

Abstract
Aminoglycoside phosphotransferases (APHs) constitute a diverse group of enzymes that are often the underlying cause of aminoglycoside resistance in the clinical setting. Several APHs have been extensively characterized, including the elucidation of the three-dimensional structure of two APH(3′) isozymes and an APH(2″) enzyme. Although many APHs are plasmid-encoded and are capable of inactivating numerous 2-deoxystreptmaine aminoglycosides with multiple regiospecificity, APH(9)-Ia, isolated from Legionella pneumophila, is an unusual enzyme among the APH family for its chromosomal origin and its specificity for a single non-2-deoxystreptamine aminoglycoside substrate, spectinomycin. We describe here the crystal structures of APH(9)-Ia in its apo form, its binary complex with the nucleotide, AMP, and its ternary complex bound with ADP and spectinomycin. The structures reveal that APH(9)-Ia adopts the bilobal protein kinase-fold, analogous to the APH(3′) and APH(2″) enzymes. However, APH(9)-Ia differs significantly from the other two types of APH enzymes in its substrate binding area and that it undergoes a conformation change upon ligand binding. Moreover, kinetic assay experiments indicate that APH(9)-Ia has stringent substrate specificity as it is unable to phosphorylate substrates of choline kinase or methylthioribose kinase despite high structural resemblance. The crystal structures of APH(9)-Ia demonstrate and expand our understanding of the diversity of the APH family, which in turn will facilitate the development of new antibiotics and inhibitors.
Keywords: Antibiotics, Drug Resistance/Bacteria, Enzymes/Kinase, Methods/X-ray Crystallography, Phosphorylation/Enzymes, Protein/Structure, Antibiotic Resistance
Introduction
Aminoglycosides are a class of commonly used broad-spectrum antibiotics that target the bacterial ribosome. They are characterized by their signature chemical structure, an aminocyclitol nucleus. These antibiotics can be further categorized into two groups: the first group includes those that contain a 2-deoxystreptamine core, such as kanamycin, and the second, smaller group includes those that contain a non-2-deoxystreptamine core (Fig. 1A). Spectinomycin has a streptamine core and it is used in the treatment of acute gonococcal infections (1). Unfortunately, the efficacy of aminoglycosides has been compromised due to the continuous rise of drug resistance in pathogens. Resistance to aminoglycosides can be attributed to several mechanisms, of which enzymatic inactivation of the aminoglycoside is the most prevalent in the clinical setting. Aminoglycosides can be inactivated by the addition of an acetyl, a nucleotidyl, or a phosphate group by acetyltransferases, nucleotidyltransferases, or phosphotransferases (kinases; APHs),5 respectively (2). Nomenclature of aminoglycoside-modifying enzymes follows the convention proposed by Shaw et al. (2): the type of modifying enzyme is identified by acetyltransferase, nucleotidyltransferase, or APH; this is followed by, in parentheses, the enzyme regiospecificity; then the substrate profile is designated by a roman numeral, and the unique amino acid sequence is denoted by a small letter.
FIGURE 1.

Diversity in aminoglycoside antibiotics and the phosphotransferases that inactivate them. A, structures of representative aminoglycosides from different classes. B, cladogram for aminoglycoside phosphotransferases (APHs). Thirty-one amino acid sequences of APHs were aligned using ClustalX version 2.0 (60) and the radial style tree was produced using FigTree version 1.2.3. GenBank protein codes are noted in parentheses. APH(9)-Ia is highlighted in a box with thick black line and the other APHs whose crystal structures have been determined (APH(3′)-IIa/IIIa and APH(2″)-IIa are highlighted in boxes with thin black lines.
APHs generally yield high levels of resistance (3) and constitute the second largest group of aminoglycoside-modifying enzymes with approximately 30 members (2, 4). The APH family of enzymes exhibits considerable diversity (Fig. 1B). Little sequence similarities can be found among the enzymes; however, they do share certain signature residues and sequences. To date, the enzymatic properties of nine APH enzymes have been characterized (5–11). They comprise enzymes of various regiospecificity, with a disparate number and range of substrate spectrum, as well as distinct preferences of nucleotide triphosphates. Only three APHs have had their crystal structures determined (12–16), two of which are isozymes belonging to the APH(3′) subgroup (types IIa and IIIa), whereas the third belongs to the APH(2″) subgroup (type IIa). These three enzymes share similar substrate specificities: 2-deoxystreptamine aminoglycosides. Despite low sequence similarities, the structures revealed that their overall fold is related to that of eukaryotic protein kinases (ePKs). However, in contrast to many ePKs that undergo a substantial conformation change from the open to closed state upon ligand binding, the structural differences between apo and ligand-bound APH(3′)-IIIa (and inferred for APH(3′)-IIa and APH(2″)-IIa) are localized (12). These crystal structures have provided crucial information for the development of new antibiotics and inhibitors (17–21).
So far, much effort has been focused on studying APH enzymes that inactivate aminoglycosides containing a 2-deoxystreptamine core. In contrast, relatively little is known about the remaining members of the APH family, i.e. those that inactivate aminoglycosides with a non-2-deoxystreptamine core. Moreover, considering the diversity in the APH enzyme family in terms of their source, location of the gene in the bacteria, amino acid sequence, regiospecificity, and the size and range of substrate specificity, the three available APH crystal structures offer a finite amount of information for the development of broad range inhibitors and effective antibiotics. We have therefore undertaken the structural studies of APH(9)-Ia from Legionella pneumophila (GenBank protein code AAB58447), a disparate APH that will broaden our understanding of aminoglycoside modifying enzymes (Fig. 1B).
Many aph genes, including aph(3′)-IIa and aph(3′)-IIIa, exist as part of a plasmid or transposable element that can be widely distributed. The enzymes they encode possess broad substrate spectrums, having the ability to detoxify multiple 2-deoxystreptamine aminoglycosides with multiple regiospecificity and high efficiency (8, 22, 23). In contrast, APH(9)-Ia is chromosomally encoded in the human pathogen L. pneumophila, and specifically phosphorylates spectinomcyin (8, 24). Two other spectinomycin phosphotransferases have recently been identified, SpcN and APH(9)-Ib (Fig. 1B) (25, 26). These two are found in spectinomycin producing streptomyces (in contrast to APH(9)-Ia) and are about 70% identical in sequence. In comparison, APH(9)-Ia shares ∼18% sequence identity with SpcN and APH(9)-Ib.
