

Abstract
It has been widely reported that the presenilin proteins PS-1 and PS-2 in extracts derived from a variety of cultured cells and from tissues are fragmented extensively by endoproteolytic processing events. It generally has been presumed that this endoproteolysis is a physiologically normal intracellular event following presenilin expression, which might play an important role in the still unknown functions of these molecules in connection with Alzheimer disease. We demonstrate herein, however, that, if a variety of cultured cells and several mouse tissues are examined under conditions minimizing cell trauma, the presenilin molecules in the extracts are found to be intact but that, if the cells and tissues are prepared under somewhat more stressful conditions, the endoproteolytic fragments are then observed. We conclude that these particular endoproteolytic events are not the result of physiologically normal processing of the presenilins but are rather artifacts occurring during the common procedures of specimen preparation.
Keywords: Alzheimer disease, anti-peptide antibodies
Molecular genetic studies of individuals afflicted with early onset familial Alzheimer disease have implicated strongly the previously unknown presenilin (PS) proteins PS-1 (1) and PS-2 (2, 3) in the genesis of the more common, nonfamilial, and late onset form of the disease (AD). PS-1 and PS-2, of 467 and 448 amino acid residues, respectively, are two closely related integral membrane proteins that have a 7-transmembrane (7-TM) spanning topography in the plasma membranes of cells, as we demonstrate in a companion paper in this issue of Proceedings (4). The normal physiological functions of the PS proteins are not yet established, nor are their roles in the genesis of AD understood. It has frequently been reported that, in extracts prepared from cultured cells and tissues, including the brain, the PS proteins, to a large extent, have been found to be endoproteolytically cleaved, mainly into two classes of fragments, a ≈30 kDa NH2-terminal fragment (NTF) and a variety of COOH-terminal fragments (CTF) of 14–29 kDa (for a recent review, see ref. 5; also, see figure 3B in ref. 4). This endoproteolytic cleavage of the PS proteins is widely thought to be a normal physiological event that occurs intracellularly while these integral proteins are located in the endoplasmic reticulum membranes. Although a function for this proteolysis has not been established, it has often been presumed to play an important role in the genesis of AD.
In the course of our studies, however, we began to have doubts about the physiological significance of the endoproteolytic cleavage of PS proteins. First, in extracts of cultured DAMI cells transfected with full length cDNAs for PS-1 or PS-2, we generally observed (6) only the full length PS molecules, and we did not observe the reported cleavage fragments, in contrast to the results obtained by others using different cultured cells and cDNA constructs (5). (The significance of the use of DAMI cells will become clear later. See Results and Discussion.) Second, if such extensive intracellular endoproteolysis occurred normally, it might be expected to impede the subsequent transport of the PS proteins to the plasma membrane of the cell whereas we have unequivocally demonstrated their ample cell surface expression (4, 6). For these and related reasons, we considered the possibility that this often-observed endoproteolytic cleavage was not a physiologically normal intracellular process, and we sought further experimental evidence to investigate its significance. In this paper, we report that, if extra precautions are taken in the manipulation of various PS-transfected and untransfected cultured cells and several mouse tissues and in the preparation of extracts from them, these cleavage products are not observed, but they do appear in proportion as less stringent preparative procedures are used. These results strongly indicate that this endoproteolytic cleavage of the PS molecules occurs only after cultured cells or tissues are structurally damaged or disrupted, whereupon one or more proteases presumably either are activated or are released to cleave the previously intact PS proteins. We therefore conclude that this particular endoproteolytic process is not a physiologically normal intracellular event accompanying PS expression, i.e., it is an artifact that probably results from a hypersensitivity to proteolysis of certain specific peptide bond(s) within the native PS molecules, bond(s) that are normally protected from intracellular cleavage.
MATERIALS AND METHODS
Anti-Peptide Antibodies.
