

Abstract
Pyranose 2-oxidase (P2O) catalyzes the oxidation by O2 of d-glucose and several aldopyranoses to yield the 2-ketoaldoses and H2O2. Based on crystal structures, in one rotamer conformation, the threonine hydroxyl of Thr169 forms H-bonds to the flavin-N5/O4 locus, whereas, in a different rotamer, it may interact with either sugar or other parts of the P2O·sugar complex. Transient kinetics of wild-type (WT) and Thr169 → S/N/G/A replacement variants show that d-Glc binds to T169S, T169N, and WT with the same Kd (45–47 mm), and the hydride transfer rate constants (kred) are similar (15.3–9.7 s−1 at 4 °C). kred of T169G with d-glucose (0.7 s−1, 4 °C) is significantly less than that of WT but not as severely affected as in T169A (kred of 0.03 s−1 at 25 °C). Transient kinetics of WT and mutants using d-galactose show that P2O binds d-galactose with a one-step binding process, different from binding of d-glucose. In T169S, T169N, and T169G, the overall turnover with d-Gal is faster than that of WT due to an increase of kred. In the crystal structure of T169S, Ser169 Oγ assumes a position identical to that of Oγ1 in Thr169; in T169G, solvent molecules may be able to rescue H-bonding. Our data suggest that a competent reductive half-reaction requires a side chain at position 169 that is able to form an H-bond within the ES complex. During the oxidative half-reaction, all mutants failed to stabilize a C4a-hydroperoxyflavin intermediate, thus suggesting that the precise position and geometry of the Thr169 side chain are required for intermediate stabilization.
Keywords: Electron Transfer, Enzymes/Catalysis, Enzymes/Flavin, Enzymes/Kinetics, Enzymes/Oxidase, Enzymes/Oxidation-Reduction
Introduction
Pyranose 2-oxidase (P2O,5 pyranose:oxygen 2-oxidoreductase, EC 1.1.3.10) from Trametes multicolor is a homotetrameric enzyme with each subunit carrying one FAD covalently linked to Nϵ2 (N3) of His167 via the FAD 8α-methyl group (1). P2O catalyzes the oxidation by molecular oxygen of d-glucose (d-Glc) and several aldopyranoses at the C2 position to yield the corresponding 2-ketoaldoses and hydrogen peroxide (2). The overall catalytic reaction can be divided into two half-reactions (Scheme 1) obeying a Ping-Pong type mechanism at pH 7 (3): a reductive half-reaction in which the protein-bound flavin receives a hydride equivalent from a sugar substrate to produce the reduced FAD (FADH−) and the 2-keto-sugar, and an oxidative half-reaction in which two electrons are transferred from the reduced flavin to O2 to form H2O2 (2, 3).
SCHEME 1.

We reported the first detection of a C4a-hydroperoxyflavin intermediate for a flavoprotein oxidase (4). This flavin intermediate has not been previously observed for flavoprotein oxidases but has been limited to reactions of flavoprotein monooxygenases (5–7). Besides P2O, an intermediate in flavoprotein oxidases has been detected only in the crystalline forms of choline oxidase (1.86 Å) (8) and nitroalkane oxidase (in the presence of cyanide) (9), and in the mutant form, C42S, of an NADH oxidase (10). In the case of glucose oxidase from Aspergillus niger, the generation of a flavin semiquinone-superoxide radical pair using pulse radiolysis resulted in formation of a putative C4a-hydroperoxyflavin intermediate (11). Studies on the P2O reductive half-reaction indicate that P2O binds d-Glc according to a two-step binding mechanism: first an initial complex is formed, followed by an isomerization step to form an active Michaelis ES complex (P2O·glucose). Interestingly, this complex shows higher absorbance at 395 nm than does the oxidized enzyme, which is unique among flavoprotein oxidases (3).