APH(9) enzymes are the only type of APH capable of inactivating spectinomycin and they exclusively phosphorylate the hydroxyl group at position 9. APH(9)-Ia has been shown to carry out its activity using the same mechanism and posses the equivalent catalytically critical residues (Lys-52 and Asp-212) as APH(3′)-IIIa (8). Although the enzyme is highly efficient with kcat/Km values in the range of 106 m−1 s−1 (8), APH(9)-Ia appears to confer only moderate levels of resistance to spectinomycin for L. pneumophila (24). Moreover, the extent of L. pneumophila exposure to spectinomycin is likely low as Legionnaires disease, the infectious manifestation of L. pneumophila, is not typically treated with spectinomycin. Therefore, it has been suggested that APH(9)-Ia might play a minor role in protecting L. pneumophila against the effects of spectinomycin or it might play a role in an as yet undetermined function in the context of L. pneumophila. Note that the genome sequences of four L. pneumophila strains (27–29) have not provided insights into such a possible role as the genes immediately flanking aph(9)-Ia are different in each genome and several translate into protein sequences of unknown function.
Efforts to circumvent bacterial resistance depend on our knowledge of the diversity in the APH family. We herein report three key crystal structures of APH(9)-Ia, an APH that differs greatly from those APHs with known structures, in terms of genetic context, sequence, and substrate spectrum and specificity. The structures of APH(9)-Ia in its apo, binary, and ternary states will be described and a comparison with other APHs and structurally homologous kinases will be presented.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification, and Crystallization
The construction of the cloning vector and protein expression and purification for APH(9)-Ia have been previously reported (30). APH(9)-Ia in which methionine residues were substituted by selenomethionines (APH(9)-IaSe-Met) was produced in a similar manner to the unmodified protein except that minimal medium was used instead of Luria broth and a mixture of amino acids (100 mg/liter of Lys, Phe, and Thr; 50 mg/liter of Val, Ile, and Leu; and 60 mg/liter of selenomethionine) were added approximately one doubling time prior to induction. This combination of metabolites serves to inhibit methionine biosynthesis while providing a ready supply of selenomethionine for incorporation into newly synthesized polypeptides. Protein purification was then carried out as described for the unmodified enzyme, with the addition of 1 mm β-mercaptoethanol in all buffers to prevent oxidation of the selenium.
Crystallization conditions for APH(9)-Ia in its apo state and in the presence of ADP and spectinomycin have been described elsewhere (30). Crystals of APH(9)-IaSe-Met were obtained under identical conditions as the apoenzyme crystals. APH(9)-Ia in complex with AMP was crystallized using the hanging drop vapor diffusion method. The APH(9)-Ia-AMP crystals were obtained in drops containing equal volumes of protein and precipitant solutions where the protein solution consisted of 7.5 mg/ml of APH(9)-Ia in 20 mm imidazole, pH 8.0, 4 mm AMP-PNP, and 10 mm MnCl2, and the precipitant solution contained 15% polyethylene glycol 3350 and 0.2 m calcium acetate.
Data Collection, Structure Determination, and Model Refinement
All diffraction data were collected at beamline X8C at the National Synchrotron Light Source, Brookhaven National Laboratory (Upton, NY), equipped with an ADSC Quantum CCD detector. Prior to data collection, crystals were flash frozen in a cold stream of gaseous nitrogen and data collection was conducted at 100 K. For the APH(9)-IaSe-Met crystals, three-wavelength anomalous diffraction data were collected for phase determination. Data were indexed, merged, and scaled using the HKL2000 program suite (31), and treating the Bijvoet pairs of multiwavelength anomalous dispersion (32) data as independent measurements.
Calculations for phase determination of the APH(9)-IaSe-Met data were performed using the multiwavelength anomalous dispersion phasing protocols incorporated into the program CNS (33). The initial model was built using O (34) and refined using the torsion angle and Cartesian simulated annealing, individual B-factor refinement and energy minimization protocols of CNS. Reciprocal space refinement was alternated with rounds of manual rebuilding, and the values of Rcryst and Rfree were used to monitor the progress of refinement. The model of APH(9)-Ia obtained was further refined against the 1.7-Å APH(9)-Ia data. The structures of APH(9)-Ia-AMP and APH(9)-Ia-ADP-spectinomycin were determined by molecular replacement with PHASER (35), using the high resolution APH(9)-Ia structure as the search model. Refinement of the ligand-bound APH(9)-Ia followed procedures analogous to that for the apoenzyme. Although AMP-PNP was used for crystallization of the binary complex, only the nucleoside monophosphate could be placed unambiguously in the available electron density, thus the β- and γ-phosphates were omitted in the final structure. Pertinent refinement statistics for the final structures are presented in Table 1.
TABLE 1.
Refinement statistics
| APH(9)-Ia | APH(9)-Ia + AMP | APH(9)-Ia + ADP + spectinomycin | |
|---|---|---|---|
| Resolution range (Å) | 50–1.7 | 50–2.8 | 50–2.4 |
| Space group | P21 | P3121 | P3121 |
| a (Å) | 47.41 | 74.47 | 75.01 |
| b (Å) | 70.70 | ||
| c (Å) | 99.31 | 135.5 | 139.9 |
| b (°) | 95.93 | ||
| Number of reflections | |||
| Working set | 57720 | 8766 | 15505 |
| Test set | 6473 | 998 | 1661 |
| Rcrysta/Rfreeb | 0.182/0.228 | 0.265/0.346 | 0.230/0.277 |
| Number of non-hydrogen atoms | |||
| Protein | 5589 | 2640 | 2615 |
| Cofactor | 23 | 27 | |
| Ion | 1 Ni2+ | 1 Ni2+/1 Mg2+ | |
| Substrate | 24 | ||
| Solvent | 860 | 16 | 48 |
| Root mean square deviation | |||
| Bond length (Å) | 0.010 | 0.008 | 0.007 |
| Bond angles (°) | 1.40 | 1.40 | 1.30 |
| Average thermal factor (Å2) | |||
| Protein | 22.02 | 62.89 | 53.41 |
| Cofactor | 62.70 | 54.02 | |
| Ion | 55.69 | 51.36 | |
| Substrate | 34.42 | ||
| Solvent | 37.97 | 48.45 | 42.22 |
| Ramachandran plotc (%) | |||
| Most favored regions | 91.0 | 78.8 | 86.4 |
| Additional allowed regions | 8.1 | 19.1 | 13.2 |
| Generously allowed regions | 0.7 | 1.8 | 0.3 |
| Disallowed regions | 0.2 | 0.4 | 0 |
a Rcryst = Σ (|Fo| − |Fc|)/Σ|Fo|, where |Fo| is the observed and |Fc| is the calculated structure factor amplitude of a reflection.
b Rfree was calculated by randomly omitting 10% of the observed reflections from the refinement.
c According to the Ramachandran plot in PROCHECK (61).