The eight polyclonal rabbit anti-peptide antibodies raised to selected short sequences within either human PS-1 or PS-2 have been described (4). Their specificities have been characterized in part by immunofluorescence microscopic labeling experiments (4) but mainly by the immunoblotting experiments described in Results. Three of the antibodies (N1 and N3 for PS-2 and N2 for PS-1) were raised to sequences within different parts of the NH2-terminal region preceding the first TM-spanning domain (see figure 1 in ref. 4); one (I1 for PS-1) was raised to a sequence within the small hydrophilic loop between the first and second TM stretches; three (L1 and L3 for PS-1 and L2 for PS-2) were raised to sequences within the large hydrophilic loop between the sixth and seventh TM stretches; and one (C1) was raised to a sequence within the COOH-terminal region of PS-2, which also reacted with the closely homologous sequence of PS-1.
Cell Cultures and Transfections.
A number of different types of cultured cell lines from several species was used, several corresponding to those used by others (5). Most were not transfected, but the DAMI cells were transfected transiently with pcDNA3 constructs of full length cDNAs of human PS-1 or PS-2, as described (7). All cells were grown attached in monolayers on plastic 25-cm2 flasks at 37°C under an atmosphere of 5% CO2 and in the following media. Human megakaryoblast DAMI cells (from American Type Culture Collection, CRL 9792): Iscove’s-modified DMEM containing 10% horse serum, penicillin (100 units/ml), and streptomycin (100 units/ml); human neuroblastoma IMR-32 cells (American Type Culture Collection, CCL-127) and human lung fibroblast IMR-90 cells (American Type Culture Collection, CCL-186): MEM supplemented with 10% fetal calf serum, penicillin (100 units/ml), and streptomycin (100 units/ml); mouse fibroblast 3T3 cells and human NTera2/CI-DI (NT2) undifferentiated embryonal carcinoma cell line (American Type Culture Collection, CRL-1973): DMEM, high glucose, supplemented with 10% fetal calf serum, penicillin (100 units/ml), and streptomycin (100 units/ml); monkey kidney Cos-1 cells (American Type Culture Collection, CRL-1650): DMEM, high glucose, supplemented with 10% fetal calf serum, penicillin (100 units/ml), streptomycin (100 units/ml), and 1 mM sodium pyruvate; and Chinese hamster ovary CHO-K1 cells (American Type Culture Collection, CCL-61): Ham’s F-12 medium containing 10% fetal calf serum, penicillin (100 units/ml), and streptomycin (100 units/ml).
Cultured Cell Extracts.
As noted in our previous studies (7), under appropriate culture conditions, DAMI cells are detached readily into suspension from monolayer flask culture simply by tapping the flasks sharply a few times. This gentle detachment is unusual. With the other adherent cell types that have been used in similar studies by others (5), more rigorous procedures, such as scraping or trypsin treatment, are ordinarily necessary to remove the attached cells. We suspected therefore that this difference in preparative procedures might be responsible for the absence of PS fragments in our DAMI cell extracts (6) of the kind found by others using a variety of different adherent cultured cells (5). This possibility was explored in two ways, by (i) deliberately subjecting DAMI cells to more vigorous detachment treatments, much like those commonly used with other cells lines, to see if, under such circumstances, PS fragments would indeed now be observed and (ii) with cell lines other than DAMI, finding a more direct procedure to prepare cell extracts than is commonly used, to ask whether, under such gentler treatment, PS fragments might no longer be observed.
Accordingly, DAMI cell extracts were prepared by two procedures, our earlier one (6), referred to herein as Method A-DAMI, and a slightly more vigorous one, Method B-DAMI. In Method A-DAMI, after removal of the culture medium, the flask was washed once with 10 ml of PBS (pH 7.4). The flask then was tapped several times to detach the cells, and the cells were collected by centrifugation. Two hundred microliters of a mixture of protease inhibitors in PBS (2 mM phenylmethylsulfonyl fluoride/2 μg/ml antipain/0.2 μg/ml leupeptin/0.2 μg/ml pepstatin A) then was added to the pellet followed by 600 μl of SDS gel loading buffer (50 mM Tris, pH 6.8/0.1 M DTT/2% SDS/0.1% bromphenol blue/10% glycerol). This mixture was then immediately boiled for 5 minutes and then briefly sonicated before being subjected to SDS/PAGE in the presence of 8 M urea. In Method B-DAMI, after removal of the culture medium from a flask, the cells were scraped with a Costar 3020 plastic cell scraper into 200 μl of PBS containing half the amounts of the protease inhibitors in Method A-DAMI above. This mixture then was immediately placed in a −20°C freezer (not quick-frozen). The frozen mixture then was thawed and sonicated with three bursts of 20 s each, and aliquots were boiled for 5 minutes in 2X concentration of the SDS gel loading buffer before being subjected to the usual SDS/PAGE as described above.
