (which in turn is at least 1,000-fold faster than the nonenzymatic rate for Grx1, and 250-fold for Grx2) (38, 81). Two observations demonstrate that Grx is highly selective for glutathionyl mixed-disulfide substrates. First, exchange reactions for mixed disulfides that do not contain glutathione (e.g., protein-SS-Cys) are not catalyzed by human Grx1 (47) or Grx2 (38). Second, amounts of Grx far above those that are effective for catalysis of deglutathionylation have little effect on the rate of glutathione oxidation by hydrogen peroxide (glutathione peroxidase activity) (38, 124).
As described earlier, the reaction is a nucleophilic displacement. Grx, due to the low pKa of its active cysteine, is a thiolate at physiologic pH and is thus primed to attack the disulfide bond of the oxidized substrate. The oxidized product of this reaction is exclusively the glutathionyl enzyme mixed disulfide (Grx-SSG), as documented by mass spectrometric analysis. Thus, reaction of Cys-SSG with a Grx1 mutant lacking the neighboring cysteine (to avoid intramolecular disulfide formation, see “Side Reaction,” later) yields only Grx-SSG, and no Grx-SS-Cys (58, 143). In contrast, experiments conducted with the analogous mutant of thioredoxin gave a mixture of glutathionyl and cysteinyl mixed disulfides (143). This specificity of attack by Grx1 also was shown for E. coli Grx1 by incubating the analogous mutant (C14S) with a peptide-glutathionyl mixed disulfide substrate (peptide-SSG). Mass spectrometric analysis showed exclusive formation of E. coli Grx1-SSG (96); however, incubation of the enzyme with a mixed disulfide of the peptide and α-glutamylcysteinylglycine (isomer of GSH that lacks the γ peptide linkage between Glu and Cys characteristic of GSH) yielded only Grx-SS-peptide. These observations suggest that the basis for selective attack of the Grx thiolate on the disulfide-adducted glutathionyl moiety is the orientation of the γ-glutamyl moiety of the molecule. The specific electrostatic interactions leading to this exclusive selectivity have not been identified, but represent an intriguing direction for future study (see later).
Other studies examined the reaction rate constants for reaction of Grx with GSSG or other RSSR compounds, which represent potential alternative first substrates for Grx (Figs. 1B and 3). In NMR line broadening experiments, only GSSG reaction rates were fast enough to be measured, giving a rate constant of 7 × 105 M/s for the reaction of GSSG with Grx (102). The implication is that the other nonglutathionyl disulfides reacted much more slowly, confirming the specificity of the enzyme for substrates containing a glutathionyl moiety.
The nonglutathionyl component of the disulfide substrate appears to be unrestricted. Many protein-SSG and small molecule-SSG mixed disulfides (i.e., Cys-SSG, and β-hydroxyethyl-SSG) are substrates (38 44, 47, 61, 75, 83). As discussed earlier, GSSG also serves as a substrate (Figs. 2 and 3B), and this alternative substrate competition explains its apparent “inhibition” of E. coli Grx1 when added in increasing concentrations to a mixture containing peptide-SSG (whose rate of deglutathionylation was measured fluorometrically) (96). However, differences are found in apparent KM and Vmax values among glutathionylated substrates when tested at the same fixed concentration of GSH (the reduced substrate). These differences likely involve steric factors, as illustrated by comparison of met- vs. oxy-hemoglobin-SSG substrates (47, 81).
Evidence suggests the presence of specific electrostatic interactions between the covalently bound glutathionyl moiety and specific residues of the Grx protein in the Grx-SSG mixed disulfide intermediate (15, 143, 145). These interactions may serve two potential roles: (a) to stabilize the adducted glutathionyl moiety at the expense of destabilizing the disulfide bond, making it more reactive in the second reaction step (see later); or (b) to decrease the rate of formation of the intramolecular disulfide of Grx by sterically or conformationally inhibiting the reaction of the second cysteine (see “Side Reaction,” later).
Under typical conditions with wild-type Grx enzyme, the Grx-SSG intermediate undergoes two disparate reactions: one leading to reduced Grx (Fig. 3A, Step 2), and the other, to an intramolecular disulfide (oxidized) form of the enzyme (Fig. 3A, Step 3).
Step 2: Reaction of the Grx-SSG intermediate with a reduced thiol substrate (Fig. 3A, Step 2)
This is the rate-determining step of the deglutathionylation reaction. For human Grx1 and Grx2, the pH rate profile of the Grx-catalyzed reaction matches that of deprotonation of the thiol on the reduced substrate, indicating the involvement of the reduced substrate thiolate in the rate-determining step (38, 122). For Grx1, this overall maximal reaction rate for Cys-SSG is ∼2,200 per minute at 0.5 mM GSH (21), and ∼10-fold lower (217 and 286 per minute) for human and mouse Grx2, respectively (38).
Many thiols (including protein thiols) may be used as reduced substrates for Grx1-SSG, and for most, the rate enhancement over noncatalyzed rates is ∼1,000-fold. This rate enhancement matches the predicted enhancement due to the difference in pKa of the leaving group in the uncatalyzed reaction compared with the pKa of the Grx-thiolate as the leaving group, according to the Bronsted theory (122, 132). Besides the leaving group effect, an additional enhancement exists for GSH as the second substrate, which suggests an enzyme-induced increase in nucleophilicity of the glutathionyl thiolate for the Grx-SSG intermediate (38, 122). As for the first step of catalysis, asymmetry is found in this nucleophilic displacement reaction, in that the reduced product in the catalyzed reaction is exclusively the Grx thiolate.
No intrinsic KM is found for Grx1 toward GSH, indicating no substrate binding (as shown for Step 1). This is further supported by experiments in which incubation of pig or human Grx1 with radiolabeled GSH yielded non-overlapping elution profiles of protein and radioactivity in size-exclusion chromatography (144). Moreover, Grx1 did not bind to GSH-columns in which GSH was covalently bound via the N-terminal amino group of the tripeptide (81). Like Grx1, the plot of 1/KM vs. 1/GSH for Grx2 exhibits an x-intercept projecting to the origin, and millimolar concentrations of S-methylglutathione did not inhibit the Grx2 enzyme. Both of these observations support a Grx1-like, encounter-type mechanism for Grx2.
Studies performed in the absence of GSH suggest that the hGrx2-SSG intermediate may be turned over by thioredoxin reductase (TR) (61), leading the authors to suggest that TR may support GRx2-catalyzed deglutathionylation during oxidative stress [i.e., low (GSH)] conditions. However, we observed that rates of deglutathionylation of the prototype substrate Cys-SSG by human Grx enzymes are much lower (>100-fold for Grx1, 20-fold for Grx2) when TR is substituted for the GSH system (GSH and GR), even when GSH and GR are reduced to one tenth of their estimated intracellular concentrations (38). Thus, although TR can support deglutathionylation of human Grx enzymes, it does so at a rate unlikely to contribute significantly to total deglutathionylation activity in vivo.
Side reaction: Formation of a Grx intramolecular disulfide from the Grx-SSG intermediate
An intramolecular reaction between the C-terminal active-site cysteine and the Grx-SSG intermediate can compete with turnover of the enzyme intermediate (Fig. 3A, Step 3). The products are oxidized Grx (i.e., Grx-S2) and reduced GSH. The intramolecular disulfide must react with 2 GSH to re-form the reduced enzyme for another cycle of catalysis, and this occurs readily. That is, assays beginning with oxidized enzyme display full activity with no lag phase, indicating that reduction of the intramolecular disulfide form of the enzyme occurs at least as rapidly as the rate-determining step (81).
Although formation of the Grx-S2 intramolecular disulfide may be favored because of the propinquity of the neighboring cysteine, such reactivity may be offset by the unfavorable orientation of this thiol relative to the mixed disulfide intermediate. Also, the conformational accommodation of the covalently adducted glutathionyl moiety in the Grx-SSG intermediate may interfere with the intramolecular reaction, either directly (sterically) or through internal movement of the protein due to the specific interactions with the adducted glutathione. Some support for this concept comes from experiments in which Grx was incubated for very short times with radiolabeled GSSG or cystine (Cys-SS-Cys), and then subjected to ion-exchange chromatography to detect radiolabel (*) incorporation into the protein fraction, reflecting detection of either Grx-SSG* or Grx-SSCys*. Although addition of [35S] GSSG to Grx resulted in some incorporation of radioactivity (30), none was associated with enzyme treated with [14C] cystine (40). These contrasting results suggest that the intramolecular reaction of the neighboring cysteine is slower in the case of the Grx-SSG intermediate, consistent with the glutathionyl specificity of the enzyme.
Formation of Grx-S2 detracts from turnover of the Grx-SSG intermediate and is nonproductive. Therefore, to the extent it occurs, it is inhibitory. Removal of the C-terminal active-site cysteine by mutagenesis resulted in a twofold increase in specific activity for Grx1 (143), indicating that intramolecular disulfide formation draws off ∼50% of the Grx-SSG intermediate at steady state during catalysis. The analogous Grx2 mutant also exhibits a twofold increase in specific activity over the unmutated enzyme (38, 61). However, this phenomenon may not be generalizable to glutaredoxins from all species. For example, the E. coli Grx1 C14S mutant is ∼75% less active than the wild-type enzyme with small glutathionyl mixed disulfide substrates (14, 96). It is not yet known whether this lesser activity relates directly to contribution of the second cysteine to catalysis [i.e., a dithiol catalytic mechanism), or it is the result of a mutation-induced conformational change that disfavors reactivity in some other way. In this regard, a dithiol mechanism for thiol-disulfide exchange has been proposed previously for Grx-catalysis of reduction of ribonucleotide reductase (RNR); however, specific kinetic data or other documentations were not reported (54). Irrespective of the catalytic mechanism, studies of E. coli mutants have indicated an interchangeable role for the Grx and Trx systems in support of RNR turnover [i.e., when thioredoxins are knocked out, the cells use Grx systems for RNR reduction, and when glutaredoxins are knocked out, the Trx systems support RNR action (reviewed in ref. 46). In mammalian cells, it appears that the Trx system may play the more prominent role in the reduction of RNR for several reasons: (a) in mice, Grx1 knockout is not embryonic lethal and appears to confer no developmental defects (52), whereas Trx knockout is lethal, indicating its importance in development and necessity for RNR function (78); (b) depletion of GSH (a co-substrate for Grx in the proposed RNR reduction mechanism) does not impair RNR activity or DNA synthesis in cultured cells (52, 121).
Evaluation of the mechanisms of the catalytic enhancement by glutaredoxin
The two human glutaredoxins characterized as thiol-disulfide oxidoreductases are dissimilar in primary sequence (<35% identity), and their active-site sequences also differ in the second amino acid, (CPYC vs. CSYC, for Grx1 vs. Grx2) (44, 75). Yet, the overall structures of the two enzymes (thioredoxin-fold) (60, 128, 143)) and their analogous catalytic features (38, 47, 122) suggest a general overall mechanism for this family of enzymes (Fig. 3). The lack of a well-defined active site with kinetically important substrate-binding modes (see earlier) has focused the study of the kinetics and structure–function relations on the highly unusual pKa of the active cysteine of these enzymes.
In human Grx1, Cys 22 has been characterized as the active catalytic principle, and its pKa (∼3.5) (40; 83) has been shown to be responsible for the majority of the catalytic advantage of Grx over the nonenzymatic reaction (122). This can be explained by the observation that the second-order rate constant of a thiol-disulfide exchange reaction increases by a factor of ∼4 for each one-pH unit decrease in the pKa of the leaving group (41). Thus, for Grx1, the fold difference in rate constant of the catalyzed reaction (in which the leaving group in the rate-determining step is Grx1-SH, pKa ∼3.5) vs. that of the uncatalyzed reaction (in which the leaving group is BSA-SH, pKa ∼8.5) is predicted to be 4ΔpKa, or 45 (∼1,000-fold) (122). For Grx2, the predicted rate enhancement is 44, or ∼250-fold, because the pKa of its catalytic cysteine is 4.6 [4ΔpKa = 44, (38)]. Indeed, rate enhancements by both glutaredoxins are consistent with these predictions for non-GSH substrates (38, 122). In contrast, thioredoxin [with an active-site pKa of 6.7 (62)] exhibits very little deglutathionylating activity (21).
The interaction of the catalytic cysteine with neighboring amino acids has been probed to understand the basis for thiolate stabilization reflecting the unusually low pKa. Examination of the NMR structure of the reduced enzyme suggested that the nearby lysine (K19) might be responsible for the low pKa of Cys-22 (58, 128). According to this premise, computational studies in which K19 in human Grx1 was replaced with glutamine and leucine, and energy minimized, predicted that cysteine-22 of the mutated enzyme would have pKa values of 7.3 and 8.3, respectively. When actual K19Q and K19L mutants were made in a form of the enzyme in which the three non–active-site cysteines were mutated to serines (C7S, C78S, C82S), the resultant pKa values for the respective catalytic cysteines were both determined to be 3.7 (i.e., little changed from wild-type enzyme). This result indicated that the neighboring lysine cannot be solely responsible for the low C22 pKa and that a more-complex set of interactions is ultimately responsible. Surprisingly, mutation of the C-terminal active-site cysteine C25 resulted in about a 1-pH-unit increase in the C22 pKa, suggesting that the C25 thiol makes some contribution to the low pKa of the catalytic cysteine, either directly or conformationally. Others have suggested multiple and varied interactions between the active cysteine and other amino acids on the protein, including H bonding within the active site (36) and ion-dipole interactions with α-helix 2 (58, 67).
Importantly, the low pKa of the Grx catalytic cysteine does not fully account for the observed rate enhancement in the presence of GSH. When GSH is used as the second substrate, second-order rate constants are further increased for Grx1 (122) and Grx2 (38) over rates with nonglutathionyl substrates. It appears that the special rate enhancement of GSH can be attributed mainly to the γ-glutamylcysteine dipeptide subset of GSH, because γ-glutamylcysteine also confers an additional, yet smaller, rate enhancement over non-GSH substrates, but cysteinylglycine (the other dipeptide subset of glutathione) does not. Although the basis for the differential enhancement of GSH nucleophilicity by human Grx isoforms is not yet known, the observation helps explain the difference in their specific activities. That is, the difference in catalytic cysteine pKa (∼1 pH unit) accounts for only about half of the tenfold lower specific activity of Grx2 compared with Grx1. However, the additional rate enhancement in the presence of GSH is 2.5-fold lower for Grx2 than for Grx1 (eightfold vs. 20-fold), explaining the remainder of the difference.