Substrate Assays
Enzyme assays to assess alternative substrates for APH(9)-Ia were performed using the procedure described by Thompson et al. (8). Briefly, the ATP-dependent phosphorylation of spectinomycin by APH(9)-Ia was monitored by coupling the release of ADP to the reactions of pyruvate kinase and lactate dehydrogenase. The rate of APH(9)-Ia activity was reflected in the decrease in absorbance at 340 nm due to the depletion of NADH in the conversion of pyruvate to lactate by lactate dehydrogenase. Assays were measured using a Varian single-cell UV-visible spectrophotometer equipped with a thermo-equilibrated cell block. Data were fit to Equation 1 with nonlinear regression using Grafit version 5.0.1 software, and Michaelis-Menten parameters, Km and kcat, were determined.
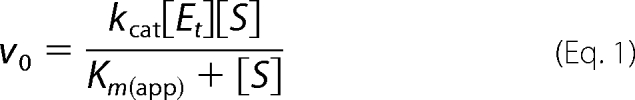 |
Choline, ethanolamine, and glycerol were obtained from Sigma; methylthioribose was generously donated by Dr. Michael Riscoe, Veterans Affairs Medical Center, Portland, OR. Kanamycin and streptomycin were used as negative controls and spectinomycin was used as the positive control. All assays were conducted in triplicates with a final reaction volume of 1 ml. The concentration of ATP was 1 mm, whereas the test substrates was 0.1 mm.
RESULTS AND DISCUSSION
APH(9)-Ia Is a Novel Member of the APH Family of Enzymes
The crystal structure of APH(9)-Ia was determined in its apo state at 1.7-Å resolution, the binary state (with nucleotide, AMP) at 2.8-Å, and the ternary complex state (with ADP and the substrate, spectinomycin) at 2.4-Å (Table 1). These three structures reveal that the 331-residue enzyme adopts a fold reminiscent of ePKs. APH(9)-Ia is a C-shaped molecule composed of two lobes, a small β-strand-rich N-terminal lobe (residues 1–101) and a larger α-helix-rich C-terminal lobe (residues 116–331), which are joined by a 12-residue linker (residues 102–115) (Fig. 2A). The core of the N-terminal lobe consists of a twisted 5-stranded anti-parallel β-sheet (β1–β5), as previously observed in ePKs. The lobe also contains two α-helices (α1 and α2), one on either face of the β-sheet. Helix α2 lies parallel to β4 and forms interactions with the C-terminal lobe.
FIGURE 2.

Schematic representation of APH(9)-Ia and its structural relatives. A, stereo image of the crystal structure of the ternary complex of APH(9)-Ia with ADP and spectinomycin in schematic representation. The N-terminal lobe is colored yellow, the linker between the N- and C-terminal lobe is colored red, the core region of the C-terminal lobe is colored dark blue, and the thumb is colored in light blue. The ADP and spectinomycin are shown as light gray sticks and the magnesium ion is shown in green. B, ternary complex of APH(3′)-IIIa with ADP and kanamycin (PDB code 1L8T); C, APH(2″)-IIa bound with gentamicin (PDB code 3HAM); D, apo form of CK (PDB code 2CKO); and E, ternary complex of MTRK with AMP-PNP and 5-methylthioribose (PDB code 2PUN). The coloring scheme for panels B–E is the same as panel A.
The C-terminal lobe can be subdivided into two intimately associated regions: an α/β“core” region consisting of four helices (α3-α6) and four strands (β6–β9), and an α-helical “thumb” region comprised of helices αA–αE. β7 runs antiparallel to β8, forming a β-sheet akin to that in ePKs. The Brenner motif (HXDXXXXN) (36), found in many enzymes catalyzing phosphoryl transfer reactions including aminoglycoside phosphotransferases and ePKs, was located in the loop connecting β6 and β7 in the core region. The extended activation loop found in ePKs was absent in the APH enzymes. The segment following the DFG motif (DWD in APH(9)-Ia) between β8 and α5 is a β-strand (β9) and forms a two-stranded anti-parallel β-sheet with β6.
To date, the structures of three aminoglycoside APH enzymes have been reported: APH(3′)-IIa (15), APH(3′)-IIIa (12, 14, 37), and APH(2″)-IIa (16). The two APH(3′) isozymes share 33% sequence identity, identical phosphorylation targets, nearly identical substrate specificity profiles, and possess an essentially identical fold. APH(2″)-IIa is 11 and 15% identical in sequence to APH(3′)-IIa and APH(3′)-IIIa, respectively. The enzyme has a comparable substrate specificity and a similarly arranged two-domain structure as the APH(3′) isozymes but differs in its phosphorylation target. In contrast, APH(9)-Ia shows low similarity in sequence to either APH(3′)-IIa/IIIa or APH(2″)-IIa, its substrate profile contains only one aminoglycoside, and its sole substrate belongs to a small group distinct from those that can be phosphorylated by the APH(3′) isozymes or APH(2″)-IIa. However, the structure reveals that APH(9)-Ia has a fold that is homologous to that of the other three APH enzymes, thus placing it in the same family (Fig. 2, A–C). The overall structure of APH(9)-Ia is less compact relative to APH(3′)-IIa/IIIa and more comparable with APH(2″)-IIa, and has a large, solvent-exposed central cavity for ligand binding. The most prominent differences are located in the thumb region of APH(9)-Ia, where there are two additional helices (αC and αE) compared with APH(3′)-IIa and -IIIa and αB is absent in APH(2″)-IIa but has a slightly longer αD helix containing a kink. However, there are numerous subtle differences in the structure throughout the fold, such as the size and topological variations of the helices and strands. This results in 8, 9, and 9% identity between APH(9)-Ia and APH(3′)-IIa, APH(3′)-IIIa, and APH(2″)-IIa, respectively. Overall, a mere 37 residues can be considered to be structurally conserved among all four APH enzymes (i.e. 11% of APH(9)-Ia), and among these only 9 are identical (i.e. 3% of APH(9)-Ia). This underscores the diversity within the APH family of enzymes.