Similarly, extracts of the cell lines other than DAMI were prepared by two procedures, one referred to as Method A-Cells, which avoided cell scraping and the other referred to as Method B-Cells, which in fact was the same as Method B-DAMI. In Method A-Cells, after removal of the culture medium, each flask was washed once with 10 ml of PBS. The attached cell layer then was dissolved directly into 200 μl of the mixture of the protease inhibitors in PBS followed by 600 μl of the SDS gel loading buffer that was used in Method A-DAMI described above. The mixture was then boiled, sonicated briefly, and subjected to SDS/PAGE, as described above.
Mouse Tissue Extracts.
A similar approach was used to investigate the significance of the reported PS endoproteolysis in whole tissues, using mouse liver, kidney, and brain. The mice were of strain B6C3F1 (Simonsen Laboratories, Gilroy, CA) and were 9 months old. Extracts were prepared by one of three procedures, in which the conditions were slightly different to subtly alter the physical state of the sample. In the first stage of each preparation, the mouse was killed by decapitation, and the liver, kidneys, and brain were rapidly removed. In Method A-Tissues, one half of each organ immediately was used fresh to prepare a 20% (wt/vol) homogenate in ice-cold lysis buffer (50 mM Tris⋅HCl, pH 8.0/150 mM NaCl/0.5% Nonidet P-40) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride/1 μg/ml antipain/0.1 μg/ml pepstatin A) by using a Dounce homogenizer. This was followed by sonication (three bursts of 20 s each). Whole extract then either was processed immediately for SDS/PAGE or was rapidly frozen in small aliquots in a methanol–dry ice freezing mixture and stored at −70°C until thawed for further analysis. In Method B-Tissues, the other half of each of the organs used in Method A-Tissues was frozen immediately in liquid N2. After freezing, the tissue then was thawed on ice and extracted to give a 20% homogenate in lysis buffer containing the protease inhibitors, exactly as described above for the fresh tissue in Method A-Tissues. In Method C-Tissues, the intact tissue was placed in a −70°C freezer (not quick frozen) within 15 minutes after death of the animal. After freezing, the tissue was thawed on ice, and a 20% homogenate in lysis buffer was prepared as in Methods A and B above, except that the protease inhibitors were not added until after the sonication step.
RESULTS
Specificities of the Anti-Peptide Antibodies.
The specificities of the antibodies used in this paper and in the companion paper (4) were determined by immunoblotting experiments on extracts of either PS-1- or PS-2-transfected DAMI cells prepared by Method A-DAMI (6). In the immunoblots shown in Fig. 1, the anti-PS-1 antibodies I1, L1, and L3 (Fig. 1A, lanes 1, 4, and 7, respectively) each mainly labeled a single protein band of ≈57 kDa in the PS-1 cell extracts. This band corresponds to intact PS-1 molecules, as was confirmed by comparison with the in vitro translation product of PS-1 mRNA (6). Usually, one or two higher molecular weight bands also were labeled, which most probably represented aggregated or modified intact PS molecules (6). These immunolabelings were specific because each was inhibited in the presence of an excess of the oligopeptide conjugate specific for the particular antibody used (Fig. 1A, lanes 3, 6, and 9, respectively). (In experiments not shown, no such inhibition of immunolabeling was observed if an excess of a nonspecific oligopeptide conjugate was used instead.)
Figure 1.