Understanding the mechanism by which the Grx enzyme enhances the nucleophilicity of GSH (122) for attack and turnover of the Grx-SSG intermediate remains a challenge in light of the lack of kinetically relevant substrate-binding modes (see earlier). Several hypotheses attempt to explain how this enhanced nucleophilicity might occur. For example, it could result from general base catalysis of proton abstraction from the attacking GSH by residues on the enzyme or on the adducted glutathionyl moiety itself. Alternatively, it could result from stabilization of the incipient thiolate of the attacking GSH by positively charged basic groups on the enzyme. For instance, mutation of the lysine close to the catalytic cysteine of human Grx1 (i.e., K19Q or K19L) caused little change in the pKa of the catalytic cysteine but resulted in substantially lower specific activity (58), suggesting that K19 might be involved in the enhancement of the nucleophilicity of GSH. Alternatively, the local environment of the Grx-SSG mixed disulfide bond may impose a particular orientation or strain that favors nucleophilic attack by GSH. It is not clear what role is played by the individual interactions of the protein residues with the adducted glutathionyl moiety in the Grx-SSG intermediate (i.e., whether they serve to orient, stabilize, or distort the scissile bond). The lack of kinetically important substrate-binding modes suggests that these interactions may form during or after covalent bond formation and, as discussed earlier, may be important in setting up the mixed disulfide for nucleophilic attack by GSH. Structure–activity relations in which these amino acids are mutated may yield important information about their roles in catalysis.
Substrate specificity and selectivity in the absence of kinetically detectable binding modes
Substrate selectivity is classically explained by specific electrostatic, hydrogen bonding, and hydrophobic interactions involved in reversible non-covalent binding of the substrate to the enzyme (ES complex). These interactions provide selectivity by increasing the lifetime of the specific substrate in the active site relative to a non-interacting compound. This view of specificity is given great support when substrate analogues bind tightly to enzymes or substrates bind tightly to inactive enzyme mutants. As described earlier, this is not the case with the glutaredoxins. Alternatively, for encounter reactions, the specificity must reside in a catalytic selection, wherein reactivity with specific substrates is enhanced. This consideration leads to an examination of the transition state for the nucleophilic displacement reaction, which for thiol-disulfide exchange involves three sulfur atoms with partial covalent bonds among them (Fig. 5). The negative charge of the incoming thiolate is distributed among the atoms, and its localization on one sulfur atom or the other affects the eventual product distribution. The three possible thiol products, corresponding to the three sulfur components of the transition state are the original thiol (nonproductive reaction), or one of the two disulfide sulfurs. For a transition state in which the thiols formed from the disulfide share the negative charge equivalently, equivalent amounts of the corresponding thiols would be formed (Fig. 5, Step 1A). Specificity on this chemical level is produced when this electronic distribution is distinctly unequal. For example, because the encounter reaction of a glutathione-containing mixed disulfide substrate with Grx1 forms only the Grx-SSG mixed disulfide intermediate (96, 102, 143), this implies that the electronic distribution of the transition state has the negative charge localized mostly on the protein thiol sulfur (Fig. 5, Step 1B).
FIG. 5.

Potential transition states in catalysis of deglutathionylation by Grx. Step 1, the Grx thiolate attacks the protein-SSG mixed-disulfide bond. (A) A symmetric transition state is depicted in which the negative charge of the attacking Grx thiolate is shared equally among all three sulfur atoms involved in the thiol-disulfide exchange reaction. (B) An asymmetric charge distribution is shown in which the negative charge is localized mainly to the protein thiolate, the exclusive leaving group in the reaction. Step 2 shows the glutathionyl thiolate attacking the Grx-SSG intermediate. (A) Steric interference is pictured, preventing interaction between the attacking glutathionyl sulfur and the catalytic cysteinyl sulfur of Grx, resulting in an asymmetric transition state. (B) An asymmetric transition state is depicted in which distortion of the mixed disulfide bond between Grx and the adducted glutathionyl moiety polarizes the disulfide bond, creating a more electrophilic site for the attack by the GS-thiolate.
The selectivity of the second reaction for GSH also involves consideration of the tri-sulfur transition state (Fig. 5). In this case, however, steric hindrance and bond distortion may be factors. As described earlier, the human Grx1 and E. coli Grx1 isoforms have specific residues identified as complementary to the adducted glutathionyl moiety in the Grx-SSG intermediate. Because these do not result in a reversible non-covalent enzyme–substrate complex (which would be detected kinetically), they apparently occur during or after the thiol disulfide exchange reaction, serving to orient or distort the disulfide bond, making it more electrophilic. Some of the possibilities are illustrated in Fig. 5 (Step 2). Orientations that make one of the sulfurs more accessible to GSH as the second substrate (Figs. 3A and Step 2), relative to other thiols, would provide a selective enhancement of its reactivity. In the case of bond distortion, incoming GSH may increase the distortion of the disulfide bond and favor the reaction of GSH with the intermediate. The intimate details of this aspect of the Grx enzymatic reaction may be discovered by further examination of the individual thiol disulfide exchange reactions, by using single turnover, stop-flow, or equilibrium perturbation experiments with appropriate substrates and Grx mutants. In the next subsection, various reports of apparent complexes of Grx proteins with GSH are discussed.
Reports of complex formation between glutaredoxins and glutathione
As described earlier for the catalytic Grx-SSG intermediate, some evidence indicates interaction of the covalently bound glutathionyl moiety with specific residues of the Grx protein. NMR and x-ray crystallographic structures of several glutaredoxins in mixed disulfides with GSH (15, 24, 48, 91, 143, 145) indicate that different components of the GS moiety are stabilized by specific residues on the Grx molecule (Fig. 6). For human, yeast, and E. coli Grx-SSG structures, the backbone of the Cys residue of GSH makes antiparallel, β-sheet–like H bond contacts with the backbone amide and carbonyl groups of a conserved Val. The carboxylate and amino groups of the γ-glutamyl moiety are stabilized by amino acids with complementary charges in most Grx-SSG structures; however, these residues vary among the Grx proteins of different species (see Fig. 6). The carboxylate of the glycyl moiety of the glutathionyl adduct is stabilized by H-bond donors or a positively charged lysine or both in most Grx-SSGs; however, a recent study of hGrx1 in which the interacting Lys was mutated to Leu or Gln resulted in retention of glutathionyl substrate specificity (58), suggesting that this specific ionic interaction is not required for reaction of Grx with the glutathionyl sulfur of the GS-containing mixed disulfide substrate. Moreover, analysis of the stability of mixed disulfides between E. coli Grx3 and various GSH analogues (28) suggested that the interaction between Grx and the glycyl group of the GS moiety contributes little to stabilization of the glutathionyl moiety in the Grx-SSG mixed disulfide intermediate.
FIG. 6.

Grx residues reported to interact with a covalently bound GS-moiety in the Grx-SSG mixed disulfide. Residues identified as making ion-pair or H bond contacts or both to the bound GS moiety are indicated next to the chemical group with which they interact. The functionality of the interacting amino acid is indicated parentheses (guan, guanidino group). Colors indicate the species from which the Grx-SSG structure was determined [red, E. coli Grx1 (15); brown, E. coli Grx3 (28, 91); green, yeast Grx1 (48, 145); orange, yeast Grx2 (24), blue, human Grx1 (143)]. For human Grx2 [represented in purple (60)], the depicted interactions correspond to those identified in a co-crystallized complex of reduced hGrx2 and GSH rather than the Grx2-SSG mixed disulfide. *A modified orientation of the γ-glutamyl group of the associated glutathionyl moiety, in comparison to the schematic representation [adapted from Nikkola et al. (89)]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Although the kinetic data described previously (i.e., encounter-type reaction mechanism with 1/KMint approaching zero, and lack of potent inhibition by glutathionyl analogues; see earlier section), indicate little or no affinity of Grx for reduced GSH, two recent reports seem to challenge this interpretation. First, Lundberg et al. (74) reported that some of the recombinant human Grx2 from a bacterial lysate could be purified via affinity chromatography with immobilized GSH-sepharose, although recombinant fusion proteins (Grx2-β-galactosidase, or Grx2-GFP) could not. Second, analysis of an x-ray crystal structure of reduced hGrx2 with reduced GSH led Johansson et al. (60) to suggest that Grx2 exhibits non-covalent, “high-affinity” binding to GSH. Specifically, cocrystallization of dimeric Grx2 under aerobic conditions in the presence of GSH resulted in monomeric Grx2 complexed with GSH. In this complex, the cysteine residues of the Grx2 active site and GSH were not sufficiently close to represent a disulfide bond. Accordingly, the cysteines were interpreted to be fully reduced, although a similar S-S distance in the x-ray structure of yeast Grx1p (C30S)-SSG was interpreted to reflect monooxidation of the mixed disulfide bond (48). The majority of the Grx2-GSH interactions were similar to those of the Grx-SSG mixed disulfides, with the glutathionyl Cys making antiparallel interactions with backbone residues of a conserved Val, and the glycyl carboxylate forming an ion pair with a conserved Lys (Fig. 6). However, the γ-Glu-Cys peptide bond was flipped relative to previously published Grx-SSG structures, creating some unique H-bonding interactions and indicating a modified “binding” mode than that of a covalently bound GS moiety, although the same region of the Grx protein was occupied. Hence, it is conceivable that the Grx-GSH complex detected in the crystal structure may represent a precursor to the (Grx2)2 2Fe2S complex.
The association of Grx2 with GSH was proposed to be “high-affinity” and inhibitory toward deglutathionylation (60), resulting in formation of a “dead-end complex” (11). The main experimental evidence for this concept appears to come from site-directed mutagenesis studies in which mutation of T95 (which contributes several electrostatic interactions with the associated GSH) to Arg was associated with decreased Grx2 dimer stabilization by GSH, and higher deglutathionylation activity toward βME-SSG. However, it was not confirmed whether the T→R mutation resulted in decreased association of reduced Grx2 with GSH. Other observations provide evidence against tight binding of GSH to Grx2. For example, Grx2 is not inhibited by GSH analogues (38), and removal of GSH from solution results in rapid dissociation of the dimeric Grx2 2Fe2S complex (11, 69). The uncertainty regarding the strength of the potential Grx2 ⋯ GSH noncovalent interaction would likely be resolved by measuring a dissociation constant directly. This approach was used by Noguera et al. (90) to estimate the relatively low affinity of P. tremula Grx C4 for GSH (Kd = 8.6 mM).
Enhancement of protein glutathionylation by Grx
The sensitivity of cells to changes in Grx content and activity suggests that reversible glutathionylation plays an important role in cellular homeostasis and in the regulation of signaling pathways (23, 80, 114). Although the mechanisms for formation of protein-SSG are not yet clear, some enzyme systems have been suggested to possess glutathionylating activities, and some preliminary data are available (reviewed in ref. 37). One perhaps unexpected enzyme that may enhance protein glutathionylation is Grx itself (124).
In certain cases, the enhancement of glutathionylating activity by Grx would be consistent with the reaction scheme presented earlier. Thus, an oxidized reactant would act as the GS-donor and a reduced protein would accept the glutathionyl moiety from the Grx-SSG mixed disulfide intermediate (Fig. 3B). This mechanism is supported by studies of human Grx-mediated glutathionylation of GAPDH by GSSG, in which the difference in glutathionylation rate enhancement between Grx1 and Grx2 reflected the differences in the pKa values of their respective active-site cysteines (38), yielding relative rate enhancements that would be expected in the presence of a non-GSH second substrate. Grx2 has also been shown to enhance glutathionylation of mitochondrial complex I in the presence of GSSG (9). The physiological relevance of these observations was uncertain, because the reactions were carried out at relatively high GSSG concentrations.
GSNO also has been characterized as a glutathionylating agent both in vitro and in cells (17, 43, 59, 65, 66, 84). Reaction of this oxidized reactant with Grx could produce the normal Grx-SSG mixed disulfide intermediate and NO−. However, this reaction may be unfavorable and therefore slow. Indeed, GSNO does not substitute for Cys-SSG in the standard coupled assay (i.e., no enzyme-mediated increase in GSSG formation is detected with GSNO as the oxidized substrate) (D.W. Starke, unpublished observations). The reaction of GSNO with small amounts of protein thiols occurs and Grx enhances the rate of formation of protein-SSG products (38, 124). The mechanistic details of this reaction are not well characterized, and the rate enhancement is relatively small.
The sequestration of glutathione thiyl radical (GS•) and subsequent delivery of the glutathionyl moiety to reduced proteins has also been observed for Grx1 and Grx2 (38, 124). In the proposed mechanism (Fig. 3C), Grx reacts with GS•, forming a disulfide anion radical on the catalytic cysteine. This intermediate is interpreted to be favored by the uniquely low pKa of Grx and its glutathionyl stabilization site. Notably, the formation of protein-SSG is inhibited by molecular oxygen, so that Grx-mediated protein-SSG formation via this mechanism is favored under hypoxic conditions (124). The rate enhancement via this Grx-mediated reaction involving GS radicals is higher than that for other glutathionylating reactants (GSNO, GSSG), even though the estimated concentration of GS• was 10- to 50-fold lower than the GSNO or GSSG concentrations. This is remarkable when it is considered that reaction to form stable protein-SSG products must involve two one-electron reactions (Fig. 3D). In addition, the GS-radical transfer reaction occurred in the presence of 0.5 mM GSH, indicating that it is likely to occur within the intracellular milieu (100). Because this reaction is not in equilibrium with the GSH/GSSG couple, it suggests a way that significant amounts of glutathionylated protein could be made without initial accumulation of GSSG (i.e., normal cellular reducing conditions). It is also conceivable that protein-SSG formation by GSNO may occur through a glutathione radical generated by homolytic cleavage of the GSNO (119).