Domain Movement
The three crystal structures of APH(9)-Ia provide four crystallographically independent views of the enzyme as the apo crystals contain two distinct protein molecules per asymmetric unit. Comparison of these four views reveals that the enzyme displays open and closed conformations. The structures of the apoenzyme are in an open conformation, whereas the binary and ternary complex structures display the closed conformation, indicating that the conformational differences are linked to the presence or absence of the nucleotide ligand (Fig. 2A). The structures provide no evidence of further domain movement triggered by the binding of the substrate spectinomycin. The conformational change in APH(9)-Ia that ensues upon the binding of the nucleotide is akin to that observed in ePKs (38, 39) (Fig. 3) and is clearly a unique departure from APH(3′)-IIa/IIIa and APH(2″)-IIa. The absence of domain motion has been most thoroughly examined in APH(3′)-IIIa (12). Here this lack of motion was attributed to the close association between the N- and C-terminal lobes via hydrogen bond interactions between α2 and the loop flanked by α4 and αA, resulting in limited lobe flexibility. Although α2 of APH(9)-Ia is in an equivalent location, the shape and position of the loop between α3 (analogous to α4 in APH(3′)-IIIa) and αA differ significantly. As a result, only one hydrogen bond is observed between the two lobes and this may contribute to the ease of domain movement in APH(9)-Ia.
FIGURE 3.

Stereo image of the comparison of the open and closed conformations of APH(9)-Ia. Representative open (apo, monomer A) and closed (nucleotide-bound) conformations of APH(9)-Ia are superposed using residues 108–323. The open conformation in backbone representation is colored in gray. The degree of movement in the main chain atoms as the enzyme shifts from open to closed conformation upon the binding of the nucleotide is demonstrated by a rainbow gradient of colors ranging, where blue represents no significant displacement and red represents displacements greater than 8 Å between corresponding main chain atoms. Root mean square deviation values were not calculated for sections of the structures not modeled in either the apo or AMP-bound APH(9)-Ia.
Examination of the domain motion using DynDom (40) shows that the five-stranded β-sheet in the N-terminal lobe and the N-terminal portion of helix α2 (residue 58–61) can rotate ∼20° with respect to the C-terminal lobe and the rest of helix α2 (residue 62–72) via three hinge regions: residues 53–62 and 79–80, which flank helix α2, and residues 101–102 in the linker region, which function as the pivot (Fig. 3). Maximum displacement in the moving domain between the open and closed conformations is ∼13 Å. Domain allocation and the extent of movement observed in APH(9)-Ia is comparable with that of protein kinase A (41). Conformational changes from an open or inactive form to a closed or active state are often observed in ePKs. This type of movement could be triggered by uncoupling of regulatory units, phosphorylation (42), and/or binding of a substrate or cofactor (41, 43). These movements are associated with regulation of enzyme activity and substrate recognition. As has been argued for APH(3′)-IIIa (12), regulation of activity for antibiotic resistance enzymes is unusual because such enzymes are expected to be continuously in an active state to rapidly thwart the effects of antibiotics. Because of this, the observed domain motion in APH(9)-Ia is unexpected and we speculate that it is perhaps a remnant of a more ePK-like ancestor. Alternatively, if APH(9)-Ia possesses another function, regulation through domain motions may be important in that context.
The Nucleotide-binding Site
Electron density maps clearly defined the binding of AMP in the binary structure and both ADP and spectinomycin in the APH(9)-Ia ternary complex (Fig. 4, A–C). The nucleotide-binding site is located between the N- and C-terminal lobes, adjacent to the fulcrum for domain rotation. Of the 9 residues that are identical between APH(3′)-IIa/IIIa, APH(2″)-IIa, and APH(9)-Ia, only two residues (Gly-32 and Lys-52) are located in the nucleotide-binding cleft. Gly-32 is located in the loop linking β1 and β2, the region analogous to the flexible nucleotide-binding loop of APH(3′)-IIIa and the glycine-rich P-loop of ePKs that serves as a structural “shield” for the phosphate group of the bound nucleotide in the active site pocket (12). In APH(9)-Ia, this loop sits over the nucleotide-binding site, as in APH(3′)-IIIa, and no direct interactions were observed between the loop residues and the phosphates of ADP. The nucleotide is confined in APH(9)-Ia via the closing of the two lobes induced by its binding to the active site. Nonetheless, the nucleotide binding pocket, even in the closed conformation, is much more solvent exposed in APH(9)-Ia compared with all other nucleotide-bound APH structures studied.
FIGURE 4.

Active site of APH(9)-Ia. A and B, schematic representations of the nucleotide-binding site of the ternary (A) and binary (B) complexes of APH(9)-Ia. ADP is shown as magenta sticks in A and AMP is shown as white sticks in B. The protein is colored according to the assignment in Fig. 2. Interactions between amino acid residues and the nucleotide are shown in dashed lines. Lys-52 is semitransparent because its interaction with the nucleotide cannot be confirmed. Solvent molecules are shown as red spheres and the magnesium ion as a light green sphere. C, schematic representation of the substrate-binding site in the ternary complex of APH(9)-Ia. Spectinomycin is shown in magenta sticks and residues interacting with the substrate and their hydrogen bond interactions with the substrate are indicated as in A and B. D, schematic representation of the hydrogen bonding interactions between spectinomycin and APH(9)-Ia. E, molecular surface of APH(9)-Ia in the vicinity of the active site illustrating the contoured spectinomycin-binding groove. ADP and spectinomycin are shown as magenta sticks. The surface is in light gray and the polar patches surrounding spectinomycin are colored red to represent negatively charged areas and blue to represent positively charged areas. The amino acids that make direct interactions with spectinomycin are mapped to the charged patches.