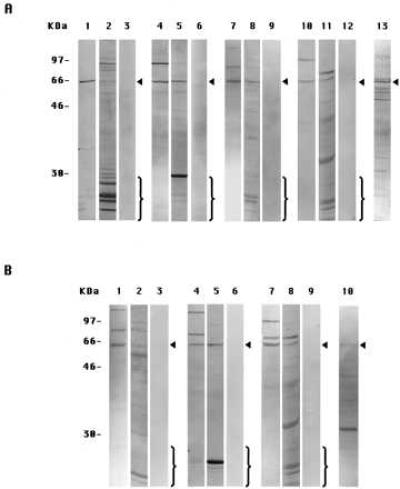
Immunoblots with eight anti-peptide antibodies to PS-1 or PS-2 of cell extracts from PS-transfected DAMI cells prepared by two methods. (A) PS-1-transfected cells. Lanes: 1, prepared by Method A-DAMI, labeled with antibody I1; 2, Method B-DAMI, antibody I1; and 3, Method A-DAMI, antibody I1 in presence of excess oligopeptide-conjugate specific for I1. The other panels of three gels each are of specimens corresponding to lanes 1, 2, and 3, respectively, except that they were labeled with different antibodies: lanes 4, 5, and 6, antibody L1; lanes 7, 8, and 9, antibody L3; and lanes 10, 11, and 12, antibody C1. Lane 13, specimen prepared by Method A-DAMI and labeled with antibody N2. (B) PS-2-transfected cells. Lanes: 1, prepared by Method A-DAMI and labeled with antibody N1; 2, Method B-DAMI, antibody N1; and 3, Method A-DAMI, antibody N1 in presence of an excess of oligopeptide-conjugate specific for N1. The other panels of three gels each are of specimens corresponding to lanes 1, 2, and 3, respectively, except that they were labeled with different antibodies: lanes 4, 5, and 6, antibody L2; and lanes 7, 8, and 9, antibody C1. Lane 10, specimen prepared by Method A-DAMI and labeled with antibody N3. The arrowheads in A denote the intact monomeric PS-1 band and in B, the intact PS-2 band. The brackets enclose the bands that represent the endoproteolytic fragments.
In similar immunoblotting experiments with extracts from PS-2-transfected DAMI cells, the anti-PS-2 antibodies N1, L2, and C1 (Fig. 1B, lanes 1, 4, and 7, respectively) labeled a single band at ≈55 kDa, corresponding to intact PS-2 molecules (6), and one or two bands of higher molecular weight, which apparently were aggregated or modified forms of intact PS-2 (6). These labelings each were inhibited by an excess of the oligopeptide conjugate specific for that antibody (Fig. 1B, lanes 3, 6, and 9, respectively). In addition, antibody C1 also specifically labeled intact PS-1 in PS-1-transfected cells (Fig. 1A, lanes 10 and 12), due to a cross-reaction between antibody C1-directed sequences of PS-1 and PS-2 molecules, as also was observed in immunofluorescence labeling experiments (4).
On the other hand, with the PS-1-transfected DAMI cell extracts also prepared by Method A-DAMI, antibody N2 labeled a large number of bands in addition to the ≈57-kDa intact PS-1 band (Fig. 1A, lane 13), and with the PS-2-transfected DAMI cell extracts similarly gently prepared, antibody N3 (Fig. 1B, lane 10) labeled a predominant band of ≈30 kDa that was not stained by any of the other anti-PS-2 antibodies. Antibodies N2 and N3 were therefore of uncertain specificity and were not examined further in this or the companion paper (4). The other six antibodies, however, were shown by the labeling and inhibition experiments in Fig. 1 each to be specific for the epitope on either PS-1 or PS-2 against which they were raised.
The Absence or the Induction of PS Endoproteolysis in Differently Prepared Extracts of Transfected DAMI Cells.
