

Abstract
Evidence indicates that the modulatory effects of the adrenergic stress hormone epinephrine as well as several other neuromodulatory systems on memory storage are mediated by activation of β-adrenergic mechanisms in the amygdala. In view of our recent findings indicating that the amygdala is involved in mediating the effects of glucocorticoids on memory storage, the present study examined whether the glucocorticoid-induced effects on memory storage depend on β-adrenergic activation within the amygdala. Microinfusions (0.5 μg in 0.2 μl) of either propranolol (a nonspecific β-adrenergic antagonist), atenolol (a β1-adrenergic antagonist), or zinterol (a β2-adrenergic antagonist) administered bilaterally into the basolateral nucleus of the amygdala (BLA) of male Sprague–Dawley rats 10 min before training blocked the enhancing effect of posttraining systemic injections of dexamethasone (0.3 mg/kg) on 48-h memory for inhibitory avoidance training. Infusions of these β-adrenergic antagonists into the central nucleus of the amygdala did not block the dexamethasone-induced memory enhancement. Furthermore, atenolol (0.5 μg) blocked the memory-enhancing effects of the specific glucocorticoid receptor (GR or type II) agonist RU 28362 infused concurrently into the BLA immediately posttraining. These results strongly suggest that β-adrenergic activation is an essential step in mediating glucocorticoid effects on memory storage and that the BLA is a locus of interaction for these two systems.
Keywords: dexamethasone, RU 28362, β-adrenergic antagonists, inhibitory avoidance
Extensive evidence from studies of memory of inhibitory avoidance training in rats indicates that several neuromodulatory systems interact with the noradrenergic system in the amygdala in modulating memory storage (1–8). Intra-amygdala infusions of the β-adrenergic antagonist propranolol or depletion of norepinephrine in the amygdala by the neurotoxin N-2-chloroethyl-N-ethyl-bromobenzylamine (DSP-4) block the memory-enhancing effect of the adrenergic stress hormone epinephrine (5, 6). Moreover, intra-amygdala infusions of propranolol as well as specific β1- or β2-adrenergic antagonists also block the effects, on memory, of drugs affecting opioid peptidergic and GABAergic systems (4, 7). Norepinephrine and other β-adrenergic agonists administered to the amygdala after training dose-dependently enhance retention (5, 9–11). These effects are also time-dependent; they affect retention when given shortly after training but have no effect if given several hours later. These findings are consistent with evidence from recent experiments using in vivo microdialysis indicating that norepinephrine is released in the amygdala by footshock stimulation of the kind typically used in inhibitory avoidance training (12). Furthermore, adrenergic and opioid peptidergic systems influence the training-induced release of norepinephrine (13, 14).
Findings of a series of recent experiments from our laboratory indicate that the amygdala is involved in mediating the memory-modulating effects of glucocorticoids (15). Excitotoxically induced lesions of the basolateral (BLA), but not the central (CEA), nucleus of the amygdala blocked the enhancing effects of systemic posttraining injections of dexamethasone, a synthetic glucocorticoid, on memory for inhibitory avoidance training (16). Furthermore, infusions of the specific glucocorticoid receptor (GR or type II) agonist RU 28362 (11β,17β-dihydroxy-6,21-dimethyl-17α-pregna-4,6-trien-20yn-3-one) into the BLA enhanced memory storage (17). Such findings suggest that glucocorticoid effects on memory storage are mediated, at least in part, by GRs in the BLA. Although the specific mode of action of glucocorticoids differs from that of the hormones and neurotransmitters just described (i.e., glucocorticoids are highly lipophylic and bind directly to intracellular receptors in the brain), the involvement of the amygdala in mediating the effects of all these compounds appears to be strikingly similar. These findings suggest that glucocorticoid-induced effects on memory storage may also depend on intact noradrenergic neurotransmission in the amygdala. In support of this view, there is extensive evidence from biochemical and electrophysiological experiments indicating interactions between glucocorticoids and the noradrenergic system in several brain structures, including the hypothalamus, hippocampus, and cerebral cortex (18, 19).
The present study examined the involvement of β-adrenoceptors in the amygdala in the modulating effects of glucocorticoids on memory storage. In a first experiment, rats received microinfusions of β-adrenergic antagonists with either a selective affinity for β1- or β2-adrenoceptors or with a nonspecific affinity for both receptor types into the BLA or CEA before training in an inhibitory avoidance task. Dexamethasone was injected subcutaneously immediately after training. In a second experiment, rats received concurrent immediate posttraining infusions of the β1-adrenergic antagonist atenolol and the specific GR agonist RU 28362 into the BLA. In both experiments the animals were tested on retention 48 h after training.