In the presence of molecular oxygen, some of the Grx-SSG•− radical intermediate is converted to the canonic Grx-SSG mixed disulfide intermediate, and reaction with GSH completes the normal catalytic cycle. This sequence of events results in net scavenging of GS radicals with concomitant formation of superoxide. The reaction occurs at physiologic concentrations of reduced GSH and demonstrates the kinetic competence of the reaction of Grx and glutathione thiyl radical. Coupled with superoxide dismutase in the physiologic setting, this reaction would represent an efficient mechanism of glutathionyl radical scavenging (38, 124), but this mechanism is yet to be documented in situ.
Human Grx1 and Grx2 promote glutathionylation of GAPDH at equal rates when GS• is the glutathionyl donor, representing a contrast to their relative deglutathionylation and GS-radical scavenging activities (10-fold lower rate for Grx2 in both cases) (38). These observations indicate that a different mechanism—or at least a different rate-determining step—governs this reaction, as proposed previously (124) (Fig. 3D).
Catalysis of deglutathionylation by other glutaredoxins and glutaredoxin domains
Glutaredoxins have been identified in most living organisms, from bacteria to plants to mammals (33). Of these glutaredoxins, human isoforms 1 and 2 are the best characterized kinetically (see earlier). Overviews of glutaredoxin proteins from different species can be found elsewhere (33, 68, 109, 112); here, we review evidence for deglutathionylation activity in glutaredoxins from prototype organisms (see Table 1), with special emphasis on newly described Grx proteins, and putative glutaredoxin domains on multidomain proteins.
Table 1.
Deglutathionylation Activities and Glutathionyl Specificity of Glutaredoxins from Prototype Organisms
| Glutaredoxin | CXXbC/S motif | Deglutathionylation activity? | Glutathionyl specificity?a | References |
|---|---|---|---|---|
| Escherichia coli | ||||
| Grx1 | CPYC | Yes (peptide-SSG,ArsC-SSG) | Yes (Step 1) | (14, 53, 96, 117) |
| Grx2 | CPYC | Yes (βME-SSG) | Yes (Step 1) | (4, 135) |
| Grx3 | CPYC | Yes (βME-SSG) | Yes (Step 1) | (4) |
| Grx4 | CGFS | No | N/A | (32) |
| Streptomyces cerevisiae | ||||
| Grx1 | CPYC | Yes (βME-SSG) | ND | (24, 39, 72) |
| Grx2 | CPYC | Yes (βME-SSG,20S proteasome-SSG) | ND | (24, 72, 118) |
| Grx3 | CGFS | Yesb | ND | (105) |
| Grx4 | CGFS | Yesb | ND | (105) |
| Grx5 | CGFS | Unresolvedc | ND | (105, 106, 116, 133) |
| Grx6 | CSYS | Yes | Yes (Step 1) | (79) |
| Grx7 | CPYS | Yes | Yes (Step 1) | (79) |
| Homo sapiens | ||||
| Grx1 | CPYC | Yes (protein-SSG,cysteine-SSG, βME-SSG) | Yes (Steps 1 and 2) | (47, 61, 122, 143) |
| Grx2 | CSYC | Yes (protein-SSG,cysteine-SSG, βME-SSG) | Yes (Steps 1 and 2) | (38, 61) |
| Grx5 | CGFS | ND | ND | (16, 85) |
| Grx domains | ||||
| TGR | CPHS | Yes (βME-SSG) | Yesd | (129, 130) |
| PICOT | CGFS | ND | ND | (57, 142) |
| TR3 | CTRC | No | N/A | (126) |
Indicated by the step of the deglutathionylation reaction for which glutathionyl specificity has been demonstrated (see Fig. 3). Step 1 indicates selectivity for glutathionyl mixed-disulfide substrates and/or for selective attack of Grx on the sulfur of the glutathionyl moiety. Step 2 indicates selectivity for GSH as the second substrate to reduce the Grx-SSG intermediate.
Inferred from studies of null and multicopy mutant strains.
Studies of null mutant strains suggest a contribution to cellular deglutathionylation activity, but assays on purified, recombinant protein show little to no activity.
Activity toward HEDS was GSH dependent (130), but it was not distinguished whether this dependence reflected a requirement for forming a glutathionyl mixed-disulfide first substrate (βME-SSG) from the pro-substrate (HEDS), for using GSH as the preferred second substrate, or both.
ND, not determined. N/A, not applicable.
Escherichia coli
E. coli contain four glutaredoxins, of which three (Grx 1–3) exhibit deglutathionylation activity toward protein, peptide, small molecule glutathionyl mixed disulfides, or a combination of these (96, 117, 135). In a comparative study of glutaredoxins 1 to 3, Grx2 was shown to have the highest activity toward βME-SSG [i.e., HEDS assay (135)], accounting for an estimated 80% of total intracellular deglutathionylation activity (136). Like human Grx1, E. coli glutaredoxins 1 to 3 exhibit glutathionyl specificity in Step 1 of the deglutathionylation mechanism, with Grx1 forming exclusively Grx1-SSG after incubation with a glutathionylated peptide (96), and glutaredoxins 2 and 3 exhibiting no GSH-dependent reductase activity toward insulin disulfide (135). Null mutant strains of glutaredoxins 1, 2, or 3 are viable, although E. coli lacking Grx2 and Grx3 exhibit somewhat increased sensitivity to certain oxidants (136). To our knowledge, a mechanistic link between decreased deglutathionylation activity and increased oxidant sensitivity has not yet been established.
Grx4 is the only monothiol glutaredoxin found in E. coli, and it exhibits considerable similarity to yeast Grx5 (see later), including 37% sequence homology and a monothiol, CGFS active site (32). Purified, recombinant Grx4 did not exhibit deglutathionylation activity in the HEDS assay (32), nor did active-site mutants of Grx4 containing a dithiol moiety (CGFC), or the classic active site of human and E. coli Grx1 (CPYC), suggesting that a structural feature outside of the monothiol active site may explain its lack of deglutathionylation activity. When treated with GSSG, Grx4 forms an intramolecular disulfide (reduced by TR in vitro), as well as a mixed disulfide with GSH (reduced by E. coli Grx1); however, whether these disulfides form under physiologic conditions—and whether Grx4 is a physiologic substrate of Grx1 or TR—remains to be seen. Analysis of the NMR structure of reduced Grx4 suggests that it may stabilize a covalently bound GS moiety similarly to Grx3 (35), but it is not yet known whether Grx4-SSG represents a catalytic intermediate or simply a posttranslational modification regulating other functions.
The physiological function of Grx4 appears to be vital, because Grx4-knockout strains are not viable. Some evidence indicates that Grx4—like Grx5 of yeast and mammals—regulates iron homeostasis, because its expression is increased after Fe depletion (32). It will be interesting to discover the role (if any) of the Grx active site in the critical functions of Grx4, given its incompatibility with catalysis of deglutathionylation.
Saccharomyces cerevisiae
To date, seven glutaredoxins have been identified in S. cerevisiae, and they have been divided into 3 groups according to structural and functional characteristics. Glutaredoxins 1 and 2 (ScGrx1-2) are the only yeast glutaredoxins containing the classic CPYC active-site motif. Analyses of lysates from mutant strains, as well as purified, recombinant enzymes, indicate that both ScGrx1 and ScGrx2 exhibit deglutathionylation activity, with ScGrx2 exhibiting approximately 15-fold higher specific activity compared with ScGrx1 (24; 72). Remarkably, mutation of the active site to CPYS has opposite effects on the activities of these two enzymes (24). Recently, the 20S proteasome was identified as a potential intracellular target for deglutathionylation by ScGrx2 (118).
Yeast glutaredoxins 3 to 5 were identified in a yeast genome-sequencing project as a family of ORFs with homology to previously identified glutaredoxins, but containing nonclassic, monothiol active sites [CGFS (105)]. Assays of lysates from null mutant strains suggest that all three glutaredoxins contribute to cellular deglutathionylation activity (i.e., decreased GSH-dependent reduction of βME-SSG by the mutant strains), with the greatest contribution from ScGrx5. Although ScGrx5 was also proposed to deglutathionylate Tdh3, the yeast homologue of GAPDH, in situ (116), studies of purified, recombinant enzyme exhibited little or no activity toward βME-SSG or a mixed disulfide between carbonic anhydrase III and GSH [i.e., CAIII-SSG (133)]. Several potential explanations may exist for this discrepancy, including inactivation of the enzyme during purification, removal of a cofactor from the cellular lysate, and an apparent lack of GSH in the assay of purified yGrx5 with CAIII-SSG (precluding turnover of the enzyme via the mechanism previously characterized for glutaredoxins, see earlier).
Although catalysis of GSH-disulfide oxidoreductase reactions by ScGrx5 remains uncertain, a catalytic mechanism of protein deglutathionylation was recently proposed (133), involving both the active site and a non–active-site cysteine (C117). Support for such a mechanism came from mass spectrometric and HPLC analyses, suggesting formation of a ScGrx5 intramolecular disulfide after incubation with GSSG, and also from the observation that a C117S mutant exhibits decreased deglutathionylation of CAIII-SSG. However, it appears that the deglutathionylation of CAIII-SSG was assayed in the absence of GSH, and if GSH is involved in the rate-determining step (i.e., reduction of the oxidized enzyme intermediate, as demonstrated for human Grx1 and Grx2), then the relative deglutathionylation rates observed for WT and C117S ScGrx5 may not represent a meaningful comparison. Thus, the deglutathionylation capacity of ScGrx5 should be determined in a standard Grx assay (i.e., in the presence of GSH), and compared with those of other Grx enzymes, to test rigorously its deglutathionylation capacity.
ScGrx6 and 7 are the most recently described glutaredoxins in yeast, characterized by unusual active site motifs (CSYS and CPYS, respectively), as well as an apparent ability to form homodimers and/or -tetramers (79). Purified, recombinant ScGrx6 and ScGrx7 exhibit deglutathionylation activity toward βME-SSG, but do not reduce insulin intramolecular disulfide, indicating glutathionyl specificity in the first step of the deglutathionylation reaction. Several lines of evidence indicate that recombinant ScGrx6 is purified from E. coli as a GSH-stabilized, tetrameric iron–sulfur cluster, which must dissociate to exhibit deglutathionylation activity (79). As for hGrx2 (60, 69), iron–sulfur cluster formation appears incompatible with deglutathionylation activity for ScGrx6, raising the important question of what percentage of endogenous ScGrx6 may exist in iron–sulfur clusters in vivo, and what stimuli trigger release of the active monomer.
Although the physiological roles of yeast glutaredoxins are not completely understood, null mutant studies suggest that ScGrx1 to 5 serve antioxidant functions (72, 105), with some isoforms exhibiting specificity for particular oxidative stimuli [e.g., ScGrx1, superoxide; ScGrx2, H2O2 (72)]. ScGrx3 to 5 appear to function in iron homeostasis, with ScGrx3 to 4 implicated in the regulation of Aft1, a transcription factor regulating genes involved in iron regulation (92), and ScGrx5 playing a critical role in iron–sulfur cluster assembly (87, 106), also a likely function of its mammalian homologue (see later). For all of the yeast glutaredoxins, understanding the relation between deglutathionylation activity and maintenance of redox balance and/or iron sulfur cluster homeostasis remains an exciting frontier for future study.
Plants
Many genes encoding putative Grx enzymes have been identified in individual plant species [e.g., 31 in Arabidopsis thaliana, (109)], but few biochemical characterizations of expressed Grx proteins have been reported. Grx from spinach (86), rice (113), fern (131), and poplar (107, 108, 110) exhibit activity toward the pro-substrate HEDS in assays containing GSH, GR, and NADPH. In contrast to human Grx enzymes, in which mutation of the C-terminal cysteine in the active site increases activity, the analogous mutation of a poplar Grx decreases deglutathionylation activity by approximately two thirds, suggesting that the side reaction involving Grx intramolecular disulfide formation (Fig. 3, step 3) does not detract substantially from the catalytic rate for poplar Grx. Alternatively, this Grx may use primarily the dithiol mechanism for catalysis of deglutathionylation, or a deactivating conformational change accompanies the mutation.
In addition to deglutathionylation of βME-SSG, poplar Grx (along with GSH and GR) supports the peroxidase activity of a type C peroxiredoxin [Prx (110)]. Although the proposed mechanism of coupling does not involve a Prx-SSG mixed-disulfide intermediate, further studies may indeed identify Prx-SSG as the substrate for Grx in this coupled reaction
Poplar Grx C1 was the first glutaredoxin to be described as existing in a dimeric, 2Fe2S cluster (31), which is coordinated by one active-site cysteine sulfhydryl from each monomer, and two GSH molecules (31, 111). As for hGrx2, the physiological role of the cluster-coordinated poplar Grx dimer is not yet known, but functions in thiol-disulfide exchange and iron–sulfur cluster assembly have been proposed (111).
Homo sapiens
hGrx5
Several lines of evidence suggest that Grx5 isoforms from higher organisms share functional similarities with the enzyme from yeast (ScGrx5; earlier). For example, (a) impaired oxidant defense in yeast mutants lacking ScGrx5 was rescued by expression of the chicken or human Grx5 genes (85); (b) a zebrafish mutant lacking Grx5 exhibited defects in hemoglobin synthesis linked to decreased iron–sulfur cluster biogenesis (141), a function proposed for ScGrx5 (87, 106); and (c) molecular changes consistent with reduced FeS cluster synthesis were observed in red blood cells from a man with a genetic mutation reducing Grx5 mRNA to ∼10% of control levels (16). To our knowledge, neither isolated nor recombinant Grx5 from higher organisms has been assayed for deglutathionylation activity. Therefore, it is not yet known whether deglutathionylation activity contributes to any of the physiological roles identified to date for the enzyme.
TGR
Trx and GSSG reductase (TGR or TR2) is a multidomain protein containing structural characteristics of TR (and GR), and Grx. Thus, the CXXXC motif, FAD- and NADPH-binding domains, dimer interface domain, and GCUG tetrapeptide are characteristic of TR proteins. The N-terminal domain contains a CXXS motif (CPHS) and some residues conserved among glutaredoxins that have been implicated in stabilization of a covalently bound glutathionyl moiety (129).