When prepared by Method A-DAMI, cell extracts, as just described, were immunolabeled for the appropriate intact PS molecule and its probable aggregates but showed no immunolabeling for lower molecular weight proteolytic breakdown products with any of the six specific antibodies (Fig. 1 A, lanes 1, 4, 7, and 10, and B, lanes 1, 4, and 7), as we had previously reported (6). On the other hand, with extracts prepared by Method B-DAMI, which included a cell scraping step, each of the six antibodies showed a decreased labeling of the band corresponding to the intact PS molecules and instead stained a generally large number of lower molecular weight bands (Fig. 1 A, lanes 2, 5, 8, and 11, and B, lanes 2, 5, and 8). The labeling of all of these lower molecular weight bands was inhibited in the presence of an excess of the oligopeptide conjugate specific for that antibody (not shown). We conclude that PS-1 and PS-2 molecules remain largely intact if the cell extracts are prepared by Method A-DAMI but are endoproteolyzed extensively in extracts prepared by Method B-DAMI.
The Absence or the Induction of PS-1 Endoproteolysis in Differently Prepared Extracts of Several Adherent Cultured Cells.
In these experiments, we show only the results for PS-1 in untransfected cultured cells. Similar results were obtained for PS-2 (not shown). When prepared by Method A-Cells, extracts of six different cell types showed labeling only or predominantly of the band at ≈57 kDa corresponding to the intact PS-1 molecule using the antibody L3 (Fig. 2A). (Apparently, this antibody cross-reacts with a similar epitope in the presumably homologous PS-1 molecule derived from the different cell donor animal species). When prepared by Method B-Cells, including a typical scraping procedure avoided in Method A-Cells, antibody L3 now labeled, in addition to the ≈57-kDa band, a number of other bands of molecular weight <30 kDa (Fig. 2B) in all of the different cells. Antibody L3 would be expected to label only those proteolytic fragments including the COOH terminus of PS-1 (CTF), so we also used antibody I1 to label mostly the proteolytic fragments (NTF) nearer the NH2-terminal of the PS-1 molecules in extracts prepared by Method B-Cells. Antibody I1 indeed labeled a different collection of small fragments (Fig. 2C) than antibody L3, as expected (5). We conclude that PS-1 molecules remain largely intact in extracts of all of the several adherent cell types if the extracts are prepared by Method A-Cells but are endoproteolyzed extensively in extracts prepared by Method B-Cells, the latter result corresponding to the findings of many other investigators (see ref. 5).
Figure 2.
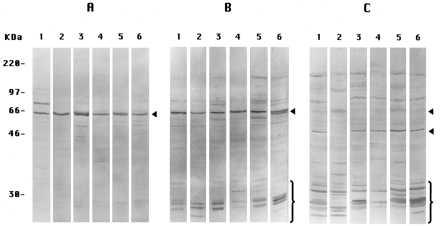
Immunoblots of PS-1 in extracts from several cultured cells. All lanes 1 are of PS-1-transfected DAMI cells; all lanes 2, Chinese hamster ovary cells; all lanes 3, IMR-32 cells; all lanes 4, IMR-90 cells; all lanes 5, NT2 cells; and all lanes 6, Cos-1 cells. (A) Extracts were prepared by Method A-Cells, labeled with antibody L3. (B) Prepared by Method B-Cells, labeled with antibody L3. (C) Prepared by Method B-Cells, labeled with antibody I1. In B, antibody L3-labeled CTF endoproteolysis products (brackets); in C, antibody I1-labeled NTF endoproteolysis products (brackets and lower of the two arrowheads). The single arrowheads in A and B and the upper arrowhead in C denote the intact monomeric PS-1 band.
The Absence or the Induction of PS-1 Endoproteolysis in Differently Prepared Extracts of Several Mouse Tissues.