The other conserved residue between APH(3′)-IIa/IIIa, APH(2″)-IIa, and APH(9)-Ia located in the nucleotide-binding cleft is Lys-52, located on β3 (Fig. 4, A and B). This residue is conserved in all APH enzymes and ePKs as either lysine or, occasionally, arginine, and has been implicated in the binding and proper positioning of the phosphate moieties for enzyme catalysis in APH(3′)-IIIa and ePKs (12). Mutation of this residue to alanine in both APH(3′)-IIIa and APH(9)-Ia results in reduced binding affinity for ATP (8, 14). The structures of APH(3′)-IIIa indicated that this lysine residue, Lys-44, makes hydrogen bond interactions with the non-bridging α and β phosphates of the nucleotide to correctly orient the nucleotide phosphates for catalysis (12). Moreover, Lys-44 in APH(3′)-IIIa is stabilized by the side chain of Glu-60, a residue that is conserved among most APH enzymes and all ePKs. In APH(9)-Ia, Lys-52 lies above the nucleotide with its side chain pointing toward the phosphate groups of the bound nucleotide. However, the interaction between NZ of Lys-52 and the phosphate oxygens cannot be validated in either the binary or ternary complexes. In the binary structure, the distance between NZ of Lys-52 and O-1 of the α-phosphate is 3.6 Å; and in the ternary complex, only partial density was observed for the side chain of Lys-52. The discrepant behavior of Lys52 can perhaps be explained by the absence of the stabilizing glutamic acid residue, which in APH(9)-Ia is substituted with an asparagine residue (Asn-62). Furthermore, helix α2, on which Asn-62 is located, is too distant from β3 to interact with Lys-52. It is reasoned that the ion pair between lysine and glutamate in kinases leads to a close association between β3 and α2, which in turn serves to stabilize the structure of the N-terminal lobe (44). The absence of this interaction in APH(9)-Ia may serve to facilitate the movement of the lobes upon the binding of a nucleotide. Nonetheless, the phosphate moieties do make several interactions with the protein. In the AMP-bound binary structure, one interaction between the enzyme and the phosphate moiety was observed, that between the O-2 of the α-phosphate and a carboxyl oxygen of Asp-230. In the ternary structure, O-2 of the α-phosphate interacts with the enzyme via the carboxyl oxygens of Asn-217 and Asp-230, as well as to the carbonyl of Gly-216 via a solvent molecule; whereas the O-1 of the β-phosphate makes interactions with the carboxyl group of Asp-230 and a water molecule.
In APH(3′)-IIIa and ePKs (12, 38, 45), the non-bridging oxygen atoms of α and β phosphate also interact with two magnesium ions that serve to balance the electrostatic charges between the negatively charged atoms of the enzyme residues and the nucleotide. In contrast, only one magnesium ion was observed in the ternary complex of APH(9)-Ia. The sole magnesium ion occupies a space between the β-phosphate oxygen and the 9-OH of spectinomycin (site of phosphorylation), making a bridging interaction between these two species. It coordinates to oxygens of both the α- and β-phosphates (1.9 and 2.5 Å) and to residues Asn-217 (2.3 Å) and Asp-230 (2.1 Å), as well as a solvent molecule (1.8 Å). Although electron density was also observed in the equivalent location in the binary APH(9)-Ia structure and shares a similar coordination pattern as that observed in the ternary complex, these interactions do not meet the distance criteria associated with a magnesium ion. Therefore, a water molecule has been assigned.
Along with the lysine residue mentioned above, an important residue that is structurally equivalent between APH(9)-Ia and APH(3′)-IIa/IIIa is Phe-50. This residue is highly conserved among the vast majority of APH enzymes as phenylalanine or tyrosine, although in APH(2″)-IIa, the equivalent residue is a valine. In the nucleotide-bound structures of both APH(9)-Ia and APH(3′)-IIIa, the adenine is stabilized and oriented by the ring structure of Phe-50 and Tyr-42, respectively, via π-π ring stacking interactions (Fig. 5). However, when the structures of these two APHs are superposed, the plane of the adenine rings differs by a rotation of ∼40°. This difference in ring orientation has been observed previously between APH(3′)-IIIa and ePKs and was attributed to the alanine in ePKs in place of a tyrosine in APH(3′)-IIIa (12). When the nucleotide-bound structures of APH(9)-Ia and cAMP-dependent protein kinase are aligned based on their conserved residues, the adenine rings superpose well with each other. Despite the presence of an aromatic residue conserved among the APHs, the adenine ring of the nucleotide is positioned in APH(9)-Ia in the same manner as that in ePKs. This observation along with the lobe movement, plus the established features shared between ePKs and APH enzymes indicate that APH(9)-Ia most closely resembles ePKs among all studied APHs.
FIGURE 5.

Comparison of nucleotide binding in APH(9)-Ia, APH(3′)-IIIa, and protein kinase A. A, APH(9)-Ia, colored in magenta, and APH(3′)-IIIa, colored in green, and (B) APH(9)-Ia and protein kinase A (PDB code 1RDQ), colored in cyan, are superposed with the conserved amino acid residues to illustrate the orientation of the adenine ring in the nucleotide. The conserved residues and the nucleotides are shown in sticks and the magnesium ions are shown as spheres.
The Aminoglycoside-binding Site
The aminoglycoside substrate of APH(9)-Ia, spectinomycin, binds in the concave area of the C-shaped enzyme, in a shallow cleft between the core and the thumb regions of the C-terminal lobe. Electron density indicates that the bound aminoglycoside is present as the gem-diol form at position 4 (Fig. 4C). The face of the three fused rings of spectinomycin lies parallel to the base of the enzyme, with the long axis of the molecule lying perpendicular with respect to the α-helices that run vertically beside it. Binding interactions come from the central core region and the α-helical region of the C-terminal lobe.
To best demonstrate the various interactions the substrate makes with the enzyme, spectinomycin can be regarded as having two sides, A and B, by dividing the molecule along the length of the molecule (Fig. 4D). The “A-side” is exposed to the α-helical thumb region, whereas the “B-side” is exposed to the catalytic cavity. Most of the interactions observed between spectinomycin and APH(9)-Ia are between the A-side of the aminoglycoside and the side chains of residues Arg-284, Asp-288, and Tyr-292, located on helix αD in the thumb region. The gem-diol conformation at position 4 of the substrate also allows for two additional water-mediated interactions between the substrate and the enzyme.
In contrast to the A-side, only two direct interactions are observed between the B-side of spectinomycin and the enzyme. The first one involves the oxane oxygen at position 10 of the antibiotic and the Nϵ-2 of His-214. The second interaction is observed between the reactive hydroxyl moiety at position 9 of spectinomycin and the Oδ2 oxygen of the catalytically important Asp-212, the invariant aspartate residue in the HXDXXXXN phosphotransferase motif (Fig. 4, C and D). This motif, also known as Brenner motif, is conserved among many enzymes that catalyze phosphoryl transfer reactions including members of the APH family and ePKs. Mutagenesis studies have well established the invariant aspartate residue to be crucial for the phosphorylation reaction, for when mutated to an alanine, both APH(9)-Ia, APH(3′)-IIIa, as well as cAMP-dependent protein kinase (46) exhibited significant decreased kcat and increased Km values (8, 14). It could be extrapolated from the studies on protein kinase A (45, 47) that this aspartate residue in APH enzymes also serves as a catalytic base to accept the proton from the substrate hydroxyl group. Similarly, the remaining two conserved residues of the phosphotransferase motif, His-210 and Asn-217, serve parallel functions to their APH(3′)-IIa/IIIa and APH(2″)-IIa equivalents, lending stabilization to Asp-212.