Both full-length TGR and the separate Grx domain are active in the HEDS assay (in which the pro-substrate HEDS is converted to βME-SSG after incubation with GSH), indicating that the CPHS motif is a functional Grx active site. Whether the Grx domain of TGR uses the same catalytic mechanism for deglutathionylation as E. coli and human glutaredoxins has not been tested directly. It has been proposed that the selenocysteine (Sec, U) residue of the TGR C-terminal CGUC motif was involved in the deglutathionylation mechanism (130). However, deglutathionylation activity of a TGR Sec → Cys mutant was measured in the absence of GSH, precluding formation of βME-SSG from HEDS. Therefore, reduction of βME-disulfide was measured rather than βME-SSG. It also was suggested that the Grx domain of TGR was responsible for the GSSG reductase activity of the enzyme, but this activity does not appear to have been tested with the Grx domain alone. Thus, whether the Grx domain of TGR is sufficient for the enzyme's deglutathionylase or GR activities or both remains an open question and will require additional experimentation.
An additional catalytic activity recently reported for the Grx domain of TGR is protein disulfide isomerization, particularly involving mixed-disulfide formation between glutathione peroxidase 4 (GPx4) and other proteins in developing sperm cells (127). The Grx domain of TGR was active in protein disulfide isomerization assays, and when immobilized on an affinity column, it formed DTT-reversible cross-links with several proteins from sperm extract. Whether protein disulfide isomerization, deglutathionylation, or Trx reductase activity represents the principal catalytic activity of TGR in vivo—and how the magnitude of those activities compares with those of other thiol-disulfide oxidoreductase enzymes—are important questions in discerning the physiological role(s) of this intriguing enzyme.
Variant 3 TR1
A recent analysis of alternative splice forms of the thioredoxin reductase 1 (TR1) gene revealed a specific isoform (named variant 3) containing an N-terminal Grx domain similar to that of TGR, but with a CTRC active-site motif (126). Kinetic analyses of variant 3 TR1 revealed an unusual profile of enzymatic activities. First, purified, recombinant variant 3 TR1 exhibited partial TR activity; that is, it reduced DTNB, but not did not support reduction of insulin with Trx. Removing the Grx domain restored insulin/Trx reductase activity, suggesting that the Grx domain may interfere with the interaction between TR and TRx. Second, when tested independently, the Grx domain of variant 3 TR1 was not active in the HEDS assay (i.e., GSH-dependent deglutathionylation of βME-SSG); however, an active-site mutant (CTRC → CPYC) exhibited some activity, suggesting that the unusual active site of the Grx domain may preclude deglutathionylation activity. The Grx domain of variant 3 TR1 exhibited no dehydroascorbate reductase, GST, peroxidase, or protein disulfide isomerase activities, leaving the catalytic role of this domain unknown. The authors of this study suggest that variant 3 TR1 may be a specific reductant for an unknown substrate, or possibly function as a protein disulfide isomerase.
PICOT
PKCθ-interacting cousin of thioredoxin (PICOT) was identified by a yeast-2 hybrid screen for interacting partners of protein kinase θ (PKCθ) in T lymphocytes (142). When overexpressed in human T cells, PICOT colocalized with PKCθ, and inhibited JNK, AP-1, and NF-κB activation. Structurally, PICOT consists of a N-terminal “Trx” domain and 1 to 3 C-terminal repeats of a “PICOT” domain, depending on the species of origin (57). PICOT's Trx domain exhibits 29% amino acid similarity to human Trx, but it is predicted not to exhibit Trx catalytic activity because it contains only one cysteine in its “active site” (APNC vs. CGPC in human Trx); thus, it has been proposed that PICOT may act as an antagonist of Trx in vivo (142).
Recently, the PICOT domain was classified as a monothiol glutaredoxin domain (10, 68), apparently based on predicted three-dimensional structure (57) and sequence similarity to other glutaredoxins, including the CGFS active site motif, which is shared by some other recently described glutaredoxins (see earlier). To our knowledge, neither the catalytic activities (including deglutathionylation), nor the mechanisms of regulation of PKCθ signaling, have been demonstrated for PICOT. Understanding the capacity of PICOT to catalyze deglutathionylation would be particularly interesting in light of its association with PKC, as some PKC isoforms may be regulated by reversible glutathionylation (114).
Conclusions about the catalytic reactions of glutaredoxin and their impact on cellular function
The glutaredoxins have been implicated as catalysts in a variety of different reactions have the common property of involving GSH in the overall reaction. Among these, the catalytic mechanism that has been characterized in the greatest detail is the reduction of glutathionylated substrates. This deglutathionylation reaction has gained increasing prominence because of the recognition of reversible glutathionylation of specific proteins as an important regulatory component of redox signal transduction, highlighting the physiological consequence of alterations in Grx activity. Perturbation of redox regulation of the glutathionylation status of specific proteins may contribute to the complications of many diseases (see ref. 80 for a recent review).
The deglutathionylation activity of Grx is also important for homeostatic defense against oxidative stress. In this context, the ability of Grx to catalyze the scavenging of glutathione-thiyl radicals may contribute to cellular defense, although evidence for this reaction in intact cells has not been reported. Under conditions such as hypoxia or radiation or chemotherapy of cancer cells with radical-generating agents, Grx may catalyze formation of protein-SSG, but evidence for this role is also limited (100). Considering the many different isoforms of Grx and fusion proteins containing Grx domains, it remains to be learned whether these different glutaredoxins exhibit different primary catalytic functions that will lead to a subclassification of this family of enzymes. For example, as more data are reported, it may become clear that some isoforms possess little deglutathionylase activity but readily promote iron–sulfur cluster synthesis.
Part II: Localization and Regulation of Grx Activity
Subcellular localization and effects on Grx activity
Both the mechanisms and the consequences of dynamic regulation of Grx activity depend partially on its intracellular localization. Specifically, localization to subcellular compartments determines substrate availability, chemical environment, and proximity to signaling factors and intermediates that may affect enzymatic activity (e.g., kinases, reactive oxygen or nitrogen species; see later). Furthermore, distinct localization patterns among different tissues suggest different levels of activity (and capacity for regulation) within the organism. The mammalian Grx enzymes exhibit distinct subcellular localizations, related in some cases to localization sequences or differential splicing of the mRNA or both.
Grx1 is primarily a cytosolic enzyme, and it has been implicated in regulation via deglutathionylation of multiple cytosolic proteins, including PTP1B-SSG, Ras-SSG, actin-SSG, and procaspase 3-SSG (1, 8, 94, 137). Recently, Grx1 was shown also to reside in the intermembrane space of mitochondria isolated from rat tissues (93), although the mechanism of its mitochondrial localization is unknown. Immunohistochemical staining of endometrial tissue suggested nuclear localization of Grx1 (125), but this has not been confirmed by confocal microscopy, colocalization studies, or studies of isolated nuclei.
Three subforms of human Grx2 have been identified, each representing the product of alternative splicing of the gene's five exons. Grx2a contains an N-terminal mitochondrial localization sequence, which is both necessary and sufficient to target Grx2 to the mitochondria (29, 44, 75). Recently, Pai et al. (93) documented endogenous Grx2 only in the matrix fraction of mitochondria isolated from rat heart and liver tissues, suggesting a specific intramitochondrial localization of Grx2a, separate from Grx1. The mRNAs encoding Grx2b and Grx2c result from alternative splicing of a distinct first exon that does not encode a mitochondrial localization sequence (71). When overexpressed in HeLa cells, both unconjugated Grx2 and GFP-Grx2 fusion proteins exhibit a diffuse staining pattern suggesting cytosolic and nuclear distribution. Unlike Grx2a, which appears to be expressed ubiquitously, mRNA encoding Grx2b and Grx2c was more restricted, detected only in cDNA libraries derived from normal testicular tissue or from certain immortalized cell lines. Levels of endogenous Grx2b and Grx2c within testes and in cancer cells have not been determined, but immunoperoxidase staining of testicular tissue sections with a nonspecific Grx2 antibody indicated cytosolic staining in spermatids, spermatogonia, and Sertoli cells, suggesting the presence of endogenous Grx2b and/or Grx2c in normal testes. Both Grx2b and Grx2c exhibit deglutathionylation activities with the pro-substrate HEDS, and Grx2c forms a dimeric 2Fe2S cluster in vitro, although the physiological significance of this observation is not yet known.
Analogous findings were reported for five variants of mouse Grx2 (mGrx2) mRNA, encoding three subforms of the enzyme (55). mRNA encoding mGrx2a was present in most tissues, whereas testicular tissue contained the highest levels of alternative splice variants Grx2c and Grx2d. However, in contrast to human Grx2 variants, Grx2c or Grx2d expression or both may not be restricted to the testes among noncancerous tissues. RT-PCR and colocalization studies suggest that these Grx2 subforms may also be expressed in specific cell types within the stomach, spleen, and other tissues. Isolated, recombinant mGrx2c (analogous to human Grx2c) exhibits activity toward HEDS and can form a dimeric 2Fe2S cluster in vitro. Isolated, renatured Grx2d did not exhibit these activities, but it is not clear whether this finding could be due to inactivation during its resolubilization from E. coli occlusion bodies, or to a potentially inhibitory domain coded by its additional exon (called IIIb).
Thus, the majority of noncancerous mammalian cells (with the exception of testicular cells) are expected to contain Grx1 in the cytosol and mitochondrial intermembrane space and Grx2 in the mitochondrial matrix. Cancerous and testicular cells may also express subforms of Grx2 in the cytosol and nucleus. These specific localizations of the Grx isoforms determine not only the capacity for protein deglutathionylation within specific subcellular compartments, but also the potential mechanisms of regulation of Grx activity.
Enzyme concentration
Global concentration
In theory, the most straightforward way to modulate Grx activity in situ is to change the content of active enzyme by altering rates of its production or degradation. Although little is known about the transcriptional regulation of Grx1, some hormonal and metabolic stimuli are associated with increased protein content and activity, including doxorubicin (Adriamycin) treatment of MCF-7 breast cancer cells (140), estradiol treatment of cardiomyoblasts (134), MPTP exposure in male mice (63), and culture of retinal Müller cells in high-glucose media (115). Recently, Grx2 mRNA was found to be elevated in peripheral blood mononuclear cells treated with the antitumor agent imexon (6), presumably via a putative antioxidant response element (ARE) in its promoter sequence.
Local concentration
In the absence of stimuli, cytosolic Grx1 concentration was determined to be ∼1 μM in cytosol from human red blood cells (82), and similar concentrations are estimated from reports of Grx content in bovine liver (51) and calf thymus (76). In rat liver mitochondria, Grx1 is estimated to be 0.1 μM in the intermembrane space, whereas Grx2 concentration is about 1 μM within the matrix (38). In both cases, calculations were performed on homogenized samples and represent an average concentration of the entire subcellular compartment. However, it is conceivable the Grx enzymes are maintained at higher local concentrations via association with structural proteins (i.e., scaffolding) within specific intracellular compartments (e.g., plasma membrane lipid rafts). Although we are not aware of direct evidence for this concept, it may apply to regulation of actin glutathionylation in fibroblasts (discussed later).
Chemical environment/milieu
pH
Kinetic analyses provide insight into the influence of environmental factors on Grx activities in vivo. Activities of human Grx1 and Grx2 are pH dependent, with inflection points ∼8.5 (38, 122) (see Mechanistic Steps of Glutaredoxin Catalysis, Step 2), predicting variations in Grx activity among subcellular compartments, where pH varies considerably. For example, analyses using pH-sensitive dyes suggest that the pH of the mitochondrial intermembrane space is close to 7.0 (99), whereas the matrix pH is approximately 8.0 (70). Taken together with the glutaredoxin pH rate profiles, these estimates predict that Grx activity would be ∼66% lower in the mitochondrial intermembrane space, and ∼2.5 times higher in the mitochondrial matrix, compared with the cytosol (pH 7.4). Of course, additional factors, such as differences in enzyme concentration across subcellular compartments (see earlier), and localization of specific substrates would also influence deglutathionylation activity in each of these compartments.
GSH concentration
Kinetic characterization of the human Grx enzymes predicts that differences in local GSH concentrations will affect their deglutathionylation activities in situ. The parallel line patterns exhibited by both isozymes on double-reciprocal plots (38, 47, 122) indicate that KM and Vmax values for protein-SSG (i.e., the first substrate, Scheme 1A) increase with increasing concentrations of the second substrate (GSH). Many pathophysiologic stimuli alter GSH content and GSH/GSSG ratio (reviewed in ref. 23); so the dependence of Grx activity on GSH concentration appears quite relevant to the in vivo condition.
Reactive oxygen species (ROS) sources
Proximity to ROS could also regulate Grx activity, either via oxidative modification (see later), or by creating reaction conditions that favor catalysis of thiyl radical scavenging or protein glutathionylation. Thus, mitochondrial electron-transport chain components as well as plasma membrane receptor–linked NADPH oxidases generate ROS (O2•−, H2O2, •OH), which can lead to GS• formation. Human Grx1 and Grx2 scavenge GS• or use it as a substrate for GS-transfer reactions, forming GSSG or protein-SSG, respectively, as products (38, 124). Hence local production of ROS may favor catalysis of glutathionylation by Grx. Grx1 was implicated as the mediator of glutathionylation of NF-κB-p65 in pancreatic cancer cells under hypoxic conditions that promote radical production (100).
Temporal regulation of glutaredoxin activity
Grx-catalyzed deglutathionylation has been implicated in catalytic control of critical processes relating to intracellular homeostasis and stress response, including cytoskeletal reorganization (137, 139), mitogenic (7) and hypertrophic signaling (1), inflammation (104, 115), and Ca2+ homeostasis (2). Although evidence suggests that Grx activity itself is regulated in both the short and long term in association with specific toxins, hormones, disease states, and extracellular stresses (later), the specific mechanisms by which Grx activity is regulated by these stimuli are not fully understood. The following section explores existing evidence for regulation of Grx activity in situ and identifies future studies to elucidate the mechanisms further.
Posttranslational modification
Potential for phosphorylation
Reversible phosphorylation of serine, threonine, and tyrosine residues is a well-documented mechanism of enzyme regulation. To our knowledge, no evidence exists for regulation of Grx by phosphorylation; however, sequence analysis (12, 13) of all three human Grx isoforms reveals several putative phosphorylation sites, and identification of these sites on published structures of reduced Grx1 (128) and Grx2 (60) suggests that they are solvent exposed (Table 2). Phosphorylation sites with the highest probability scores (i.e., 0.8–0.9) were found in Grx2, and included T41 (equivalent to T81 in the FASTA sequence), immediately after the CPYC active-site motif; T97 (T137 FASTA), located on helix 4 adjacent to the active site; and S118 (S158 FASTA). All three were consensus sites for protein kinase C (PKC), and T97 also represents a generic kinase target. A high-probability, nonkinase–specific phosphorylation site was also identified near the C terminus of Grx5 (S156 in its FASTA sequence).