MATERIALS AND METHODS
Subjects.
Male Sprague–Dawley rats (270–300 g at time of surgery) from Charles River Laboratories were used. They were individually housed in a temperature-controlled (22°C) colony room and maintained on a standard 12-h light/12-h dark cycle (0700–1900 h lights on) with food and water available ad libitum. Training and testing were performed between 1000 and 1500 h.
Surgery.
The animals were adapted to the vivarium for at least 1 week before surgery. They were anesthetized with sodium pentobarbital (50 mg/kg of body weight, i.p.) and given atropine sulfate (0.4 mg/kg, i.p.). The skull was fixed to a stereotaxic frame (Kopf Instruments, Tujunga, CA), and stainless-steel guide cannulae (15 mm; 23 gauge) were implanted bilaterally with the cannula tips 2 mm above the BLA [coordinates: anteroposterior (AP), −2.8 mm from bregma; mediolateral (ML), ±5.0 mm from midline; dorsoventral (DV), −6.5 mm from skull surface] or CEA [coordinates: AP, −2.2 mm; ML, ±4.3 mm; DV, −6.0 mm] according to the atlas of Paxinos and Watson (20). The cannulas and two anchoring screws were affixed to the skull with dental cement. Stylets (15 mm long 00 insect dissection pins) were inserted into the cannulae to maintain patency and were removed only for the infusion of drugs. The rats were allowed to recover 7 days before training and were handled three times for 1 min during this recovery period.
Drugs and Infusion Procedures.
The nonspecific β-adrenergic antagonist dl-propranolol (0.5 μg per side; Sigma) or the specific β1- or β2-adrenergic antagonists atenolol (0.5 μg per side; Sigma) or zinterol (0.5 μg per side; Bristol-Myers), respectively, were dissolved in 0.9% saline and infused either into the BLA or the CEA 10 min before training. The doses were selected on the basis of previous experiments conducted in this laboratory (21). Bilateral intra-amygdala nuclei infusions of either of the β-adrenergic antagonists or a saline control solution were made by using 30-gauge injection needles connected to a 10-μl Hamilton syringe by polyethylene (PE-20) tubing. The injection needles protruded 2 mm beyond the tips of the cannulas. A 0.2-μl injection volume per hemisphere was infused over a period of 25 s by an automated syringe pump (Sage Instruments, Boston). The infusion volume was based on our findings that selective neurotoxically induced lesions of the BLA and CEA are produced with an infusion volume of 0.2 μl (16). The injection needles were retained within the cannulas for an additional 30 s following drug infusion to maximize diffusion. After completion of the infusion the animals were returned to their home cages until the start of the inhibitory avoidance training.
Dexamethasone (Sigma) was injected subcutaneously at a dose of 0.3 mg/kg in a volume of 2.0 ml/kg immediately after training. This dose was selected on the basis of our previous experiments (16). Dexamethasone was first dissolved in 100% ethanol and subsequently diluted in 0.9% saline to reach the appropriate concentration. The final concentration of ethanol was 2%. Solutions of all drugs were freshly prepared before the experiments.
For the second experiment, atenolol (0.5 μg) and the specific GR agonist RU 28362 (1.0, 3.0, or 10.0 ng) were infused together into the BLA immediately after training. The drugs were first dissolved in 100% ethanol and subsequently diluted with saline to reach a 2% ethanol solution. The infusion volume and procedures used were identical to those described above.
Inhibitory Avoidance Apparatus and Procedure.
The rats were trained in an inhibitory avoidance apparatus (8) consisting of a trough-shaped alley (91 cm long, 15 cm deep, 20 cm wide at the top, 6.4 cm wide at the floor) divided into two compartments separated by a sliding door that opened by retracting into the floor. The starting compartment (31 cm long) was illuminated and the shock compartment (60 cm long) was dark. Training was conducted in a sound and light-attenuated room.