A general remark could be made regarding its restricted substrate range of APH(9)-Ia relative to APH(3′)-IIIa. Spectinomycin is unique among the aminoglycsoide antibiotics family. It is structurally, chemically, and functionally very different from the disubstituted 2-deoxystreptamine aminoglycosides, which are substrates for APH(3′)-IIa and IIIa and APH(2″)-IIa. Therefore, a priori one would not expect to observe any structural similarity in the aminoglycoside binding pocket between APH(9)-Ia and APH(3′)-IIa/IIIa and APH(2″)-IIa. Indeed, comparison of the crystal structures reveals that although the location of the aminoglycoside-binding site is effectively the same between the aminoglycoside phosphotransferases, except for the Brenner motif there are essentially no commonality in the residues that line the binding sites. For example, a conserved property that has been observed in disubstituted 2-deoxystreptamine aminoglycoside modifying enzymes whose crystal structures have been determined, namely a negatively charged antibiotic binding pocket, is not observed in APH(9)-Ia. Although isolated polar patches exist in the binding pocket of spectinomycin, it is relatively neutral in its overall charge, corresponding to the uncharged aminoglycoside substrate at physiological pH. It should be noted that the charged areas of the spectinomycin-binding site coincide specifically with the polar groups of the substrate and are produced by residues of the Brenner motif (Asp-212 and His-214) and residues from αD (Arg-284, Asp-288, and Tyr-292). The equivalent of αD in APH(3′)-IIIa is the C-terminal helix and residues from this region have been shown to be critical for aminoglycoside binding (48).
Binding of various aminoglycosides in APH(3′)-IIIa is achieved by the presence of a spacious binding site and closing the loop between αA and αB toward the substrate binding pocket. This loop is highly flexible and can adopt different shapes to accommodate the structurally different substrates bound to different zones of the active site (13, 37). It has also been indicated that aminoglycoside binding in APH(3′)-IIIa does not make use of distinguishing functional groups of its various substrates for binding. These factors have been suggested to confer APH(3′)-IIIa in its ability to bind and detoxify a large number of aminoglycosides. In contrast, in APH(9)-Ia the flexibility of the shape and characteristics of the aminoglycoside-binding site are significantly restricted. In addition to the precise pairing of polar areas of the active site to the polar groups of the substrate, the shape of the aminoglycoside-binding site complements closely to the more compact fused ring structure of spectinomycin. This is achieved by the presence of a number of non-polar residues from the core region of the enzyme and αE, the C-terminal helix. These residues together delineate a fitted groove contoured for spectinomycin (Fig. 4E). Intriguingly, the groove extends beyond spectinomycin, which suggests that if APH(9)-Ia possesses another function, the associated substrate might be large.
Structural Relatives of APH(9)-Ia
It has been reported that CK (49, 50) and MTRK (51, 52) are structurally related to the APH and ePK families. Particularly, the structures of APH(9)-Ia reported here show that it more closely resembles CK and MTRK than APH(3′)-IIa/IIIa (Fig. 2, D and E). This is further supported by the comparison of APH(9)-Ia to published crystal structures in the Protein Data Bank using Dali (53), which reveals that APH(9)-Ia is most similar to human CK (49) (Protein Data Bank code 2CKO; Z-score 18.5) and MTRK from Bacillus subtilis (52) (PDB code 2PUN; Z-score 17.2). In addition to the overall structural homology between APH(9)-Ia and the human CK, the latter also undergoes a similar domain movement upon the binding of its substrate.
However, the topological similarities between APH(9)-Ia, CK, and MTRK share with other APH members, and ePKs are limited to the N-terminal lobe and the core domain in the C-terminal. They share the same N-terminal characteristics of the five-stranded anti-parallel β-sheet sandwiched between two helices. In particular, the β3 strand contains the conserved lysine/arginine residue, as mentioned above. Similar to nearly all other members of the ePK superfamily, in MTRK, this lysine residue is stabilized by a glutamic acid. Although a corresponding glutamate residue (Glu180) is noted in the amino acid sequence of CK, the helix on which it is located is too far away from β3 to interact with Arg-146, as in APH(9)-Ia. Moreover, the side chain of this residue was not observed in the crystal structure.
Despite the differences in length and absolute position of the α-helices, the topology of the secondary structures in the C-terminal lobe of APH(9)-Ia, CK, and MTRK are very similar. The only major difference is the presence of an additional helix at the C terminus of MTRK. The core domain is conserved among CK, MTRK, and the APH and ePK families of enzymes and contains Brenner phosphotransferase motif, HXDXXXXN (36). The remainder of the C-terminal lobe is involved in substrate binding and hence they generally differ in structure. Although the substrates of APH(9)-Ia, CK, and MTRK vary greatly, the structural arrangement of the substrate-binding region are remarkably similar. When the structures of the three enzymes are superposed based on the conserved residues in the C-terminal lobe, the substrate-binding sites distinctly overlap. Furthermore, the reactive hydroxyl group of the substrates also superpose in proximity to each other. Therefore, the catalytic activity of APH(9)-Ia with known substrates of CK (choline and ethanolamine) (54) and MTRK (glycerol and methylthioribose) (55) were tested. Results indicated that no phosphorylation activity by APH(9)-Ia was detected for any of the substrates of CK or MTRK. This can be explained by the absence of the specific amino acids required for interactions with the substrates despite a comparable topology in the substrate-binding area.
Concluding Remarks
The crystal structures of an atypical aminoglycoside phosphotransferase APH(9)-Ia in its apo, AMP-bound, and ADP- and spectinomycin-bound forms were reported. The structures revealed that APH(9)-Ia retains the overall fold of the APH family but differs significantly in the substrate-binding region. APH(9)-Ia exhibits a conformation change upon binding of a ligand, the first to be reported for the APH family of enzymes. This conformational change brings about specific binding of the nucleotide and is likely to be essential for catalysis.
The structural resemblance between APH(9)-Ia, CK, and MTRK affirms the ubiquitous existence of the protein kinase-like motif in all super kingdoms of life (44). The comparison of the architecture of the substrate-binding sites of the three enzymes, together with enzyme kinetics experiments using alternate substrates demonstrate the extensive degree of specificity required for ligand binding. Stringent ligand recognition poses challenges for the design of an inhibitor that would impede the enzyme with the same or higher efficiency as the intended substrate. Alternatively, tremendous efforts have been focused on designing inhibitors that target the nucleotide binding pocket of kinases. Although structural similarity among nucleotide-binding sites is also a concern for the development of therapeutics that aim to inhibit a single enzyme or a restricted group of kinases, there exists subtle distinctions to confer specificity for successful drug development (56–58).