Table 2.
Potential Phosphorylation Sites of Human Grx Enzymes
| Grx isoform | Residue (FASTA) | Kinase | Probability score | aLocation (relative to active site) |
|---|---|---|---|---|
| Human Grx1 | S34 | DNAPK | 0.6 | Opposite end of α-helix 2 |
| S88 | PKC | 0.66 | Neighboring α-helix 4 | |
| Human Grx2 | S16 | General | 0.73 | b |
| S20 | General, PKA | 0.91, 0.68 | b | |
| S39 | General | 0.69 | b | |
| S48 | General | 0.99 | b | |
| S73 | PKC | 0.61 | Facing active site (β-sheet 1) | |
| T81 | PKC | 0.8 | Immediately after active site (α-helix 2) | |
| Y113 | General | 0.73 | Opposite side of protein(α-helix 3) | |
| T137 | General, PKC | 0.9, 0.84 | Neighboring α-helix(α-helix 4) | |
| S158 | General, PKC | 0.9, 0.84 | b | |
| Human Grx5 | S41 | General | 0.69 | c |
| S156 | General | 0.87 | c |
FASTA sequences of human Grx1, Grx2, and Grx5 were entered into NetPhos 2.0 (12) and NetPhosK 1.0 (13) search engines to identify potential phosphorylation sites. Sites with probability scores ≥0.6 are included.
DNAPK, DNA-dependent protein kinase; PKC, protein kinase C; PKG, cyclic GMP–dependent protein kinase.
The hydroxyl groups of all of the indicated residues are solvent exposed and outward facing, with the exception of Grx2 T137, which faces the protein interior.
Not included in published x-ray structure.
No structural information available.
None of the predicted phosphorylation sites appeared to be conserved among the human Grx isoforms (see Fig. 7), suggesting that phosphorylation could regulate the enzymes in an isozyme-specific manner. If phosphorylation is to be documented as a mode of regulation of Grx activity, then it must be demonstrated in situ, under physiologic conditions, and associated with a functional change, analogous to the criteria for regulation by glutathionylation proposed by Shelton et al. (114).
FIG. 7.

Predicted phosphorylation sites for human Grx isoforms. General [NetPhos 2.0 (12)] and kinase-specific [NetPhosK 1.0 (13)] phosphorylation sites were predicted using FASTA sequences of human Grx1, Grx2, and Grx5. All sites with probability scores >0.8 are boxed. The CXXC active-site sequence is underlined. The sequence alignment was adapted from Johansson et al. (60).
Oxidative modification
Although Grx has been characterized as a regulator of protein thiol-disulfide status, some investigators suggest that its own cysteines may be modified by oxidation, with concomitant changes in activity (see later). Human Grx1 has three cysteines outside its active site (C7, C78, and C82). NMR structural analysis predicts that they are all solvent accessible (128), and thus susceptible to oxidative modification. In contrast, the two non–active-site cysteines of Grx2 (C28, C100) appear to be involved in a structural disulfide bond (60, 112). Although these observations support the possibility of posttranslational redox regulation of Grx, little is known about the specific modifications or their relevance to redox homeostasis in vivo.
Grx1 treated with HEDS is converted to multiple intramolecular and mixed-disulfide forms according to mass spectrometric analysis, and fully active enzyme is regained by adding it to the assay mix containing GSH (95). Likewise, both Grx1 and Grx2 treated with GSSG exhibited a near total loss of thiol content, attributable to intramolecular or mixed disulfides or both by mass spectrometry (50), and this was associated with a 25% loss of activity for Grx1 that could be fully reversed by DTT. The in vivo relevance of this relatively small deactivation by GSSG is questionable. First, the oxidative challenge (10 mM GSSG in the absence of GSH) does not represent a physiologic condition. Second, the absence of GSH may allow oxidations that would not occur in its presence, resulting in “false-positive” modifications. These considerations are useful in interpreting the results of additional oxidation experiments (later).
H2O2
Human Grx1 and Grx2 undergo partial inactivation at millimolar concentrations of H2O2 in vitro, with Grx1 being slightly more sensitive to inactivation (44, 50, 101). H2O2-treated Grx1 could be reactivated by DTT, GSH, or the Trx/TR system (101). In contrast, high concentrations of GSH (5 mM) but not low GSH concentrations (0.5 mM), DTT, or the Trx/TR system restored Grx2 activity after H2O2 treatment (44). The potential effect of H2O2 on Grx activity is an important consideration because H2O2 may reach relatively high concentrations in subcellular compartments where Grx isoforms are found. It will be critical to document the dose dependence of H2O2-mediated inactivation in the presence of GSH because physiologic levels of GSH may offset the oxidative effects of H2O2 on the enzymes, thus diminishing the relevance of the modifications.
•OH
Hydroxyl radicals are formed by reaction of H2O2 with metal ions such as Fe2+ and Cu+ (49), and are widely implicated in oxidative damage in disease states such as ischemia/reperfusion injury. We (123) observed inactivation of Grx1 in a dose-dependent manner by a hydroxyl radical–generating system in vitro. However, stop-flow ischemia/reperfusion of intact rabbit heart did not affect Grx1 activity, suggesting either that ischemia/reperfusion did not cause sufficient oxidative stress to inhibit the enzyme, or that the inhibition was reversed by a component absent from in vitro assays (e.g., GSH; see earlier).
Reactive nitrogen species (RNS)
Reactive nitrogen species, including peroxynitrite (ONOO−, a product of O2•− and NO), and S-nitrosoglutathione (GSNO) are produced under redox signaling and oxidative stress conditions (reviewed in ref. 37) and are associated with both glutathionylation (2, 22) and nitrosylation (43, 65, 84) of protein cysteines. To date, it appears that mammalian Grx isozymes exhibit distinct sensitivities to RNS. For example, rat liver Grx1 (5) and recombinant Grx1 (50) were largely inactivated after ONOO− exposure in vitro, whereas Grx2 activity was not affected (50). The ONOO−-dependent inactivation of the recombinant Grx1 was not reversed by DTT, suggesting a nondisulfide modification (cysteine sulfinic or sulfonic acid formation, or tyrosine nitration). The lack of reversal precludes this modification as a regulatory mechanism. For GSNO, the inhibition of Grx1 was influenced by O2, with greater inhibition and refractoriness to reversal under anaerobic conditions (50). The GSNO-mediated inhibition correlated with appearance of an absorption band characteristic of protein-SNO, but a specific site of Grx1 nitrosylation was not determined. Monomeric Grx2, like Grx1, exhibited a near total loss of thiol content on exposure to GSNO; however, Grx2 activity was not affected by treatment. Dimeric Grx2 dissociated into active monomers in the presence of GSNO, suggesting a potential mechanism for regulation in situ (50).
Proteolysis
Many splice variants of human Grx2 have been identified (71, 75); and one (termed Grx2a) codes for an N-terminal mitochondrial signal sequence. The functionality of the signal sequence was confirmed by demonstrating that Grx2-GFP fusion proteins were targeted to the mitochondria (44; 75), and that immunoreactive Grx2 was isolated from the matrix fraction of mitochondria from rat tissues (93). The mature, truncated form of Grx2 is about 4 times more active than the full-length form, possibly due to steric interference by the signal sequence (60, 75).
Analysis of the predicted sequence of human Grx5 by using ProP1.0 software (25) reveals two putative cleavage sites for proprotein convertase (PC). PC enzymes cleave pre-pro-proteins at specific sites containing single or paired basic amino acids, often before secretion. No PC cleavage site was detected for Grx1, although it is speculated to be a secreted protein (73, 97).
Metal coordination
Cd2+ is a non–redox active, toxic heavy metal ion that characteristically binds to vicinal dithiols, and Cd2+ inhibits Grx1 in a dose-dependent manner in vitro, apparently by coordinating to the thiolate forms of the neighboring active-site cysteine moieties. In HT4 cells, Cd2+ treatment was correlated with increased protein-SSG content (34); and Cd2+dose-dependent inhibition of deglutathionylation activity was observed in Jurkat and H9 cells (21). In contrast, a mutant strain of Streptomyces cerevisiae lacking yeast Grx2 exhibited decreased protein-SSG content after Cd2+ exposure (45), suggesting that yeast Grx2 may exhibit primarily glutathionylating activity in situ, as observed for the human Grx enzymes in vitro under glutathionyl radical–generating conditions (38, 124). Because Cd2+ is not likely available to coordinate to the Grx enzymes naturally, and confers an essentially irreversible inactivation (21), this does not represent a likely mechanism of regulation.
Recent studies on human Grx2 (11, 60, 69) and poplar GrxC1 (31, 111) suggest that these isozymes may exist naturally within dimeric 2Fe2S clusters, and that enzyme activity may be directly influenced by dimer integrity. Structural analysis of recombinant Grx from both species suggests that coordination to the 2Fe2S center uses two sulfhydryl groups per monomer: one from the catalytic cysteine, and the other from a non-covalently associated GSH molecule (31, 60, 111). Because an iron-coordinated cysteine thiolate is not available for nucleophilic attack (i.e., it is complexed to the positively charged metal ion), it is not surprising that the Grx2 dimer exhibits no detectable catalytic activity (69). In HeLa and BL30 cells cultured with 55Fe, the majority of immunoprecipitated Grx2 was radioactive, indicating association with the radiolabeled iron and suggesting that Grx2 may be predominantly dimerized (i.e., inactive) under nonstressed conditions in these cells. Clearly, identifying factors that lead to the release of active enzyme is critical for predicting Grx2 activity within the mitochondrial matrix. In vitro, a variety of thiol oxidants [e.g., GSSG (69)], reductants [e.g., dithionite, ascorbate (69)], and RNS [e.g., GSNO, SIN-1 (50)] disrupt the Grx2 dimer, but intracellular regulators of dimer status remain unknown.
Finally, it has been suggested that Grx2 may function to regulate iron sulfur cluster homeostasis (69), but evidence supporting such a role—including whether its thioltransferase activity is required—represents a direction for future investigation.
Protein–protein interactions
ASK1
Association of Grx1 with apoptosis signal-regulating kinase (ASK1) has been proposed to regulate ASK1 activity—and subsequently apoptosis—in multidrug-resistant MCF7 (MCF7/ADR) cells subjected to metabolic stress (120). Specifically, the ability to co-precipitate endogenous Grx1 and ASK1 was lost with glucose deprivation, and the dissociation of the Grx1-ASK complex was associated with ASK1 activation and increased apoptosis. The observation that Grx1-ASK1 dissociation was blocked by mutation of either or both of the Grx1 active-site cysteines led the authors to propose that fully reduced Grx1 is the form that associates with ASK1, and formation of the active-site intramolecular disulfide of Grx1 is required for its dissociation from the heterodimer (Fig. 8A). If the heterodimerized Grx1 is truly in its reduced form, then it should be catalytically competent; however, it is possible that the site of interaction with ASK1 (or the subcellular localization of ASK1) could restrict its availability to protein-SSG substrates. Conversely, the released Grx1-disulfide should be quickly reduced by GSH so long as GSH is not depleted. Clearly, testing cytosolic Grx1 activity before and after glucose deprivation would help answer this question. The result of such experiments would be particularly interesting, given the apparently opposing effects of Grx1 on apoptotic signaling [i.e., Grx1 overexpression protects H9c2 cells from apoptosis via regulation of Akt activity (88), but Grx1-catalyzed deglutathionylation of caspase-3 supports apoptosis in TNF-α–treated endothelial cells, see later (94)]. Thus, the increase in ASK1 activity attributed to its dissociation from Grx1 could be either potentiated or countered by the increase in Grx1 activity after its release.
FIG. 8.

Proposed interactions between Grx1 and cytosolic enzymes. (A) Reduced Grx1 is shown bound to ASK1 in MCF-7/ADR (multidrug-resistant breast cancer) cells under resting conditions (120). On glucose deprivation, the GSH/GSSG ratio decreases, resulting in oxidation of thiols in the Grx1 active site and disruption of its association with ASK1. Dissociation of Grx1 and ASK1 leads to ASK1 activation and increased apoptosis. (B) Grx1 and caspase-3 are shown associated under nonstressed conditions in BAECs. TNF-α–induced activation of Grx leads to pro-caspase-3 deglutathionylation, release from the complex, and cleavage by caspase-8 to active caspase-3, triggering apoptosis (94).
Also to be considered is the percentage of total Grx1 in complex with ASK1 under resting conditions. The heterodimerization status of Grx1, if inactive in this bound form, would determine the deglutathionylase activity available for cytosolic protein-SSG targets. Resolving these important questions warrants further study.
Pro-caspase-3
Another protein that appears to colocalize with Grx1 is pro-caspase-3. Immunoprecipitation of Grx1 from bovine aortic endothelial cells pulled down a protein that reacts with an anti-caspase-3 antibody. This interaction was almost completely lost after TNF-α/cycloheximide treatment, leading Pan and Berk (94) to propose a model shown in Fig. 8B. That is, Grx1 and caspase-3 are associated under resting conditions, but TNF-α stimulation causes Grx1 to deglutathionylate pro-caspase 3-SSG, leading to its cleavage and activation, which promotes downstream apoptotic events. As discussed later, the specific mechanism of Grx1 activation by TNF-α has not yet been identified.
Examples of apparent rapid activation of Grx
FGF-induced actin-SSG deglutathionylation
Treatment of NIH 3T3 fibroblasts with FGF (Fig. 9A) resulted in robust deglutathionylation of β-actin within minutes (139). Although steady-state levels of actin-SSG were not sensitive to manipulation of Grx1 content, its FGF-induced deglutathionylation was completely blocked by knockout of Grx1 by stable transfection of siRNA targeting Grx1. In addition, actin-SSG status was correlated inversely to actin polymerization (137) and cytoskeletal reorganization (139), suggesting that FGF-induced actin deglutathionylation may aid cytoskeletal changes involved in the proliferation response. Although these observations could be explained by acute activation of Grx1, several conceivable explanations must be distinguished, including (a) activation of Grx1 deglutathionylation activity (via posttranslational modification, conformational change, or some other mechanism); (b) exposure of the actin-SSG disulfide bond to already active Grx1 (via conformational change in actin or other cytoskeletal components); and (c) rearrangement of Grx1 and actin-SSG into close proximity via scaffolding or protein–protein interactions. Experiments that would address these possibilities include testing the effect of FGF treatment on global Grx1 activity (i.e., distinguishing changes in activity from changes in substrate availability), investigating the subcellular localization of Grx1 before and after FGF stimulus, and performing pull-down experiments with antibodies to actin and Grx to determine whether the two proteins may be colocalized through mutual protein–protein interactions.