The rat was placed in the starting compartment of the apparatus, facing away from the door, and was allowed to enter the dark compartment. As the rat stepped completely into the dark compartment, the door was closed and a footshock was delivered. For the animals given pretraining infusions of drug or control solution into the BLA, the footshock level used was 0.45 mA with a duration of 1 s. Preliminary data revealed that comparable retention latencies in CEA-infused animals were obtained with a slightly lower footshock intensity. Therefore, CEA-infused animals received a footshock intensity of 0.40 mA for 1 s. The animals were removed from the shock compartment 15 s after termination of the footshock and, after drug treatment, returned to their home cages. On the retention test 48 h after training, the rat was placed in the starting compartment, as in the training session, and the latency to re-enter the dark compartment (maximum latency of 600 s) was recorded. Longer latencies were interpreted as indicating better retention. Shock was not administered on the retention test trial.
Histology.
The rats were anesthetized with an overdose of sodium pentobarbital (100 mg/kg, i.p.) and perfused intracardially with a 0.9% saline solution followed by 10% formaldehyde (vol/vol). Following decapitation, the brains were removed and placed in 10% formaldehyde. At least 24 h before sectioning, the brains were submerged in a 20% sucrose solution (wt/vol) for cryoprotection. Sections of 40 μm were made (using a freezing microtome) and stained with cresyl violet. The sections were examined under a light microscope. Determination of the location of the cannula tips was made according to the standardized atlas plates of Paxinos and Watson (20).
Statistics.
Retention data were analyzed with a two-way ANOVA with β-adrenergic antagonist treatment and glucocorticoid treatment both as between-subject variables. Because different footshock intensities were used for the BLA-infused and CEA-infused animals, data of the two sets of groups were analyzed with separate ANOVAs. Further analysis used Fisher’s post-hoc tests to determine the source of the detected significances in the ANOVAs. A probability level of less than 0.05 was accepted as statistical significance. The number of animals per group is indicated in the figure legends.
RESULTS
Fig. 1 A and B show representative brain slices showing the location of tips of cannulas aimed at the BLA and CEA, respectively. Behavioral data from 29% of the animals (108 of 371) were excluded from statistical analysis either because the cannulae in these animals were not placed bilaterally in the specific amygdala nuclei or because the infusion induced extensive damage to the targeted areas.
Figure 1.
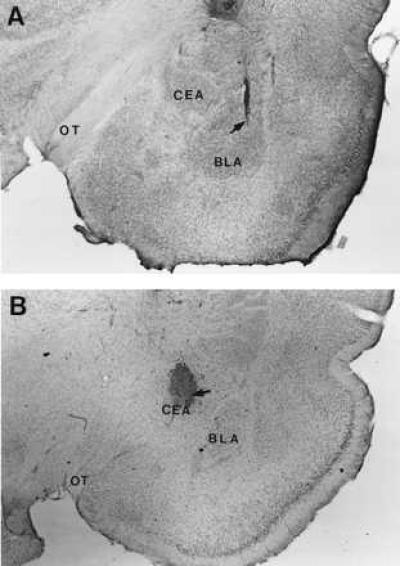
Photomicrographs illustrating representative location of cannulas and injection needle tips within the BLA (A) and CEA (B). Arrows point to the cannula tips. (The injection needles were inserted through the surgically implanted guide cannula and protruded 2 mm beyond the tip of each cannula to reach the BLA or CEA.) OT, optic tract.
The retention test latencies of rats given pretraining infusions of β-adrenergic antagonists into either the BLA or the CEA followed by immediate posttraining systemic injections of the glucocorticoid dexamethasone are shown in Fig. 2. The retention test latencies of animals that received control treatments were relatively short, as was intended. A two-way ANOVA for the data of BLA-infused animals revealed a significant dexamethasone × β-adrenergic antagonist interaction [F(3,80) = 10.08; P < 0.0001]. Post-hoc comparisons between groups indicated that none of the β-adrenergic antagonists affected retention latencies in otherwise untreated animals. Systemic posttraining injections of dexamethasone enhanced the retention of rats given saline infusions into the BLA compared with that of the corresponding vehicle-treated animals (P < 0.01). However, dexamethasone did not increase the retention latencies of animals given infusions of either of the β-adrenergic antagonists in the BLA. In fact, in these animals dexamethasone showed a nonsignificant impairment in retention performance. Further, retention latencies of dexamethasone-treated rats given any of the β-adrenergic antagonists into the BLA were significantly shorter than retention latencies of dexamethasone-treated rats infused with saline (all P < 0.01).
Figure 2.