Because proteolytic fragments of PS molecules also have been observed in extracts of whole tissues (including human brain) (5), we extended these experiments to several tissues of the mouse, whose preparation and extraction we could manipulate reproducibly. When prepared by Method A-Tissues and stained with the anti-PS-1 antibody L3, extracts of mouse liver (Fig. 3A, lane 1) and mouse kidney (Fig. 4, lane 3) each showed only the one labeled band corresponding to the ≈57-kDa intact PS-1 molecule. None of the smaller proteolytic fragments generally seen by others (5) was observed. The similarly prepared (Method A-Tissues) extracts of mouse brain (Figs. 3A, lane 2, and 4, lane 4) exhibited the same ≈57 kDa-labeled band of intact PS-1 but also a prominent band at ≈40 kDa, not seen in the other mouse tissue extracts (Figs. 3A, lane 1, and 4, lanes 2 and 3) or in extracts of cultured mouse 3T3 fibroblast cells (Fig. 4, lane 1) or in human preneuronal NT2 cells (Fig. 2A, lane 5) or human neuroblastoma cells (Fig. 2A, lane 3) prepared by Method A-Cells. The labeling of this ≈40 kDa band in the brain extracts was specific for PS-1 as it was inhibited by the presence of an excess of the oligopeptide conjugate specific for antibody L3 (not shown). If this unique brain band is a fragment of the PS-1 molecule, it is an entirely different molecular species from any of the smaller fragments produced by the often observed endoproteolysis of PS-1 (5) that we have been investigating in the present studies. These endoproteolytic fragments are not observed in brain extracts prepared by Method A-Tissues (Figs. 3A, lane 1, and 4, lane 4), but the multiple small CTF, as detected by antibody L3, do appear in brain extracts prepared either by Method B-Tissues (Fig. 3B, lane 2) or by Method C-Tissues (Fig. 3C, lane 2). The endoproteolytic fragments also are seen in extracts of mouse liver prepared by Method B-Tissues (Fig. 3B, lane 1) and even more extensively with Method C-Tissues (Fig. 3C, lane 1). Leaving aside the separate matter of the 40-kDa band in the brain extracts (see Discussion), we conclude that, in extracts of the several mouse tissues prepared by Method A-Tissues, PS-1 molecules remain largely intact (as do PS-2 molecules; not shown) but are endoproteolyzed extensively when either Method B-Tissues or Method C-Tissues is used.
Figure 3.
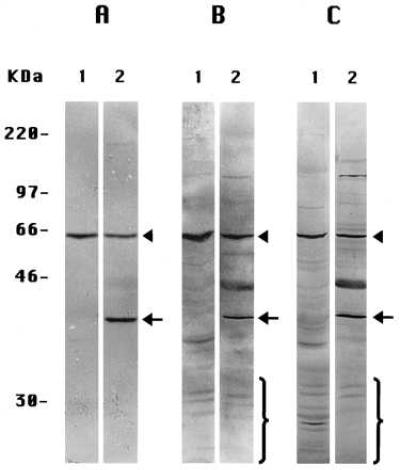
Immunoblots of extracts of mouse liver (all lanes 1) and brain (all lanes 2) with anti-PS-1 antibody L3. (A) Specimens prepared by Method A-Tissues. (B) Method B-Tissues. (C) Method C-Tissues. Note the ≈40-kDa brain band in A, lane 2, also is present in B, lane 2, and C, lane 2. The arrowheads signify the intact monomeric PS-1 band; the arrows signify the brain-specific ≈40-kDa species; and the brackets signify some of the CTF endoproteolytic products of PS-1.
Figure 4.
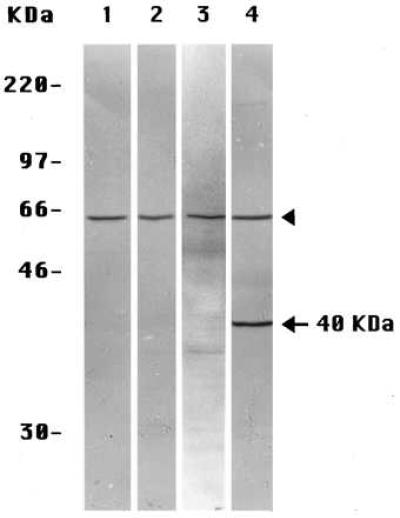
Immunoblots of extracts each labeled with anti-PS-1 antibody L3 of mouse 3T3 cultured cells prepared by Method A-Cells (lane 1) and of mouse liver (lane 2), kidney (lane 3), and brain (lane 4), all prepared by Method A-Tissues. The arrowheads indicate the intact monomeric PS-1 species, and the arrow indicates the brain-specific, ≈40-kDa, PS-1-related species.