The same principles can be applied to the design of APH inhibitors. It has been shown that APH(3′)-IIIa can be inhibited by several ePK inhibitors (59). It was thought that based on the similarities in sequence, and possibly in structure, in the nucleotide-binding area among APH enzymes, an inhibitor competing with the binding of the nucleotide could be developed to block the activity of the APH family of enzymes. However, the crystal structures of APH(9)-Ia show that its nucleotide binding mode resembles more closely that of ePKs than of the known APH enzymes. The success of developing a useful inhibitor against APH enzymes depends on the collective knowledge of the diverse group of APH enzymes. Structural studies presented here will advance such efforts.
Acknowledgments
We thank Dr. Nicholas P. Cianciotto (Norwestern University Medical School, Chicago, IL) for providing the plasmid containing APH(9)-Ia, Dr. Michael Riscoe (VA Medical Center, Portland, OR) for providing the methylthioribose for alternative substrate analysis, and Dr. Clyde Smith (Stanford Synchrotron Radiation Laboratory, Stanford University, Menlo Park, CA) for early access to the coordinates of APH(2")-IIa. Beamline X8C at the NSLS-BNL, Upton, NY, was supported in part from the Natural Sciences and Engineering Council of Canada and the Canadian Institutes of Health Research.
This work was supported in part by Canadian Institutes of Health Research Grant MOP-13107.
The atomic coordinates and structure factors (codes 3I1A, 3I0Q, and 3I0O) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- APH
- aminoglycoside phosphotransferases
- APH(9)-Ia
- 9-aminoglycoside phosphotransferase type Ia
- APH(3′)-IIIa
- 3′,5″-aminoglycoside phosphotransferase type IIIa
- CK
- choline kinase
- MTRK
- methylthioribose kinase
- ePK
- eukaryotic protein kinase
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate
- PDB
- Protein Data Bank.
REFERENCES
- 1.Mingeot-Leclercq M. P., Glupczynski Y., Tulkens P. M. (1999) Antimicrob. Agents Chemother. 43, 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw K. J., Rather P. N., Hare R. S., Miller G. H. (1993) Microbiol. Rev. 57, 138–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vakulenko S. B., Mobashery S. (2003) Clin. Microbiol. Rev. 16, 430–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright G. D., Thompson P. R. (1999) Front. Biosci. 4, D9–21 [DOI] [PubMed] [Google Scholar]
- 5.McKay G. A., Wright G. D. (1995) J. Biol. Chem. 270, 24686–24692 [DOI] [PubMed] [Google Scholar]
- 6.Siregar J. J., Lerner S. A., Mobashery S. (1994) Antimicrob. Agents Chemother. 38, 641–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siregar J. J., Miroshnikov K., Mobashery S. (1995) Biochemistry 34, 12681–12688 [DOI] [PubMed] [Google Scholar]
- 8.Thompson P. R., Hughes D. W., Cianciotto N. P., Wright G. D. (1998) J. Biol. Chem. 273, 14788–14795 [DOI] [PubMed] [Google Scholar]
- 9.Toth M., Chow J. W., Mobashery S., Vakulenko S. B. (2009) J. Biol. Chem. 284, 6690–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toth M., Zajicek J., Kim C., Chow J. W., Smith C., Mobashery S., Vakulenko S. (2007) Biochemistry 46, 5570–5578 [DOI] [PubMed] [Google Scholar]
- 11.Nakamura A., Takakura Y., Sugimoto N., Takaya N., Shiraki K., Hoshino T. (2008) Biosci. Biotechnol. Biochem. 72, 2467–2471 [DOI] [PubMed] [Google Scholar]
- 12.Burk D. L., Hon W. C., Leung A. K., Berghuis A. M. (2001) Biochemistry 40, 8756–8764 [DOI] [PubMed] [Google Scholar]
- 13.Fong D. H., Berghuis A. M. (2002) EMBO J. 21, 2323–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hon W. C., McKay G. A., Thompson P. R., Sweet R. M., Yang D. S., Wright G. D., Berghuis A. M. (1997) Cell 89, 887–895 [DOI] [PubMed] [Google Scholar]
- 15.Nurizzo D., Shewry S. C., Perlin M. H., Brown S. A., Dholakia J. N., Fuchs R. L., Deva T., Baker E. N., Smith C. A. (2003) J. Mol. Biol. 327, 491–506 [DOI] [PubMed] [Google Scholar]
- 16.Young P. G., Walanj R., Lakshmi V., Byrnes L. J., Metcalf P., Baker E. N., Vakulenko S. B., Smith C. A. (2009) J. Bacteriol. 191, 4133–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanessian S., Szychowski J., Campos-Reales Pineda N. B., Furtos A., Keillor J. W. (2007) Bioorg. Med. Chem. Lett. 17, 3221–3225 [DOI] [PubMed] [Google Scholar]
- 18.Bastida A., Hidalgo A., Chiara J. L., Torrado M., Corzana F., Pérez-Cañadillas J. M., Groves P., Garcia-Junceda E., Gonzalez C., Jimenez-Barbero J., Asensio J. L. (2006) J. Am. Chem. Soc. 128, 100–116 [DOI] [PubMed] [Google Scholar]
- 19.Li J., Chen H. N., Chang H., Wang J., Chang C. W. (2005) Org. Lett. 7, 3061–3064 [DOI] [PubMed] [Google Scholar]
- 20.Fridman M., Belakhov V., Yaron S., Baasov T. (2003) Org. Lett. 5, 3575–3578 [DOI] [PubMed] [Google Scholar]
- 21.Russell R. J., Murray J. B., Lentzen G., Haddad J., Mobashery S. (2003) J. Am. Chem. Soc. 125, 3410–3411 [DOI] [PubMed] [Google Scholar]
- 22.Davies J., Wright G. D. (1997) Trends Microbiol. 5, 234–240 [DOI] [PubMed] [Google Scholar]
- 23.McKay G. A., Thompson P. R., Wright G. D. (1994) Biochemistry 33, 6936–6944 [DOI] [PubMed] [Google Scholar]
- 24.Suter T. M., Viswanathan V. K., Cianciotto N. P. (1997) Antimicrob. Agents Chemother. 41, 1385–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim K. R., Kim T. J., Suh J. W. (2008) Curr. Microbiol. 57, 371–374 [DOI] [PubMed] [Google Scholar]