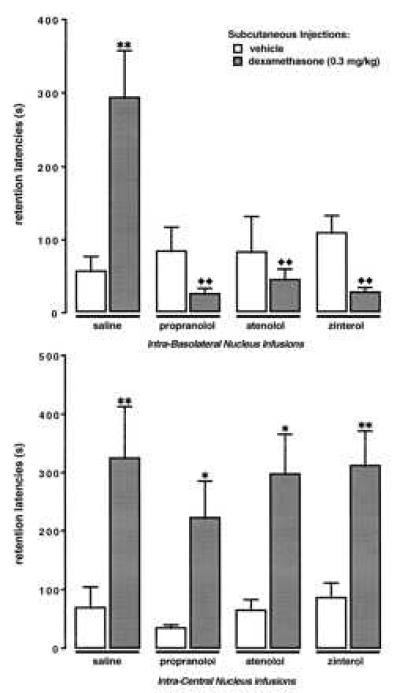
Inhibitory avoidance retention latencies of animals that received pretraining infusions of either the nonspecific β-adrenergic antagonist propranolol (0.5 μg), the β1-adrenergic antagonist atenolol (0.5 μg), or the β2-adrenergic antagonist zinterol (0.5 μg) into the BLA or CEA and immediate posttraining subcutaneous injections of dexamethasone (0.3 mg/kg). Bars represent mean (±SEM) latency in seconds. ∗, P < 0.05; ∗∗, P < 0.01 as compared with the corresponding vehicle group; ♦♦, P < 0.01 as compared with the vehicle-dexamethasone group (n = 8–14/group).
A two-way ANOVA for the data of CEA-infused animals revealed a significant dexamethasone effect [F(1,81) = 24.55; P < 0.0001], but no β-adrenergic antagonist effect [F(3,81) = 0.17; NS] or dexamethasone × β-adrenergic antagonist interaction [F(3,81) = 0.56; NS]. Post-hoc comparisons indicated that none of the β-adrenergic antagonists influenced retention performance of otherwise untreated animals and that dexamethasone enhanced retention in saline-treated rats (P < 0.01), as well as in all of the β-adrenergic antagonist-treated rats (propranolol and atenolol, P < 0.05; zinterol, P < 0.01).
Fig. 3 shows retention test latencies for rats infused concurrently with the specific GR agonist RU 28362 and the β1-adrenergic antagonist atenolol into the BLA immediately after training. A two-way ANOVA revealed significant glucocorticoid [F(3,80) = 6.39; P < 0.001] and atenolol effects [F(1,80) = 14.34; P < 0.001] and a significant glucocorticoid × atenolol interaction [F(3,80) = 3.63; P < 0.05]. Post-hoc analysis revealed that posttraining infusions of the GR agonist into the BLA dose-dependently enhanced retention performance. Retention latencies of rats treated with the two lower doses of RU 28362 (i.e., 1.0 and 3.0 ng) were significantly longer than those of vehicle-treated rats (both P < 0.01). Infusion of the highest dose of RU 28362 did not significantly enhance retention. When RU 28362 was infused together with atenolol the memory-enhancing effect of RU 28362 was blocked. Retention latencies of animals treated simultaneously with RU 28362 and atenolol were significantly shorter than those of animals treated with RU 28362 alone (1.0 ng, P < 0.01; 3.0 ng, P < 0.05).
Figure 3.
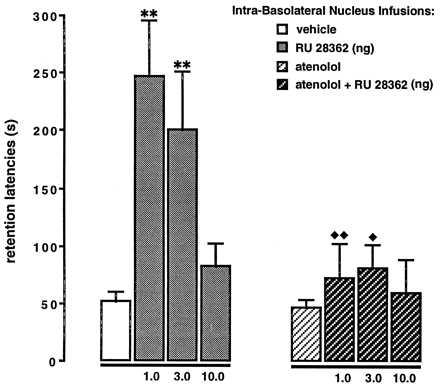
Inhibitory avoidance retention latencies of animals that received posttraining concurrent administration of the GR agonist RU 28362 (1.0, 3.0, or 10.0 ng) and the β1-adrenergic antagonist atenolol (0.5 μg) into the BLA. Bars represent mean (±SEM) latency in seconds. ∗∗, P < 0.01 as compared with the vehicle group; ♦, P < 0.05; ♦♦, P < 0.01 as compared with the corresponding RU 28362 group (n = 9–13/group).