DISCUSSION
Our results clearly establish that the widely observed extensive endoproteolysis of PS proteins was not detected in extracts of cells or tissues that were prepared by the mildest and most direct procedures but did appear copiously in all of the specimens that we subjected to slightly more stressful procedures of the kind usually applied in others’ studies (5). This explains our “failure” to observe the endoproteolytic fragments in our initial experiments with PS-transfected DAMI cells (6). DAMI cells are unusual among the variety of adherent cells that we and others have studied in the ease of their detachment from their monolayers. [Our original choice of these cells was based on their unusual lack of expression of the human β-amyloid precursor protein (β-APP) (8) and not on their adherence properties.] However, if DAMI cells were detached by the alternative process of scraping, the extracts now also exhibited the endoproteolytic fragments (Fig. 1). Conversely, with all of the more strongly adherent cells that we examined, if the cells were removed directly into lysis buffer, avoiding scraping or other stresses, the extracts now showed only intact PS molecules (Figs. 2A and 4, lane 1) and little or none of the usual endoproteolytic fragments (Fig. 2 B and C).
Analogous results were obtained with several mouse tissues that were similarly investigated. The use of fresh specimens and the avoidance of slow freezing steps in the preparation yielded tissue extracts of liver, kidney, and brain (Figs. 3A and 4) that did not contain the usual PS-1 endoproteolytic fragments that, however, could be observed under more stressful preparative conditions (Fig. 3 B and C). (The brain extracts contained a unique band discussed below.)
The main conclusion from all of these results is inescapable: The often-detected endoproteolysis of PS proteins in cell and tissue extracts (5) is a preparative artifact. In live cells in culture and in most tissues, PS molecules are generally present in intact form. Our results confirm that PS molecules are unusually sensitive to endoproteolysis, which occurs, however, only when cells or tissues are disturbed physically or disrupted. This endoproteolysis may be involved physiologically in the turnover of PS proteins, but it is clearly unlikely to have any bearing on their normal cellular functions.
The status of PS molecules in the brain is unusual, however, and may be of considerable potential interest to AD. Even with normal mouse brain prepared and extracted under the most favorable conditions, which showed none of the NTF and CTF breakdown products that only appeared under more stressful conditions, a prominent PS-1-related band of ≈40 kDa in addition to the band of intact PS-1 molecules was detected by antibody L3 (Figs. 3A, lane 2, and 4, lane 4) but was not present in liver or kidney (Fig. 4, lanes 2 and 3). This PS-1-related, brain-specific species is clearly distinct in size and homogeneity from the fragments generated in the usual endoproteolytic breakdown of PS-1 (5) and appears indeed to be normally present in live brain cells. If so, our preliminary evidence does not distinguish between whether it is a proteolytic fragment derived from intact PS-1 molecules or it represents some type of alternative form of PS-1 expression. This 40-kDa, PS-1-specific molecular species may be closely similar to the species observed in extracts of rat brain by Hartmann et al. (9), who also found it in neuronal cells in culture but only after the cells had been induced to differentiate in vitro into neurons. Correspondingly, we did not find this band in preneuronal NT2 cells (Fig. 2A, lane 5) or in neuroblastoma cells (Fig. 2A, lane 3). This brain-specific molecular species clearly merits detailed structural and functional investigation.