P2O belongs to the large family of glucose-methanol-choline oxidoreductases (12, 13) with the catalytic side chains His548–Asn593 positioned at the re-face of the flavin isoalloxazine ring (13). The active site is gated by a dynamic active-site loop (residues 451–461), which has been shown to harbor several residues important for substrate binding and catalysis (13–15). Comparison of P2O structures in sugar-free and sugar-bound forms reveals the presence of at least two well defined active-site loop conformations, each of which is relevant to one of the half-reactions (14). The structure of a less active P2O mutant (H167A), in complex with a slow substrate (2-deoxy-2-fluoro-d-glucose (14)) or the reaction product (2-keto-d-glucose (16)), shows the active-site loop in an open conformation. The open loop state is able to accommodate spatially the sugar substrate, and thus this conformation is relevant for the reductive half-reaction (3, 14). The wild-type structure of P2O in complex with the small competitive inhibitor acetate revealed the loop in a different well defined conformational state (13). This state is characterized as fully closed, and the active site is solvent-inaccessible and provides a more hydrophobic substrate environment than does the open state. Thus, this structural state provides a suitable environment for the reaction of the reduced enzyme and oxygen, and can spatially accommodate the C4a-hydroperoxy-FAD intermediate during the oxidative half-reaction (4, 13, 14).
We have discussed previously that the conserved residue Thr169 occupies an important position in the P2O active site, positioned immediately “above” the flavin, on the flavin si-side, where it forms a hydrogen bond to the flavin N5 and O4 atoms when the loop is in the closed conformation (13, 14). Flavin-N5-protein interactions are recurrent features in structures of many flavoprotein oxidases (17), and little is known about the functional effects of this H-bonding interaction. When the active-site loop assumes the open conformation in P2O, Thr169 discards its flavin interaction by means of a rotamer change (14). The position of the Thr169 side chain relative to the sugar substrate and FAD also provides a simple, straightforward explanation for the poor performance of d-galactose (d-Gal) as a substrate. The oxidation of d-Gal by P2O is industrially useful, because it produces the intermediate for further synthesis of d-tagatose, a low caloric and non-glycemic sweetener (18). In wild-type P2O (WT), the axial O4 hydroxyl group of d-Gal is probably unfavorable due to steric clashes with the Thr169 side chain (14). The kinetics of the binding and reaction mechanism of P2O with d-Gal have not been rationalized in detail, because transient kinetics of the reaction have not been investigated.
In this report, steady-state and transient kinetics of Thr169 variants, T169S, T169N, T169G, and T169A, with d-Glc and d-Gal were investigated. Rapid kinetics of WT with d-Gal are also reported. These results provide mechanistic insights regarding the role of Thr169 in the reductive and oxidative half-reactions. In addition, three-dimensional structures of T169G and T169S were solved and used in conjunction with kinetic data for interpretation of the roles of Thr169 in catalysis of P2O.
EXPERIMENTAL PROCEDURES
Reagents
d-Glc (99.5% purity), d-Gal (99% purity), and horseradish peroxidase were purchased from Sigma-Aldrich. ABTS (2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt) was purchased from Roche Applied Science (Germany). Concentrations of the following compounds were determined using the known absorption coefficients at pH 7.0: ϵ403 = 105 m−1cm−1 for horseradish peroxidase; ϵ458 = 1.3 × 104 m−1 cm−1 for the value of FAD per each subunit of T169S; ϵ458 = 1.2 × 104 m−1 cm−1 for T169N; ϵ458 = 1.1 × 104 m−1 cm−1 for T169G; and ϵ453 = 1.18 × 104 m−1 cm−1 for T169A. All mutants were expressed in Escherichia coli without His6 tag, and purified according to the protocol described earlier (14).
Site-directed Mutagenesis
Site-directed mutagenesis at position Thr169 of P2O was performed using the QuikChange® II site-directed mutagenesis kit. All primers were from Sigma-Proligo (Singapore). The plasmid pET11a was used as a template for the mutagenic PCR (GeneAmp PCR system, Applied Biosystems, model 2004 or MyCyclerTM, Thermal Cycler, Bio-Rad). All plasmids were confirmed using T7 forward and reverse primers, as well as a primer to an internal region of the p2o gene. The DNA sequencing was performed by MACROGEN (Korea). The nucleotide sequences of the synthetic oligonucleotides are listed in supplemental Table S1.