DISCUSSION
These findings provide additional evidence that glucocorticoids and the noradrenergic system are involved in modulating memory storage (5, 9–11, 16, 17, 22–29). Further, they indicate that the BLA is a locus of interaction between these two systems in regulating memory. Systemic injections of dexamethasone enhanced inhibitory avoidance retention when administered immediately after training. As dexamethasone has a high affinity for GRs (30), these findings are consistent with the hypothesis that long-term storage of information is strengthened by posttraining activation of GR-sensitive pathways (23, 25, 27, 28, 31). Similarly, in support of our previous findings (17), posttraining infusions of the specific GR agonist RU 28362 into the BLA enhanced memory storage. These findings, together with the finding that the BLA has a moderate density of GRs (32, 33), strongly suggest that glucocorticoid effects on memory storage are mediated, at least in part, by binding to GRs in the BLA. The finding that the highest dose of RU 28362 (10.0 ng) was ineffective in enhancing retention performance indicates that glucocorticoids, via GRs, have biphasic effects on memory storage. These findings are consistent with those of a recent study examining the effects of systemic administration of different doses of corticosterone to adrenalectomized rats on spatial memory in a Y-maze task (31). It was found that the level of GR occupancy, as measured by a binding assay, was significantly correlated with spatial memory performance following an inverted-U shape curve, whereas the level of mineralocorticoid receptor (MR or type I) occupancy was not. The effects of doses of glucocorticoids used also depend on the specific training conditions used. In the present experiment the animals received a relative low-intensity footshock which resulted in a weak memory that was enhanced by dexamethasone. However, when animals are trained under more stressful conditions that induce high circulating levels of endogenous glucocorticoids, such as in a water-maze spatial task, posttraining administration of a similar dose of dexamethasone impairs memory (34). Administration of a specific GR antagonist also impairs memory formation in the water maze (17, 25, 27).
The main finding of the present experiments is that infusions of β-adrenergic antagonists into the BLA blocked the memory-enhancing effects of systemically or intra-amygdally administered glucocorticoids. Moreover, the finding that higher doses of the GR agonist were ineffective in enhancing memory in animals given atenolol concurrently suggests that β-adrenergic receptor activation in the BLA is required in order for glucocorticoids to modulate memory storage processes. Atenolol did not simply induce a shift in the dose-response effects of glucocorticoids. No differential effects were observed following the administration of the specific β1- or β2-adrenergic antagonists atenolol or zinterol, respectively, or the nonspecific β-adrenergic antagonist propranolol. The finding that pretraining infusions of β-adrenergic antagonists administered alone did not impair memory for inhibitory avoidance training is consistent with the results of previous studies as well as with our finding that excitotoxic lesions of the BLA induced before training do not impair inhibitory avoidance learning (6, 8, 16). Previous findings indicate that a much higher dose of propranolol (i.e., 5.0 μg) administered posttraining induces memory impairment (5, 9).
Although the CEA has a higher density of GRs than does the BLA (32, 33) and the CEA is also richly innervated by noradrenergic fibers (35), antagonism of β-adrenoceptors in the CEA did not block the memory-enhancing effects of systemically administered dexamethasone. Such a selective involvement of the BLA in mediating hormonal effects on memory storage is consistent with previous findings that neurotoxically induced lesions of the BLA, but not the CEA, blocked the memory enhancement induced by systemically administered dexamethasone (16) as well as with evidence that microinfusion of glucocorticoids into the BLA, but not the CEA, enhanced retention (17). The adrenal steroid receptors in the CEA are considered to be involved in feedback mechanisms of the hypothalamic–pituitary–adrenal cortex axis and behavioral responsiveness to stress (36). The present findings indicate that it is very unlikely that the modulatory effects of hormones on cognition involve information flowing from the BLA to the CEA as has been described for processes of fear conditioning (37). In general, the present results are highly consistent with findings of our previous studies examining the role of amygdaloid β-adrenergic mechanisms in mediating the effects of other neuromodulatory systems on memory storage (7, 38).
As GRs are intracellular receptors, they are not present presynaptically on the noradrenergic terminals in the BLA (39). Thus, it seems likely that glucocorticoids bind postsynaptically of the noradrenergic receptor system in the BLA, possibly in the same neurons expressing the β-adrenergic receptors. Although the possibility that glucocorticoids modulate the activity and/or efficacy of the noradrenergic system in the BLA via postsynaptic mechanisms has not been investigated, there is an extensive literature concerning studies of the acute effects of glucocorticoids on the noradrenergic system in hypothalamic, hippocampal, and cerebral cortical areas (18, 19, 40–43).