FIG. 9.

Examples of acute regulation of Grx1. (A) Treatment of NIH 3T3 fibroblasts with FGF resulted in robust deglutathionylation of actin within 15 min (139). Actin deglutathionylation and associated cytoskeletal changes were blocked by knockdown of Grx1. (B) Bovine aortic endothelial cells (BAECs) exposed to shear stress in culture exhibited increased Grx activity within the first 5–10 min of treatment (138). Increased Grx activity was correlated to increases in phosphorylation of eNOS and Akt. Changes in Grx activity were blocked by treatment with BCNU, whereas the changes in downstream events were blocked by Grx1 knockdown or transfection with catalytically inactive mutant enzyme. (C) Treatment of BAECs with TNF-α and cycloheximide was correlated with increased Grx activity within 3–6 h, deglutathionylation of pro-caspase-3, increased caspase-3 activity, and increased apoptosis (94). Events downstream of caspase-3 deglutathionylation were blocked by Grx1 knockdown or transfection of a catalytically inactive mutant.
Shear stress
Flow, or fluid shear stress, represents the frictional force of blood acting on the surface of vascular endothelial cells (138) (Fig. 9B). Disruption of physiologic flow rates appears to alter signal-transduction and gene-expression patterns in endothelial cells, although the specific mechanisms are not yet fully elucidated (20). Wang et al. (138) observed that cultured bovine aortic endothelial cells exhibited a brief (5 to 10 min) but robust increase in total Grx activity on exposure to shear stress. Grx activation was correlated to increases in Akt and eNOS phosphorylation, and it was blocked by both siRNA directed to Grx1 and transfection of a catalytically inactive Grx1 mutant (C22S/C25S). This short time course precludes induction of the enzyme and suggests an acute, reversible mechanism of regulation of Grx1. The authors propose that the increase in Grx1 activity is explained by a similarly brief increase in the activity of GSSG reductase (GR), the enzyme that reduces GSSG, the second product of Grx-mediated protein deglutathionylation (Fig. 3A). This model is based on observations that shear stress also resulted in increased GR activity, and BCNU (a GR inhibitor) blocked the downstream effects of shear stress–induced Grx1 activity. Some assumptions related to this interpretation should be tested explicitly: that the BCNU did not inhibit Grx1 directly; and that GR activity is limiting in this system (i.e., that an increase in Grx activity can be “realized” only if GR activity is increased). If GR activity is not limiting, then future studies should focus on events such as posttranslational modification of Grx1, etc. (see earlier).
TNF-α–induced caspase-3 deglutathionylation
As discussed earlier, treatment of bovine aortic endothelial cells with TNF-α and cycloheximide (CHX) resulted in an increase in total Grx activity (94). Although the time course of activation by TNF-α/CHX (3–6 h) is longer than those observed for FGF and shear-stress exposures (Fig. 9C), it is unlikely to be explained by changes in protein expression or degradation because the time course is much shorter than the half-life of the enzyme [Grx1 t1/2 ∼ 1.5 days (139)]. Because Grx1 and its substrate (pro-caspase 3-SSG) appear to be physically associated at baseline (see earlier), a likely mechanism of activation would involve a post-translational modification or conformational change allowing active Grx1 to access the glutathionylated cysteine of the pro-caspase 3 substrate.
Conclusions
The functional consequences of the deglutathionating activity of the glutaredoxins are likely profound. The high catalytic rate and rate enhancement over the low uncatalyzed reaction rate imply that Grx controls the glutathionylation status of proteins that are specifically glutathionylated, analogous to the action of phosphatases. The specificity of Grx for protein-SSG substrates combined with the enhanced reactivity of Grx-SSG with reduced GSH suggest that reversible glutathionylation is an organized and systematic signaling system, analogous to the kinase/phosphatase signaling cascades. Modulation of intracellular Grx activity by hormones, stress factors, and disease states suggests an additional level of regulation of protein glutathionylation status via Grx in vivo. The biochemical features that this catalytic system exhibits may uniquely poise glutaredoxin as a sensitive and important cellular effector.
Abbreviations
2Fe2S, 2-iron, 2-sulfur cluster; βME, β-mercaptoethanol; βME-SSG, hydroxyethyl-glutathione mixed disulfide; Akt, protein kinase B; AP-1, activator protein-1; ARE, antioxidant response element; ASK1, apoptosis signal-regulating kinase; BCNU, bis-chloronitrosourea; CA-III, carbonic anhydrase III; DTT, dithiothreitol; eNOS, endothelial nitric oxide synthase; FAD, flavin adenine dinucleotide; FGF, fibroblast growth factor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GR, glutathione disulfide reductase; GPx, glutathione peroxidase; Grx, glutaredoxin; GS•, glutathione thiyl radical; GS-, glutathionyl; GSH, glutathione; GST, glutathione-S-transferase; GSNO, S-nitrosoglutathione; GSSG, glutathione disulfide; h, human; H2O2, hydrogen peroxide; HEDS, hydroxyethyl disulfide; HPLC, high-performance liquid chromatography; JNK, Jun-N-terminal kinase; KM, Michaelis constant (substrate concentration at which v = 1/2Vmax); KMint, intrinsic (or “true”) KM; Kox, the GSH/GSSG ratio at which protein/SH:protein-SSG = 1; m, mouse; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NADPH, nicotinamide adenine dinucleotide phosphate (reduced); NMR, nuclear magnetic resonance; NF-κB, nuclear factor κB; O2•−, superoxide; •OH, hydroxyl radical; ONOO−, peroxynitrite; PC, proprotein convertase; PICOT, PKCθ-interacting cousin of thioredoxin; PKC, protein kinase C; protein-SSG, protein-glutathione mixed disulfide; Prx, peroxiredoxin; PSH, protein with a reduced cysteine moiety; PSSG, protein-glutathione mixed disulfide; PTP1B, protein tyrosine phosphatase 1B; RNR, ribonucleotide reductase; RNS, reactive nitrogen species; ROS, reactive oxygen species; RT-PCR, reverse transcriptase polymerase chain reaction; Sc, Streptomyces cerevisiae; SIN-1, 3-morpholinosydnonimine; Tdh3, glyceraldehyde 3-phosphate dehydrogenase, isozyme 3 (S. cerevisiae); TGR, thioredoxin and GSSG reductase; TNF-α, tumor necrosis factor α; TR, thioredoxin reductase; Trx, thioredoxin; Vmax, maximum velocity; Vmaxint, intrinsic (or “true”) Vmax.
Acknowledgments
This work was supported in part by NIH research grants R01 AG024413 and P01 AG 15885 (J.J.M.), and a Merit Review research grant from the Department of Veteran's Affairs (J.J.M.); and by NIH training grants F30 AG029687, T32 GM008803, and T32 GM07250 (M.M.G.).
References
- 1.Adachi T. Pimentel DR. Heibeck T. Hou X. Lee YJ. Jiang B. Ido Y. Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 2.Adachi T. Weisbrod RM. Pimentel DR. Ying J. Sharov VS. Schoneich C. Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 3.Askelof P. Axelsson K. Eriksson S. Mannervik B. Mechanism of action of enzymes catalyzing thiol-disulfide interchange: thioltransferases rather than transhydrogenases. FEBS Lett. 1974;38:263–267. doi: 10.1016/0014-5793(74)80068-2. [DOI] [PubMed] [Google Scholar]
- 4.Aslund F. Ehn B. Miranda-Vizuete A. Pueyo C. Holmgren A. Two additional glutaredoxins exist in Escherichia coli: glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc Natl Acad Sci U S A. 1994;91:9813–9817. doi: 10.1073/pnas.91.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aykac-Toker G. Bulgurcuoglu S. Kocak-Toker N. Effect of peroxynitrite on glutaredoxin. Hum Exp Toxicol. 2001;20:373–376. doi: 10.1191/096032701680350578. [DOI] [PubMed] [Google Scholar]
- 6.Baker AF. Landowski T. Dorr R. Tate WR. Gard JM. Tavenner BE. Dragovich T. Coon A. Powis G. The antitumor agent imexon activates antioxidant gene expression: evidence for an oxidative stress response. Clin Cancer Res. 2007;13:3388–3394. doi: 10.1158/1078-0432.CCR-06-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrett WC. DeGnore JP. Keng YF. Zhang ZY. Yim MB. Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 8.Barrett WC. DeGnore JP. Konig S. Fales HM. Keng YF. Zhang ZY. Yim MB. Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 9.Beer SM. Taylor ER. Brown SE. Dahm CC. Costa NJ. Runswick MJ. Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant defense. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 10.Belli G. Polaina J. Tamarit J. De La Torre MA. Rodriguez-Manzaneque MT. Ros J. Herrero E. Structure-function analysis of yeast Grx5 monothiol glutaredoxin defines essential amino acids for the function of the protein. J Biol Chem. 2002;277:37590–37596. doi: 10.1074/jbc.M201688200. [DOI] [PubMed] [Google Scholar]
- 11.Berndt C. Hudemann C. Hanschmann EM. Axelsson R. Holmgren A. Lillig CH. How does iron-sulfur cluster coordination regulate the activity of human glutaredoxin 2? Antioxid Redox Signal. 2007;9:151–157. doi: 10.1089/ars.2007.9.151. [DOI] [PubMed] [Google Scholar]
- 12.Blom N. Gammeltoft S. Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 13.Blom N. Sicheritz-Ponten T. Gupta R. Gammeltoft S. Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 14.Bushweller JH. Aslund F. Wuthrich K. Holmgren A. Structural and functional characterization of the mutant Escherichia coli glutaredoxin (C14—S) and its mixed disulfide with glutathione. Biochemistry. 1992;31:9288–9293. doi: 10.1021/bi00153a023. [DOI] [PubMed] [Google Scholar]
- 15.Bushweller JH. Billeter M. Holmgren A. Wuthrich K. The nuclear magnetic resonance solution structure of the mixed disulfide between Escherichia coli glutaredoxin (C14S) and glutathione. J Mol Biol. 1994;235:1585–1597. doi: 10.1006/jmbi.1994.1108. [DOI] [PubMed] [Google Scholar]
- 16.Camaschella C. Campanella A. De FL. Boschetto L. Merlini R. Silvestri L. Levi S. Iolascon A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–1358. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 17.Chandra A. Srivastava S. Petrash JM. Bhatnagar A. Srivastava SK. Modification of aldose reductase by S-nitrosoglutathione. Biochemistry. 1997;36:15801–15809. doi: 10.1021/bi9714722. [DOI] [PubMed] [Google Scholar]
- 18.Chen FC. Ogut O. Decline of contractility during ischemia-reperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am J Physiol Cell Physiol. 2006;290:C719–C727. doi: 10.1152/ajpcell.00419.2005. [DOI] [PubMed] [Google Scholar]
- 19.Chen YR. Chen CL. Pfeiffer DR. Zweier JL. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 20.Chiu JJ. Usami S. Chien S. Vascular endothelial responses to altered shear stress: pathologic implications for atherosclerosis. Ann Med. 2009;41:19–28. doi: 10.1080/07853890802186921. [DOI] [PubMed] [Google Scholar]
- 21.Chrestensen CA. Starke DW. Mieyal JJ. Acute cadmium exposure inactivates thioltransferase (glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J Biol Chem. 2000;275:26556–26565. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- 22.Clavreul N. Adachi T. Pimental DR. Ido Y. Schoneich C. Cohen RA. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006;20:518–520. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- 23.Dalle-Donne I. Milzani A. Gagliano N. Colombo R. Giustarini D. Rossi R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid Redox Signal. 2008;10:445–473. doi: 10.1089/ars.2007.1716. [DOI] [PubMed] [Google Scholar]
- 24.Discola KF. de Oliveira MA. Rosa C Jr. Monteiro G. Barcena JA. Porras P. Padilla CA. Guimaraes BG. Soares Netto LE. Structural aspects of the distinct biochemical properties of glutaredoxin 1 and glutaredoxin 2 from Saccharomyces cerevisiae. J Mol Biol. 2009;385:889–901. doi: 10.1016/j.jmb.2008.10.055. [DOI] [PubMed] [Google Scholar]
- 25.Duckert P. Brunak S. Blom N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel. 2004;17:107–112. doi: 10.1093/protein/gzh013. [DOI] [PubMed] [Google Scholar]
- 26.Eaton P. Wright N. Hearse DJ. Shattock MJ. Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J Mol Cell Cardiol. 2002;34:1549–1560. doi: 10.1006/jmcc.2002.2108. [DOI] [PubMed] [Google Scholar]
- 27.Eklund H. Cambillau C. Sjoberg BM. Holmgren A. Jornvall H. Hoog JO. Branden CI. Conformational and functional similarities between glutaredoxin and thioredoxins. EMBO J. 1984;3:1443–1449. doi: 10.1002/j.1460-2075.1984.tb01994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elgan TH. Berndt KD. Quantifying Escherichia coli glutaredoxin-3 substrate specificity using ligand-induced stability. J Biol Chem. 2008;283:32839–32847. doi: 10.1074/jbc.M804019200. [DOI] [PubMed] [Google Scholar]
- 29.Enoksson M. Fernandes AP. Prast S. Lillig CH. Holmgren A. Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem Biophys Res Commun. 2005;327:774–779. doi: 10.1016/j.bbrc.2004.12.067. [DOI] [PubMed] [Google Scholar]