In three recent papers, we have demonstrated conclusively: (i) that the PS proteins are indeed expressed at the surfaces of cells, including neurons (6), whereas they have been regarded widely as exclusively intracellular proteins (5, 10); (ii) that they exhibit a 7-TM spanning topography (4) rather than the 6-TM (11) or 8-TM (10) spanning topography that commonly has been accepted for them (5); and (iii) in this paper, that the often-observed endoproteolytic cleavage of the PS molecules (5), which is widely thought to be physiologically significant to PS functions, is an artifact of the preparative conditions used. Having cleared away this underbrush of misconceptions about the PS proteins, we can now get on with the cultivation of our original proposal about the role of the PS proteins in AD (12), namely, that the PS proteins serve as plasma membrane-bound cell surface receptors for their specific ligand, the β-APP, expressed on the surface of a neighboring neuron in the brain; that, as a result of this intercellular binding of β-APP to either PS-1 or PS-2, a cell adhesion forms (13) between the two cells; that, at this adhesion site, a pinching off of double membrane-bounded vesicles is induced into the β-APP-expressing cell (with precedents described in refs. 14 and 15); and that these vesicles fuse with intracellular multivesicular bodies (14, 15) in which it is proposed that the internalized β-APP undergoes proteolysis to form the β-amyloid (Aβ) oligopeptides that are widely thought to be a key component involved in the onset of AD. We subsequently have demonstrated (7) with transfected cultured cells that indeed specific cell–cell interactions did occur between cells expressing β-APP on their surfaces and cells expressing either PS-1 or PS-2 on theirs. It remains to investigate whether these specific cell–cell interactions are required for the production of the forms of Aβ that are relevant to AD.
Acknowledgments
This work was supported by the National Institutes of Health Grant 2RO1-NS27850 to N.N.D.
ABBREVIATIONS
- PS
presenilin
- AD
Alzheimer disease
- TM
transmembrane
- NTF
NH2-terminal fragment
- CTF
COOH-terminal fragments
- β-APP
β-amyloid precursor protein
References
- 1.Sherrington R, Rogaev E I, Liang Y, Rogaeva E A, Levesque G, et al. Nature (London) 1995;375:754–760. [Google Scholar]
- 2.Levy-Lahad E, Wijsman E M, Nemens E, Andertson L, Goddard K A, Weber J L, Bird T D, Schellenberg G D. Science. 1995;269:970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- 3.Rogaev E I, Sherrington R, Rogaeva E A, Levesque G, Ikeda M, et al. Nature (London) 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 4.Dewji N N, Singer S J. Proc Natl Acad Sci USA. 1997;94:14025–14030. doi: 10.1073/pnas.94.25.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haass C. Neuron. 1977;18:687–690. doi: 10.1016/s0896-6273(00)80309-8. [DOI] [PubMed] [Google Scholar]
- 6.Dewji N N, Singer S J. Proc Natl Acad Sci USA. 1997;94:9926–9931. doi: 10.1073/pnas.94.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dewji N N, Singer S J. Proc Natl Acad Sci USA. 1996;93:12575–12580. doi: 10.1073/pnas.93.22.12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Overfurth H W, Selkoe D J. Biochemistry. 1994;33:4450–4461. [Google Scholar]
- 9.Hartmann H, Busciglio J, Baumann K-H, Staufenbiel M, Yankner B A. J Biol Chem. 1997;272:14505–14508. doi: 10.1074/jbc.272.23.14505. [DOI] [PubMed] [Google Scholar]
- 10.Doan A, Thinakaran G, Borchett D R, Slunt H H, Ratovitsky T, Podlisney M, Selkoe D J, Seeger M, Gandy S E, Price D L, Sisodia S S. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 11.Lehmann S, Chiesa R, Harris D A. J Biol Chem. 1997;272:12047–12051. doi: 10.1074/jbc.272.18.12047. [DOI] [PubMed] [Google Scholar]
- 12.Dewji N N, Singer S J. Science. 1996;271:159–160. doi: 10.1126/science.271.5246.159. [DOI] [PubMed] [Google Scholar]
- 13.Singer S J. Science. 1992;255:1671–1677. doi: 10.1126/science.1313187. [DOI] [PubMed] [Google Scholar]
- 14.Cagan R L, Kramer H, Hart A C, Zipursky S L. Cell. 1992;69:393–399. doi: 10.1016/0092-8674(92)90442-f. [DOI] [PubMed] [Google Scholar]
- 15.Bailey C H, Chen M, Keller F, Kandel E. Science. 1992;256:645–647. doi: 10.1126/science.1585177. [DOI] [PubMed] [Google Scholar]