Systemically administered glucocorticoids bind to GRs in many brain regions. It is of particular interest that GRs are abundantly present in the cell bodies of the A1–A7 noradrenergic cell groups (32, 44), and that the locus coeruleus (LC) and the nucleus of the solitary tract (NTS) densely innervate the amygdala (45, 46). Most noradrenergic nerve terminals reach the CEA, but a considerable density of fibers is found in the BLA (35). Thus, glucocorticoids might modulate memory storage by influencing the activity of the central noradrenergic system at both pre- and postsynaptic sites (18). It has been reported that glucocorticoids do not affect tyrosine hydroxylase (47) or norepinephrine synthesis rate in the LC of adult rodents (19). However, we recently found that activation of GRs in the NTS with posttraining infusions of the specific agonist RU 28362 dose-dependently enhanced memory for inhibitory avoidance training (B.R., C. L. Williams, and J.L.M., unpublished observation). Furthermore, the memory enhancement was blocked in animals given infusions of atenolol into the BLA. These findings suggest that activation of GRs in the NTS, and possibly the LC, may increase the release of norepinephrine in the BLA. We have not as yet examined this implication experimentally.
Finally, there might be an indirect way for β-adrenergic antagonists administered to the BLA to block the memory-enhancing effects of dexamethasone. Systemically administered dexamethasone also binds and activates hippocampal GRs (30, 48). However, the present findings indicate that a blockade of β-adrenergic mechanisms in the BLA blocked the dexamethasone effect, suggesting that any contribution of GR activation in the hippocampus to memory storage is also blocked. Such an interaction between the BLA and the hippocampus in glucocorticoid-induced memory modulation is supported by recent findings from our laboratory indicating that lesions of the BLA block the memory-enhancing effects of the GR agonist RU 28362 infused directly into the dorsal hippocampus (49). These findings, together with evidence that lesions of the BLA as well as infusions of propranolol into the BLA inhibit the effects of electrical stimulation of the perforant path on the induction of long-term potentiation in the dentate gyrus granule cell synapses (50, 51), suggest that β-adrenergic activation in the BLA may be essential for inducing glucocorticoid-mediated plasticity in the hippocampus. These findings are consistent with our hypothesis that activation of the BLA may modulate or comodulate memory consolidation processes in other brain regions, including the hippocampus and caudate nucleus (52).
Acknowledgments
We thank Drs. Béla Bohus and Bruce S. McEwen for helpful comments on an earlier version of the manuscript, and Cindy Quach and Bichngoc Nguyen for excellent technical assistance. RU 28362 was generously provided by Rousell Uclaf (Romainville, France). This research was supported by Dirección General de Asuntos del Personal Académico-Universitat Nacional Autonome de Mexico (G.L.Q.), an R. W. and L. Gerard Trust Fellowship (B.R.), and National Institute of Mental Health/National Institute on Drug Abuse Research Grant MH12526 (J.L.M.).
ABBREVIATIONS
- BLA
basolateral amygdala
- CEA
central amygdala
- GR
glucocorticoid receptor
- RU 28362
11β,17β-dihydroxy-6,21-dimethyl-17α-pregna-4,6-trien-20yn-3-one
References
- 1.Introini-Collison I B, Dalmaz C, McGaugh J L. Neurobiol Learn Mem. 1996;65:57–64. doi: 10.1006/nlme.1996.0006. [DOI] [PubMed] [Google Scholar]