- 30.Eriksson S. Askelof P. Axelsson K. Mannervik B. Evidence for a glutathione-dependent interconversion of two forms of a thioltransferase from rat liver catalyzing thiol-disulfide interchange. Acta Chem Scand B. 1974;28:931–936. doi: 10.3891/acta.chem.scand.28b-0931. [DOI] [PubMed] [Google Scholar]
- 31.Feng Y. Zhong N. Rouhier N. Hase T. Kusunoki M. Jacquot JP. Jin C. Xia B. Structural insight into poplar glutaredoxin C1 with a bridging iron-sulfur cluster at the active site. Biochemistry. 2006;45:7998–8008. doi: 10.1021/bi060444t. [DOI] [PubMed] [Google Scholar]
- 32.Fernandes AP. Fladvad M. Berndt C. Andresen C. Lillig CH. Neubauer P. Sunnerhagen M. Holmgren A. Vlamis-Gardikas A. A novel monothiol glutaredoxin (Grx4) from Escherichia coli can serve as a substrate for thioredoxin reductase. J Biol Chem. 2005;280:24544–24552. doi: 10.1074/jbc.M500678200. [DOI] [PubMed] [Google Scholar]
- 33.Fernandes AP. Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 34.Figueiredo-Pereira ME. Yakushin S. Cohen G. Disruption of the intracellular sulfhydryl homeostasis by cadmium-induced oxidative stress leads to protein thiolation and ubiquitination in neuronal cells. J Biol Chem. 1998;273:12703–12709. doi: 10.1074/jbc.273.21.12703. [DOI] [PubMed] [Google Scholar]
- 35.Fladvad M. Bellanda M. Fernandes AP. Mammi S. Vlamis-Gardikas A. Holmgren A. Sunnerhagen M. Molecular mapping of functionalities in the solution structure of reduced Grx4, a monothiol glutaredoxin from Escherichia coli. J Biol Chem. 2005;280:24553–24561. doi: 10.1074/jbc.M500679200. [DOI] [PubMed] [Google Scholar]
- 36.Foloppe N. Nilsson L. Stabilization of the catalytic thiolate in a mammalian glutaredoxin: structure, dynamics and electrostatics of reduced pig glutaredoxin and its mutants. J Mol Biol. 2007;372:798–816. doi: 10.1016/j.jmb.2007.05.101. [DOI] [PubMed] [Google Scholar]
- 37.Gallogly MM. Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 38.Gallogly MM. Starke DW. Leonberg AK. Ospina SM. Mieyal JJ. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry. 2008;47:11144–11157. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan ZR. Cloning and sequencing of a gene encoding yeast thioltransferase. Biochem Biophys Res Commun. 1992;187:949–955. doi: 10.1016/0006-291x(92)91289-3. [DOI] [PubMed] [Google Scholar]
- 40.Gan ZR. Wells WW. Identification and reactivity of the catalytic site of pig liver thioltransferase. J Biol Chem. 1987;262:6704–6707. [PubMed] [Google Scholar]
- 41.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 42.Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 43.Giustarini D. Milzani A. Aldini G. Carini M. Rossi R. Dalle-Donne I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid Redox Signal. 2005;7:930–939. doi: 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- 44.Gladyshev VN. Liu A. Novoselov SV. Krysan K. Sun QA. Kryukov VM. Kryukov GV. Lou MF. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J Biol Chem. 2001;276:30374–30380. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 45.Gomes DS. Pereira MD. Panek AD. Andrade LR. Eleutherio EC. Apoptosis as a mechanism for removal of mutated cells of Saccharomyces cerevisiae: the role of Grx2 under cadmium exposure. Biochim Biophys Acta. 2008;1780:160–166. doi: 10.1016/j.bbagen.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 46.Gon S. Faulkner MJ. Beckwith J. In vivo requirement for glutaredoxins and thioredoxins in the reduction of the ribonucleotide reductases of Escherichia coli. Antioxid Redox Signal. 2006;8:735–742. doi: 10.1089/ars.2006.8.735. [DOI] [PubMed] [Google Scholar]
- 47.Gravina SA. Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32:3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 48.Hakansson KO. Winther JR. Structure of glutaredoxin Grx1p C30S mutant from yeast. Acta Crystallogr D Biol Crystallogr. 2007;63:288–294. doi: 10.1107/S0907444906051675. [DOI] [PubMed] [Google Scholar]
- 49.Halliwell B. Mechanisms involved in the generation of free radicals. Pathol Biol (Paris) 1996;44:6–13. [PubMed] [Google Scholar]
- 50.Hashemy SI. Johansson C. Berndt C. Lillig CH. Holmgren A. Oxidation and S-nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: effects on structure and activity. J Biol Chem. 2007;282:14428–14436. doi: 10.1074/jbc.M700927200. [DOI] [PubMed] [Google Scholar]
- 51.Hatakeyama M. Tanimoto Y. Mizoguchi T. Purification and some properties of bovine liver cytosol thioltransferase. J Biochem. 1984;95:1811–1818. doi: 10.1093/oxfordjournals.jbchem.a134794. [DOI] [PubMed] [Google Scholar]
- 52.Ho YS. Xiong Y. Ho DS. Gao J. Chua BH. Pai H. Mieyal JJ. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic Biol Med. 2007;43:1299–1312. doi: 10.1016/j.freeradbiomed.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmgren A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc Natl Acad Sci U S A. 1976;73:2275–2279. doi: 10.1073/pnas.73.7.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holmgren A. Glutathione-dependent synthesis of deoxyribonucleotides: characterization of the enzymatic mechanism of Escherichia coli glutaredoxin. J Biol Chem. 1979;254:3672–3678. [PubMed] [Google Scholar]
- 55.Hudemann C. Lonn ME. Godoy JR. Avval FZ. Capani F. Holmgren A. Lillig CH. Identification, expression pattern, and characterization of mouse glutaredoxin 2 isoforms. Antioxid Redox Signal. 2009;11:1–14. doi: 10.1089/ars.2008.2068. [DOI] [PubMed] [Google Scholar]
- 56.Hurd TR. Requejo R. Filipovska A. Brown S. Prime TA. Robinson AJ. Fearnley IM. Murphy MP. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Isakov N. Witte S. Altman A. PICOT-HD: a highly conserved protein domain that is often associated with thioredoxin and glutaredoxin modules. Trends Biochem Sci. 2000;25:537–539. doi: 10.1016/s0968-0004(00)01685-6. [DOI] [PubMed] [Google Scholar]
- 58.Jao SC. English Ospina SM. Berdis AJ. Starke DW. Post CB. Mieyal JJ. Computational and mutational analysis of human glutaredoxin (thioltransferase): probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry. 2006;45:4785–4796. doi: 10.1021/bi0516327. [DOI] [PubMed] [Google Scholar]
- 59.Ji Y. Akerboom TP. Sies H. Thomas JA. S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Arch Biochem Biophys. 1999;362:67–78. doi: 10.1006/abbi.1998.1013. [DOI] [PubMed] [Google Scholar]
- 60.Johansson C. Kavanagh KL. Gileadi O. Oppermann U. Reversible sequestration of active site cysteines in a 2Fe-2S-bridged dimer provides a mechanism for glutaredoxin 2 regulation in human mitochondria. J Biol Chem. 2007;282:3077–3082. doi: 10.1074/jbc.M608179200. [DOI] [PubMed] [Google Scholar]
- 61.Johansson C. Lillig CH. Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J Biol Chem. 2004;279:7537–7543. doi: 10.1074/jbc.M312719200. [DOI] [PubMed] [Google Scholar]
- 62.Kallis GB. Holmgren A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J Biol Chem. 1980;255:10261–10265. [PubMed] [Google Scholar]
- 63.Kenchappa RS. Diwakar L. Annepu J. Ravindranath V. Estrogen and neuroprotection: higher constitutive expression of glutaredoxin in female mice offers protection against MPTP-mediated neurodegeneration. FASEB J. 2004;18:1102–1104. doi: 10.1096/fj.03-1075fje. [DOI] [PubMed] [Google Scholar]
- 64.Khojasteh-Bakht SC. Chen W. Koenigs LL. Peter RM. Nelson SD. Metabolism of (R)-(+)-pulegone and (R)-(+)-menthofuran by human liver cytochrome P-450s: evidence for formation of a furan epoxide. Drug Metab Dispos. 1999;27:574–580. [PubMed] [Google Scholar]
- 65.Klatt P. Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 66.Klatt P. Molina EP. De Lacoba MG. Padilla CA. Martinez-Galesteo E. Barcena JA. Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 67.Kortemme T. Creighton TE. Ionisation of cysteine residues at the termini of model alpha-helical peptides: relevance to unusual thiol pKa values in proteins of the thioredoxin family. J Mol Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 68.Lillig CH. Berndt C. Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1980:1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 69.Lillig CH. Berndt C. Vergnolle O. Lonn ME. Hudemann C. Bill E. Holmgren A. Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc Natl Acad Sci U S A. 2005;102:8168–8173. doi: 10.1073/pnas.0500735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Llopis J. McCaffery JM. Miyawaki A. Farquhar MG. Tsien RY. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci U S A. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lonn ME. Hudemann C. Berndt C. Cherkasov V. Capani F. Holmgren A. Lillig CH. Expression pattern of human glutaredoxin 2 isoforms: identification and characterization of two testis/cancer cell-specific isoforms. Antioxid Redox Signal. 2008;10:547–557. doi: 10.1089/ars.2007.1821. [DOI] [PubMed] [Google Scholar]
- 72.Luikenhuis S. Perrone G. Dawes IW. Grant CM. The yeast Saccharomyces cerevisiae contains two glutaredoxin genes that are required for protection against reactive oxygen species. Mol Biol Cell. 1998;9:1081–1091. doi: 10.1091/mbc.9.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lundberg M. Fernandes AP. Kumar S. Holmgren A. Cellular and plasma levels of human glutaredoxin 1 and 2 detected by sensitive ELISA systems. Biochem Biophys Res Commun. 2004;319:801–809. doi: 10.1016/j.bbrc.2004.04.199. [DOI] [PubMed] [Google Scholar]
- 74.Lundberg M. Holmgren A. Johansson M. Human glutaredoxin 2 affinity tag for recombinant peptide and protein purification. Protein Expr Purif. 2006;45:37–42. doi: 10.1016/j.pep.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 75.Lundberg M. Johansson C. Chandra J. Enoksson M. Jacobsson G. Ljung J. Johansson M. Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J Biol Chem. 2001;276:26269–26275. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 76.Luthman M. Holmgren A. Glutaredoxin from calf thymus: purification to homogeneity. J Biol Chem. 1982;257:6686–6690. [PubMed] [Google Scholar]
- 77.Martin JL. Thioredoxin: a fold for all reasons. Structure. 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 78.Matsui M. Oshima M. Oshima H. Takaku K. Maruyama T. Yodoi J. Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178:179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 79.Mesecke N. Mittler S. Eckers E. Herrmann JM. Deponte M. Two novel monothiol glutaredoxins from Saccharomyces cerevisiae provide further insight into iron-sulfur cluster binding, oligomerization, and enzymatic activity of glutaredoxins. Biochemistry. 2008;47:1452–1463. doi: 10.1021/bi7017865. [DOI] [PubMed] [Google Scholar]
- 80.Mieyal JJ. Gallogly MM. Qanungo S. Sabens EA. Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mieyal JJ. Srinivasan U. Starke DW. Gravina S A. Mieyal P A. Packer L. Cadenas E. In Biothiols in Health and Disease. New York: Marcel Dekker; 1995. Glutathionyl specificity of the thioltransferases: mechanistic and physiological implications; pp. 305–372. [Google Scholar]
- 82.Mieyal JJ. Starke DW. Gravina SA. Dothey C. Chung JS. Thioltransferase in human red blood cells: purification and properties. Biochemistry. 1991;30:6088–6097. doi: 10.1021/bi00239a002. [DOI] [PubMed] [Google Scholar]
- 83.Mieyal JJ. Starke DW. Gravina SA. Hocevar BA. Thioltransferase in human red blood cells: kinetics and equilibrium. Biochemistry. 1991;30:8883–8891. doi: 10.1021/bi00100a023. [DOI] [PubMed] [Google Scholar]
- 84.Mohr S. Hallak H. de Boitte A. Lapetina EG. Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 85.Molina-Navarro MM. Casas C. Piedrafita L. Belli G. Herrero E. Prokaryotic and eukaryotic monothiol glutaredoxins are able to perform the functions of Grx5 in the biogenesis of Fe/S clusters in yeast mitochondria. FEBS Lett. 2006;580:2273–2280. doi: 10.1016/j.febslet.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 86.Morell S. Follmann H. Haberlein I. Identification and localization of the first glutaredoxin in leaves of a higher plant. FEBS Lett. 1995;369:149–152. doi: 10.1016/0014-5793(95)00690-b. [DOI] [PubMed] [Google Scholar]
- 87.Muhlenhoff U. Gerber J. Richhardt N. Lill R. Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. EMBO J. 2003;22:4815–4825. doi: 10.1093/emboj/cdg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murata H. Ihara Y. Nakamura H. Yodoi J. Sumikawa K. Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 89.Nikkola M. Gleason FK. Saarinen M. Joelson T. Bjornberg O. Eklund H. A putative glutathione-binding site in T4 glutaredoxin investigated by site-directed mutagenesis. J Biol Chem. 1991;266:16105–16112. [PubMed] [Google Scholar]
- 90.Noguera V. Walker O. Rouhier N. Jacquot JP. Krimm I. Lancelin JM. NMR reveals a novel glutaredoxin-glutaredoxin interaction interface. J Mol Biol. 2005;353:629–641. doi: 10.1016/j.jmb.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 91.Nordstrand K. Eslund F. Holmgren A. Otting G. Berndt KD. NMR structure of Escherichia coli glutaredoxin 3-glutathione mixed disulfide complex: implications for the enzymatic mechanism. J Mol Biol. 1999;286:541–552. doi: 10.1006/jmbi.1998.2444. [DOI] [PubMed] [Google Scholar]