- 26.Thapa L. P., Oh T. J., Liou K., Sohng J. K. (2008) J. Appl. Microbiol. 105, 300–308 [DOI] [PubMed] [Google Scholar]
- 27.Cazalet C., Rusniok C., Brüggemann H., Zidane N., Magnier A., Ma L., Tichit M., Jarraud S., Bouchier C., Vandenesch F., Kunst F., Etienne J., Glaser P., Buchrieser C. (2004) Nat. Genet. 36, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 28.Chien M., Morozova I., Shi S., Sheng H., Chen J., Gomez S. M., Asamani G., Hill K., Nuara J., Feder M., Rineer J., Greenberg J. J., Steshenko V., Park S. H., Zhao B., Teplitskaya E., Edwards J. R., Pampou S., Georghiou A., Chou I. C., Iannuccilli W., Ulz M. E., Kim D. H., Geringer-Sameth A., Goldsberry C., Morozov P., Fischer S. G., Segal G., Qu X., Rzhetsky A., Zhang P., Cayanis E., De Jong P. J., Ju J., Kalachikov S., Shuman H. A., Russo J. J. (2004) Science 305, 1966–1968 [DOI] [PubMed] [Google Scholar]
- 29.Glöckner G., Albert-Weissenberger C., Weinmann E., Jacobi S., Schunder E., Steinert M., Hacker J., Heuner K. (2008) Int. J. Med. Microbiol. 298, 411–428 [DOI] [PubMed] [Google Scholar]
- 30.Lemke C. T., Hwang J., Xiong B., Cianciotto N. P., Berghuis A. M. (2005) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 606–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 32.Hendrickson W. A. (1991) Science 254, 51–58 [DOI] [PubMed] [Google Scholar]
- 33.Brünger A., Adams P., Clore G., DeLano W., Gros P., Grosse-Kunstleve R., Jiang J. S., Kuszewski J., Nilges M., Pannu N., Read R., Rice L., Simonson T., Warren G. (1998) Acta Crystallogr. Sect. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 34.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Acta Crystallogr. Sect. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 35.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brenner S. (1987) Nature 329, 21. [DOI] [PubMed] [Google Scholar]
- 37.Fong D. H., Berghuis A. M. (2009) Antimicrob. Agents Chemother. 53, 3049–3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narayana N., Cox S., Nguyen-huu X., Ten Eyck L. F., Taylor S. S. (1997) Structure 5, 921–935 [DOI] [PubMed] [Google Scholar]
- 39.Zheng J., Knighton D. R., Xuong N. H., Taylor S. S., Sowadski J. M., Ten Eyck L. F. (1993) Protein Sci. 2, 1559–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayward S., Kitao A., Berendsen H. J. C. (1997) Proteins Struct. Funct. Genet. 27, 425–437 [DOI] [PubMed] [Google Scholar]
- 41.Hayward S. (2004) J. Mol. Biol. 339, 1001–1021 [DOI] [PubMed] [Google Scholar]
- 42.Huse M., Kuriyan J. (2002) Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 43.Kannan N., Neuwald A. F. (2005) J. Mol. Biol. 351, 956–972 [DOI] [PubMed] [Google Scholar]
- 44.Scheeff E. D., Bourne P. E. (2005) PLoS Comput. Biol. 1, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng J., Knighton D. R., ten Eyck L. F., Karlsson R., Xuong N., Taylor S. S., Sowadski J. M. (1993) Biochemistry 32, 2154–2161 [DOI] [PubMed] [Google Scholar]
- 46.Gibbs C. S., Zoller M. J. (1991) J. Biol. Chem. 266, 8923–8931 [PubMed] [Google Scholar]
- 47.Cheng Y., Zhang Y., McCammon J. A. (2005) J. Am. Chem. Soc. 127, 1553–1562 [DOI] [PubMed] [Google Scholar]
- 48.Thompson P. R., Schwartzenhauer J., Hughes D. W., Berghuis A. M., Wright G. D. (1999) J. Biol. Chem. 274, 30697–30706 [DOI] [PubMed] [Google Scholar]
- 49.Malito E., Sekulic N., Too W. C., Konrad M., Lavie A. (2006) J. Mol. Biol. 364, 136–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peisach D., Gee P., Kent C., Xu Z. (2003) Structure 11, 703–713 [DOI] [PubMed] [Google Scholar]
- 51.Ku S. Y., Cornell K. A., Howell P. L. (2007) BMC Struct. Biol. 7, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ku S. Y., Yip P., Cornell K. A., Riscoe M. K., Behr J. B., Guillerm G., Howell P. L. (2007) J. Biol. Chem. 282, 22195–22206 [DOI] [PubMed] [Google Scholar]
- 53.Holm L., Kääriäinen S., Rosenström P., Schenkel A. (2008) Bioinformatics 24, 2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gee P., Kent C. (2003) Biochim. Biophys. Acta 1648, 33–42 [DOI] [PubMed] [Google Scholar]
- 55.Sekowska A., Mulard L., Krogh S., Tse J. K., Danchin A. (2001) BMC Microbiol. 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller J. R., Dunham S., Mochalkin I., Banotai C., Bowman M., Buist S., Dunkle B., Hanna D., Harwood H. J., Huband M. D., Karnovsky A., Kuhn M., Limberakis C., Liu J. Y., Mehrens S., Mueller W. T., Narasimhan L., Ogden A., Ohren J., Prasad J. V., Shelly J. A., Skerlos L., Sulavik M., Thomas V. H., VanderRoest S., Wang L., Wang Z., Whitton A., Zhu T., Stover C. K. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 1737–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weisberg E., Manley P. W., Breitenstein W., Brüggen J., Cowan-Jacob S. W., Ray A., Huntly B., Fabbro D., Fendrich G., Hall-Meyers E., Kung A. L., Mestan J., Daley G. Q., Callahan L., Catley L., Cavazza C., Azam M., Mohammed A., Neuberg D., Wright R. D., Gilliland D. G., Griffin J. D. (2005) Cancer Cell 7, 129–141 [DOI] [PubMed] [Google Scholar]
- 58.Toledo L. M., Lydon N. B., Elbaum D. (1999) Curr. Med. Chem. 6, 775–805 [PubMed] [Google Scholar]
- 59.Daigle D. M., McKay G. A., Wright G. D. (1997) J. Biol. Chem. 272, 24755–24758 [DOI] [PubMed] [Google Scholar]
- 60.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 61.Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]