- 2.Introini-Collison I B, Ford L, McGaugh J L. Neurobiol Learn Mem. 1995;63:200–205. doi: 10.1006/nlme.1995.1021. [DOI] [PubMed] [Google Scholar]
- 3.Introini-Collison I B, Nagahara A H, McGaugh J L. Brain Res. 1989;476:94–101. doi: 10.1016/0006-8993(89)91540-0. [DOI] [PubMed] [Google Scholar]
- 4.Introini-Collison I B, Saghafi D, Novack G, McGaugh J L. Brain Res. 1992;572:81–86. doi: 10.1016/0006-8993(92)90454-h. [DOI] [PubMed] [Google Scholar]
- 5.Liang K C, Chen L L, Huang T-E. Chin J Physiol. 1995;38:81–91. [PubMed] [Google Scholar]
- 6.Liang K C, Juler R G, McGaugh J L. Brain Res. 1986;368:125–133. doi: 10.1016/0006-8993(86)91049-8. [DOI] [PubMed] [Google Scholar]
- 7.McGaugh J L, Cahill L, Parent M B, Mesches M H, Coleman-Mesches K, Salinas J A. In: Plasticity in the Central Nervous System: Learning and Memory. McGaugh J L, Bermúdez-Rattoni F, Prado-Alcalá R A, editors. Mahwah, NJ: Lawrence Erlbaum; 1995. pp. 17–39. [Google Scholar]
- 8.McGaugh J L, Introini-Collison I B, Nagahara A H. Brain Res. 1988;446:37–49. doi: 10.1016/0006-8993(88)91294-2. [DOI] [PubMed] [Google Scholar]
- 9.Gallagher M, Kapp B S, Musty R E, Driscoll P A. Science. 1977;198:423–425. doi: 10.1126/science.20664. [DOI] [PubMed] [Google Scholar]
- 10.Introini-Collison I B, Miyazaki B, McGaugh J L. Psychopharmacology. 1991;104:541–544. doi: 10.1007/BF02245663. [DOI] [PubMed] [Google Scholar]
- 11.Liang K C, McGaugh J L, Yao H-Y. Brain Res. 1990;508:225–233. doi: 10.1016/0006-8993(90)90400-6. [DOI] [PubMed] [Google Scholar]
- 12.Galvez R, Mesches M H, McGaugh J L. Neurobiol Learn Mem. 1996;66:253–257. doi: 10.1006/nlme.1996.0067. [DOI] [PubMed] [Google Scholar]
- 13.Galvez R, Quirarte G L, McGaugh J L. Soc Neurosci Abstr. 1996;22:1869. [Google Scholar]
- 14.Williams C L, Gold P E, Men D. Soc Neurosci Abstr. 1996;22:1127. [Google Scholar]
- 15.Roozendaal B, Cahill L, McGaugh J L. In: Brain Processes and Memory. Ishikawa K, McGaugh J L, Sakata H, editors. Amsterdam: Elsevier; 1996. pp. 39–54. [Google Scholar]
- 16.Roozendaal B, McGaugh J L. Neurobiol Learn Mem. 1996;65:1–8. doi: 10.1006/nlme.1996.0001. [DOI] [PubMed] [Google Scholar]
- 17.Roozendaal B, McGaugh J L. Neurobiol Learn Mem. 1997;67:176–179. doi: 10.1006/nlme.1996.3765. [DOI] [PubMed] [Google Scholar]
- 18.de Kloet E R. Front Neuroendocrinol. 1991;12:94–164. [Google Scholar]
- 19.McEwen B S. Biochem Pharmacol. 1987;36:1755–1763. doi: 10.1016/0006-2952(87)90234-6. [DOI] [PubMed] [Google Scholar]
- 20.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- 21.McGaugh J L, Introini-Collison I B, Cahill L, Kim M, Liang K C. In: The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction. Aggleton J P, editor. New York: Wiley–Liss; 1992. pp. 431–451. [Google Scholar]
- 22.Cottrell G A, Nakajima S. Pharmacol Biochem Behav. 1977;7:277–280. doi: 10.1016/0091-3057(77)90146-0. [DOI] [PubMed] [Google Scholar]
- 23.de Kloet E R, de Kock S, Schild V, Veldhuis D H. Neuroendocrinology. 1988;47:109–115. doi: 10.1159/000124900. [DOI] [PubMed] [Google Scholar]
- 24.Flood J F, Vidal D, Bennett E L, Orme A E, Rosenzweig M R, Jarvik M E. Pharmacol Biochem Behav. 1978;8:81–87. doi: 10.1016/0091-3057(78)90127-2. [DOI] [PubMed] [Google Scholar]
- 25.Kovacs G L, Telegdy G, Lissak K. Horm Behav. 1977;8:155–165. doi: 10.1016/0018-506x(77)90032-0. [DOI] [PubMed] [Google Scholar]
- 26.Oitzl M S, de Kloet E R. Behav Neurosci. 1992;106:62–71. doi: 10.1037//0735-7044.106.1.62. [DOI] [PubMed] [Google Scholar]
- 27.Pugh C R, Tremblay D, Fleshner M, Rudy J W. Behav Neurosci. 1997;111:503–511. [PubMed] [Google Scholar]