- 92.Ojeda L. Keller G. Muhlenhoff U. Rutherford JC. Lill R. Winge DR. Role of glutaredoxin-3 and glutaredoxin-4 in the iron regulation of the Aft1 transcriptional activator in Saccharomyces cerevisiae. J Biol Chem. 2006;281:17661–17669. doi: 10.1074/jbc.M602165200. [DOI] [PubMed] [Google Scholar]
- 93.Pai HV. Starke DW. Lesnefsky EJ. Hoppel CL. Mieyal JJ. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid Redox Signal. 2007;9:2027–2033. doi: 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
- 94.Pan S. Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- 95.Papov VV. Gravina SA. Mieyal JJ. Biemann K. The primary structure and properties of thioltransferase (glutaredoxin) from human red blood cells. Protein Sci. 1994;3:428–434. doi: 10.1002/pro.5560030307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peltoniemi MJ. Karala AR. Jurvansuu JK. Kinnula VL. Ruddock LW. Insights into deglutathionylation reactions: different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the gamma-linkage present in glutathione. J Biol Chem. 2006;281:33107–33114. doi: 10.1074/jbc.M605602200. [DOI] [PubMed] [Google Scholar]
- 97.Peltoniemi MJ. Rytila PH. Harju TH. Soini YM. Salmenkivi KM. Ruddock LW. Kinnula VL. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir Res. 2006;7:133. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pimentel DR. Adachi T. Ido Y. Heibeck T. Jiang B. Lee Y. Melendez JA. Cohen RA. Colucci WS. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J Mol Cell Cardiol. 2006;41:613–622. doi: 10.1016/j.yjmcc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 99.Porcelli AM. Ghelli A. Zanna C. Pinton P. Rizzuto R. Rugolo M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem Biophys Res Commun. 2005;326:799–804. doi: 10.1016/j.bbrc.2004.11.105. [DOI] [PubMed] [Google Scholar]
- 100.Qanungo S. Starke DW. Pai HV. Mieyal JJ. Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 101.Qiao F. Xing K. Liu A. Ehlers N. Raghavachari N. Lou MF. Human lens thioltransferase: cloning, purification, and function. Invest Ophthalmol Vis Sci. 2001;42:743–751. [PubMed] [Google Scholar]
- 102.Rabenstein DL. Millis KK. Nuclear magnetic resonance study of the thioltransferase-catalyzed glutathione/glutathione disulfide interchange reaction. Biochim Biophys Acta. 1995;1249:29–36. doi: 10.1016/0167-4838(95)00067-5. [DOI] [PubMed] [Google Scholar]
- 103.Racker E. Glutathione-homocystine transhydrogenase. J Biol Chem. 1955;217:867–874. [PubMed] [Google Scholar]
- 104.Reynaert NL. van der Vliet A. Guala AS. McGovern T. Hristova M. Pantano C. Heintz NH. Heim J. Ho YS. Matthews DE. Wouters EF. Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rodriguez-Manzaneque MT. Ros J. Cabiscol E. Sorribas A. Herrero E. Grx5 glutaredoxin plays a central role in protection against protein oxidative damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:8180–8190. doi: 10.1128/mcb.19.12.8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rodriguez-Manzaneque MT. Tamarit J. Belli G. Ros J. Herrero E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol Biol Cell. 2002;13:1109–1121. doi: 10.1091/mbc.01-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rouhier N. Gelhaye E. Jacquot JP. Exploring the active site of plant glutaredoxin by site-directed mutagenesis. FEBS Lett. 2002;511:145–149. doi: 10.1016/s0014-5793(01)03302-6. [DOI] [PubMed] [Google Scholar]
- 108.Rouhier N. Gelhaye E. Jacquot JP. Glutaredoxin-dependent peroxiredoxin from poplar: protein-protein interaction and catalytic mechanism. J Biol Chem. 2002;277:13609–13614. doi: 10.1074/jbc.M111489200. [DOI] [PubMed] [Google Scholar]
- 109.Rouhier N. Gelhaye E. Jacquot JP. Plant glutaredoxins: still mysterious reducing systems. Cell Mol Life Sci. 2004;61:1266–1277. doi: 10.1007/s00018-004-3410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rouhier N. Gelhaye E. Sautiere PE. Brun A. Laurent P. Tagu D. Gerard J. De FE. Meyer Y. Jacquot JP. Isolation and characterization of a new peroxiredoxin from poplar sieve tubes that uses either glutaredoxin or thioredoxin as a proton donor. Plant Physiol. 2001;127:1299–1309. [PMC free article] [PubMed] [Google Scholar]
- 111.Rouhier N. Unno H. Bandyopadhyay S. Masip L. Kim SK. Hirasawa M. Gualberto JM. Lattard V. Kusunoki M. Knaff DB. Georgiou G. Hase T. Johnson MK. Jacquot JP. Functional, structural, and spectroscopic characterization of a glutathione-ligated [2Fe-2S] cluster in poplar glutaredoxin C1. Proc Natl Acad Sci U S A. 2007;104:7379–7384. doi: 10.1073/pnas.0702268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sagemark J. Elgan TH. Burglin TR. Johansson C. Holmgren A. Berndt KD. Redox properties and evolution of human glutaredoxins. Proteins. 2007;68:879–892. doi: 10.1002/prot.21416. [DOI] [PubMed] [Google Scholar]
- 113.Sha S. Minakuchi K. Higaki N. Sato K. Ohtsuki K. Kurata A. Yoshikawa H. Kotaru M. Masumura T. Ichihara K. Tanaka K. Purification and characterization of glutaredoxin (thioltransferase) from rice (Oryza sativa L.) J Biochem. 1997;121:842–848. doi: 10.1093/oxfordjournals.jbchem.a021663. [DOI] [PubMed] [Google Scholar]
- 114.Shelton MD. Chock PB. Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 115.Shelton MD. Kern TS. Mieyal JJ. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Muller cells: model of diabetic retinopathy. J Biol Chem. 2007;282:12467–12474. doi: 10.1074/jbc.M610863200. [DOI] [PubMed] [Google Scholar]
- 116.Shenton D. Perrone G. Quinn KA. Dawes IW. Grant CM. Regulation of protein S-thiolation by glutaredoxin 5 in the yeast Saccharomyces cerevisiae. J Biol Chem. 2002;277:16853–16859. doi: 10.1074/jbc.M200559200. [DOI] [PubMed] [Google Scholar]
- 117.Shi J. Vlamis-Gardikas A. Aslund F. Holmgren A. Rosen BP. Reactivity of glutaredoxins 1, 2, and 3 from Escherichia coli shows that glutaredoxin 2 is the primary hydrogen donor to ArsC-catalyzed arsenate reduction. J Biol Chem. 1999;274:36039–36042. doi: 10.1074/jbc.274.51.36039. [DOI] [PubMed] [Google Scholar]
- 118.Silva GM. Netto LE. Discola KF. Piassa-Filho GM. Pimenta DC. Barcena JA. Demasi M. Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J. 2008;275:2942–2955. doi: 10.1111/j.1742-4658.2008.06441.x. [DOI] [PubMed] [Google Scholar]
- 119.Singh SP. Wishnok JS. Keshive M. Deen WM. Tannenbaum SR. The chemistry of the S-nitrosoglutathione/glutathione system. Proc Natl Acad Sci U S A. 1996;93:14428–14433. doi: 10.1073/pnas.93.25.14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Song JJ. Rhee JG. Suntharalingam M. Walsh SA. Spitz DR. Lee YJ. Role of glutaredoxin in metabolic oxidative stress: glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 121.Spyrou G. Holmgren A. Deoxyribonucleoside triphosphate pools and growth of glutathione-depleted 3T6 mouse fibroblasts. Biochem Biophys Res Commun. 1996;220:42–46. doi: 10.1006/bbrc.1996.0353. [DOI] [PubMed] [Google Scholar]
- 122.Srinivasan U. Mieyal PA. Mieyal JJ. pH profiles indicative of rate-limiting nucleophilic displacement in thioltransferase catalysis. Biochemistry. 1997;36:3199–3206. doi: 10.1021/bi962017t. [DOI] [PubMed] [Google Scholar]
- 123.Starke DW. Chen Y. Bapna CP. Lesnefsky EJ. Mieyal JJ. Sensitivity of protein sulfhydryl repair enzymes to oxidative stress. Free Radic Biol Med. 1997;23:373–384. doi: 10.1016/s0891-5849(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 124.Starke DW. Chock PB. Mieyal JJ. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase): potential role in redox signal transduction. J Biol Chem. 2003;278:14607–14613. doi: 10.1074/jbc.M210434200. [DOI] [PubMed] [Google Scholar]
- 125.Stavreus-Evers A. Masironi B. Landgren BM. Holmgren A. Eriksson H. Sahlin L. Immunohistochemical localization of glutaredoxin and thioredoxin in human endometrium: a possible association with pinopodes. Mol Hum Reprod. 2002;8:546–551. doi: 10.1093/molehr/8.6.546. [DOI] [PubMed] [Google Scholar]
- 126.Su D. Gladyshev VN. Alternative splicing involving the thioredoxin reductase module in mammals: a glutaredoxin-containing thioredoxin reductase 1. Biochemistry. 2004;43:12177–12188. doi: 10.1021/bi048478t. [DOI] [PubMed] [Google Scholar]
- 127.Su D. Novoselov SV. Sun QA. Moustafa ME. Zhou Y. Oko R. Hatfield DL. Gladyshev VN. Mammalian selenoprotein thioredoxin-glutathione reductase: roles in disulfide bond formation and sperm maturation. J Biol Chem. 2005;280:26491–26498. doi: 10.1074/jbc.M503638200. [DOI] [PubMed] [Google Scholar]
- 128.Sun C. Berardi MJ. Bushweller JH. The NMR solution structure of human glutaredoxin in the fully reduced form. J Mol Biol. 1998;280:687–701. doi: 10.1006/jmbi.1998.1913. [DOI] [PubMed] [Google Scholar]
- 129.Sun QA. Kirnarsky L. Sherman S. Gladyshev VN. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc Natl Acad Sci U S A. 2001;98:3673–3678. doi: 10.1073/pnas.051454398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sun QA. Su D. Novoselov SV. Carlson BA. Hatfield DL. Gladyshev VN. Reaction mechanism and regulation of mammalian thioredoxin/glutathione reductase. Biochemistry. 2005;44:14528–14537. doi: 10.1021/bi051321w. [DOI] [PubMed] [Google Scholar]
- 131.Sundaram S. Rathinasabapathi B. Ma LQ. Rosen BP. An arsenate-activated glutaredoxin from the arsenic hyperaccumulator fern Pteris vittata L. regulates intracellular arsenite. J Biol Chem. 2008;283:6095–6101. doi: 10.1074/jbc.M704149200. [DOI] [PubMed] [Google Scholar]
- 132.Szajewski RP. Whitesides GM. Rate constants and equilibrium-constants for thiol-disulfide interchange reactions involving oxidized glutathione. J Am Chem Soc. 1980;102:2011–2026. [Google Scholar]
- 133.Tamarit J. Belli G. Cabiscol E. Herrero E. Ros J. Biochemical characterization of yeast mitochondrial Grx5 monothiol glutaredoxin. J Biol Chem. 2003;278:25745–25751. doi: 10.1074/jbc.M303477200. [DOI] [PubMed] [Google Scholar]
- 134.Urata Y. Ihara Y. Murata H. Goto S. Koji T. Yodoi J. Inoue S. Kondo T. 17Beta-estradiol protects against oxidative stress-induced cell death through the glutathione/glutaredoxin-dependent redox regulation of Akt in myocardiac H9c2 cells. J Biol Chem. 2006;281:13092–13102. doi: 10.1074/jbc.M601984200. [DOI] [PubMed] [Google Scholar]
- 135.Vlamis-Gardikas A. Aslund F. Spyrou G. Bergman T. Holmgren A. Cloning, overexpression, and characterization of glutaredoxin 2, an atypical glutaredoxin from Escherichia coli. J Biol Chem. 1997;272:11236–11243. doi: 10.1074/jbc.272.17.11236. [DOI] [PubMed] [Google Scholar]
- 136.Vlamis-Gardikas A. Potamitou A. Zarivach R. Hochman A. Holmgren A. Characterization of Escherichia coli null mutants for glutaredoxin 2. J Biol Chem. 2002;277:10861–10868. doi: 10.1074/jbc.M111024200. [DOI] [PubMed] [Google Scholar]
- 137.Wang J. Boja ES. Tan W. Tekle E. Fales HM. English S. Mieyal JJ. Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 138.Wang J. Pan S. Berk BC. Glutaredoxin mediates Akt and eNOS activation by flow in a glutathione reductase-dependent manner. Arterioscler Thromb Vasc Biol. 2007;27:1283–1288. doi: 10.1161/ATVBAHA.107.144659. [DOI] [PubMed] [Google Scholar]
- 139.Wang J. Tekle E. Oubrahim H. Mieyal JJ. Stadtman ER. Chock PB. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc Natl Acad Sci U S A. 2003;100:5103–5106. doi: 10.1073/pnas.0931345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wells WW. Rocque PA. Xu DP. Meyer EB. Charamella LJ. Dimitrov NV. Ascorbic acid and cell survival of Adriamycin resistant and sensitive MCF-7 breast tumor cells. Free Radic Biol Med. 1995;18:699–708. doi: 10.1016/0891-5849(94)00188-p. [DOI] [PubMed] [Google Scholar]
- 141.Wingert RA. Galloway JL. Barut B. Foott H. Fraenkel P. Axe JL. Weber GJ. Dooley K. Davidson AJ. Schmid B. Paw BH. Shaw GC. Kingsley P. Palis J. Schubert H. Chen O. Kaplan J. Zon LI. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 142.Witte S. Villalba M. Bi K. Liu Y. Isakov N. Altman A. Inhibition of the c-Jun N-terminal kinase/AP-1 and NF-kappaB pathways by PICOT, a novel protein kinase C-interacting protein with a thioredoxin homology domain. J Biol Chem. 2000;275:1902–1909. doi: 10.1074/jbc.275.3.1902. [DOI] [PubMed] [Google Scholar]
- 143.Yang Y. Jao S. Nanduri S. Starke DW. Mieyal JJ. Qin J. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry. 1998;37:17145–17156. doi: 10.1021/bi9806504. [DOI] [PubMed] [Google Scholar]
- 144.Yang YF. Wells WW. Catalytic mechanism of thioltransferase. J Biol Chem. 1991;266:12766–12771. [PubMed] [Google Scholar]
- 145.Yu J. Zhang NN. Yin PD. Cui PX. Zhou CZ. Glutathionylation-triggered conformational changes of glutaredoxin Grx1 from the yeast Saccharomyces cerevisiae. Proteins. 2008;72:1077–1083. doi: 10.1002/prot.22096. [DOI] [PubMed] [Google Scholar]