Reduction Potential Measurement
Measurements of reduction potential values (Emo′) of the mutant enzymes were carried out according to the protocol described in a previous study (19), using xanthine/xanthine oxidase as a reduction system. Reactions for all mutants were carried out in reaction mixtures containing 500 μm xanthine, 3 nm xanthine oxidase (side arm), standard dye (26.8 μm indigo carmine (Emo′ = −116 mV), 18 μm cresyl violet (Emo′ = −166 mV), or 20 μm methylene blue (Emo′ = +10 mV)), and Thr169 mutants (26.8 μm T169S, 26.8 μm T169N, 18 μm T169A, or 20 μm T169G) in 50 mm sodium phosphate (pH 7.0). Details of the procedure are provided in supplemental Procedures.
Steady-state Kinetics
Initial velocities at various concentrations of oxygen with d-Glc or d-Gal were measured at 4 °C and 25 °C using a stopped-flow spectrophotometer (3). Steady-state kinetics of the P2O variants was studied by coupling to the reaction of horseradish peroxidase and ABTS. Progress of the reaction was followed at 420 nm (ϵ420 = 42.3 mm−1cm−1) where the oxidized ABTS absorbs (3). The assay reaction typically contained 20–40 nm of a mutant enzyme, 0.6 μm peroxidase, 2 mm ABTS, varying concentrations of d-Glc or d-Gal, and varying oxygen concentrations in 50 mm sodium phosphate (pH 7.0).
Rapid Reaction Experiments
Reductive and oxidative half-reactions of the Thr169 variants were studied using a stopped-flow spectrophotometer (Hi-Tech Scientific). Experimental protocols are similar to those in the previous report (3, 4) and described in detail in supplemental Procedures. The reduction of the enzyme-bound FAD was monitored at 395 and 458 nm. For studying the oxidative half-reaction, solutions containing the reduced enzyme were prepared as described previously (3, 4) and then mixed with buffers equilibrated with oxygen ([O2] = 0.13, 0.31, 0.61, and 0.96 mm) using the stopped-flow spectrophotometer. The reactions were performed in 50 mm sodium phosphate (pH 7.0), and monitored at 5 nm intervals in the λ range between 300 and 550 nm.
Data Analysis: Transient Kinetics
Analyses of observed rate constants (kobs) from kinetic traces were conducted by fitting data to exponential equations using the software Kinetic Studio (Hi-Tech Scientific, Salisbury, UK) or Program A (developed by C. J. Chiu, R. Chung, J. Diverno, and D. P. Ballou at the University of Michigan, Ann Arbor, MI). Rate constants were determined from plots of kobs versus sugar or oxygen concentrations using Marquardt-Levenberg nonlinear fitting algorithms included in KaleidaGraph (Synergy Software) according to Equations 1 and 2 (3). Rate constants are defined as in Table 2.
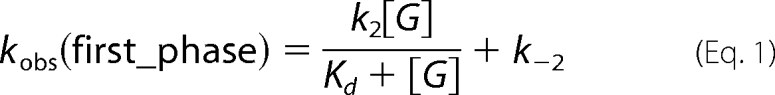 |
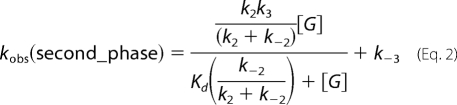 |
TABLE 2.
Summary of rate constants for the flavin reduction of Thr169 variants at pH 7
| Substrate | Parameter | WT | T169S | T169N | T169G | T169A |
|---|---|---|---|---|---|---|
| d-Glc, 4 °C | kred (s−1) | 15.3 ± 0.4a | 13.8 ± 0.4 | 9.7 ± 0.1 (fast) 0.002 (slow) | 0.7 ± 0.01 | ND |
| Kd (mm) | 45a | 45 | 47 | NDb | ND | |
| d-Glc, 25 °C | kred (s−1) | 48 ± 2 | ND | ND | ND | 0.03 ± 0.007c |
| Kd (mm) | ND | ND | ND | ND | ND | |
| d-Gal, 4 °C | kred (s−1) | 0.3 ± 0.03 | 0.5 ± 0.04 | 0.9 ± 0.01 (fast) 0.006 (slow) | 2.7 ± 0.02 | ND |
| Kd (mm) | 3.7 ± 0.2 | 5.9 ± 0.3 | 6.6 ± 0.3 | 4.2 ± 0.2 | ND | |
| d-Gal, 25 °C | kred (s−1) | 1.2 ± 0.04 | ND | ND | ND | 0.005 ± 0.0008c |
| Kd (mm) | ND | ND | ND | ND | ND |
a Rate constant and Kd are from the previous report (3).