- 28.Roozendaal B, Portillo-Marquez G, McGaugh J L. Behav Neurosci. 1996;110:1074–1083. doi: 10.1037//0735-7044.110.5.1074. [DOI] [PubMed] [Google Scholar]
- 29.Sandi C, Rose S P R. Brain Res. 1994;647:106–112. doi: 10.1016/0006-8993(94)91404-4. [DOI] [PubMed] [Google Scholar]
- 30.Reul J M H M, de Kloet E R. Endocrinology. 1985;117:2505–2512. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 31.Lupien S J, McEwen B S. Brain Res Rev. 1997;24:1–27. doi: 10.1016/s0165-0173(97)00004-0. [DOI] [PubMed] [Google Scholar]
- 32.Honkaniemi J, Pelto-Huikko M, Rechardt L, Isola J, Lammi A, Fuxe K, Gustafsson J Å, Wikström A-C, Hökfelt T. Neuroendocrinology. 1992;55:451–459. doi: 10.1159/000126156. [DOI] [PubMed] [Google Scholar]
- 33.Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. Neurosci Res. 1996;26:235–269. doi: 10.1016/s0168-0102(96)01105-4. [DOI] [PubMed] [Google Scholar]
- 34.Roozendaal B, Bohus B, McGaugh J L. Psychoneuroendocrinology. 1996;21:681–693. doi: 10.1016/s0306-4530(96)00028-5. [DOI] [PubMed] [Google Scholar]
- 35.Fallon J H, Ciofi P. In: The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction. Aggleton J P, editor. New York: Wiley–Liss; 1992. pp. 431–451. [Google Scholar]
- 36.Schulkin J, McEwen B S, Gold P W. Neurosci Biobehav Rev. 1994;18:385–396. doi: 10.1016/0149-7634(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 37.Sarter M, Markowitsch H J. Behav Neurosci. 1985;99:342–380. doi: 10.1037//0735-7044.99.2.342. [DOI] [PubMed] [Google Scholar]
- 38.McGaugh J L, Cahill L, Roozendaal B. Proc Natl Acad Sci USA. 1996;93:13508–13514. doi: 10.1073/pnas.93.24.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joëls M, de Kloet E R. Prog Neurobiol. 1994;43:1–36. doi: 10.1016/0301-0082(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 40.Duman R S, Strada S J, Enna S J. Brain Res. 1989;477:166–171. doi: 10.1016/0006-8993(89)91404-2. [DOI] [PubMed] [Google Scholar]
- 41.Joëls M, de Kloet E R. Science. 1989;245:1503–1505. [Google Scholar]
- 42.Nakagawa K, Kuriyama K. J Neurochem. 1976;27:609–612. doi: 10.1111/j.1471-4159.1976.tb12290.x. [DOI] [PubMed] [Google Scholar]
- 43.Stone E A, McEwen B S, Herrera A S, Carr E D. Eur J Pharmacol. 1987;141:347–356. doi: 10.1016/0014-2999(87)90551-6. [DOI] [PubMed] [Google Scholar]
- 44.Härfstrand A, Fuxe K, Cintra A, Agnati L F, Zini I, Wikström A C, Okret S, Yu Z Y, Goldstein M, Steinbusch H, Verhofstad A, Gustafsson J-Å. Proc Natl Acad Sci USA. 1986;83:9779–9783. doi: 10.1073/pnas.83.24.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fallon J H, Koziell D A, Moore R Y. J Comp Neurol. 1978;180:509–532. doi: 10.1002/cne.901800308. [DOI] [PubMed] [Google Scholar]
- 46.Zardetto-Smit A M, Gray T S. Brain Res Bull. 1990;25:875–887. doi: 10.1016/0361-9230(90)90183-z. [DOI] [PubMed] [Google Scholar]
- 47.Markey K A, Towle A C, Sze P Y. Endocrinology. 1982;111:1519–1523. doi: 10.1210/endo-111-5-1519. [DOI] [PubMed] [Google Scholar]
- 48.McEwen B S, Weiss J M, Schwartz L S. Nature (London) 1968;220:911–912. doi: 10.1038/220911a0. [DOI] [PubMed] [Google Scholar]
- 49.Roozendaal B, McGaugh J L. Eur J Neurosci. 1997;9:76–83. doi: 10.1111/j.1460-9568.1997.tb01355.x. [DOI] [PubMed] [Google Scholar]
- 50.Ikegaya Y, Saito H, Abe K. Brain Res. 1994;656:157–174. doi: 10.1016/0006-8993(94)91377-3. [DOI] [PubMed] [Google Scholar]
- 51.Ikegaya Y, Nakanishi K, Saito H, Abe K. NeuroReport. 1997;8:3143–3146. doi: 10.1097/00001756-199709290-00027. [DOI] [PubMed] [Google Scholar]
- 52.Packard M G, Cahill L, McGaugh J L. Proc Natl Acad Sci USA. 1994;91:8477–8481. doi: 10.1073/pnas.91.18.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]