b ND, not determined. The Kd value could not be determined because the absorbance increase at 395 nm of the first phase was not significant.
c These numbers contain large errors, because the highest concentrations of d-Glc and d-Gal used were not in a range that gives the saturating rate.
Data Analysis: Steady-state Kinetics
Initial rates of steady-state measurements were calculated using Program A. The initial rates were analyzed for steady-state kinetic parameters of a two-substrate enzyme reaction according to a double-reciprocal plot of a Ping-Pong (Equation 3) or a ternary complex mechanism (Equation 4) (20). In addition, data were analyzed according to direct plots (Equations 5 and 6) using the EnzFitter program (Biosoft, Cambridge, UK).
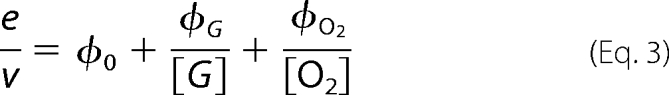 |
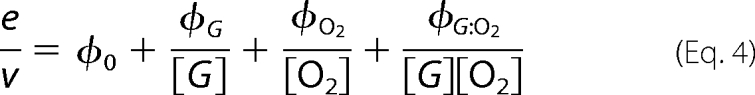 |
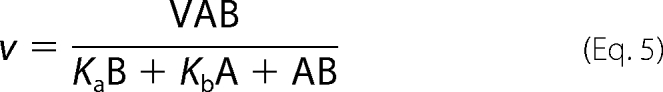 |
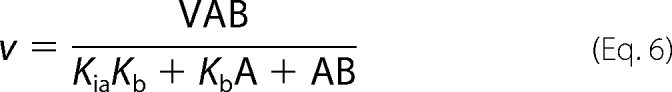 |
Crystallographic Analysis
Expression and purification of the Thr169 mutants used for crystallization have been reported previously (21). For all proteins, crystals grew from microseeded hanging drops containing a 1:1 mixture of 0.1 m Mes (pH 5.2), 50 mm MgCl2, 10% (w/v) monomethyl ether polyethylene glycol 2000 and protein at a concentration of 20 mg/ml in 20 mm Mes (pH 5.2). Crystals were stabilized in mother liquor with 28 (w/v) % polyethylene glycol 2000, followed by vitrification in liquid nitrogen. Data were collected at 100 K using synchrotron radiation at beam line I911-2, MAX-lab, Lund, Sweden, and processed and scaled using the XDS package (22). Details of data processing are provided in supplemental Procedures. Data collection and refinement statistics are given in supplementary Table S2.
RESULTS
Reduction Potentials of Thr169 Variants
The absorbances at 458 nm, an isosbestic point for indigo carmine, and at 610 nm where P2O has no absorbance, were used for calculating the amount of the enzyme (E) and dye (D), respectively (19). Plots of log(Dred/Dox) versus log(Ered/Eox) gave a slope of ∼1.2 (supplemental Fig. S1), indicating that both T169S and T169N were reduced by xanthine/xanthine oxidase via a two-electron process (indigo carmine as the reference dye). No absorption was detected at the long wavelength or 370 nm regions, which suggests that no semiquinone was stabilized during the reduction of FAD. The midpoint potentials (Emo′) calculated from these plots are −106 mV for T169S and −95 mV for T169N, which are similar to that of the wild-type enzyme (WT), −105 mV (14). T169A and cresyl violet were also reduced by xanthine/xanthine oxidase via a two-electron process (absorbance at 414 and 585 nm was used for calculating concentrations of the oxidized enzyme and dye, respectively). The slope of the plot was ∼0.92 (data not shown), and Emo′ was calculated to be −197 mV, which is significantly lower than WT. For T169G (methylene blue as the standard dye), calculations using absorbance at 458 and 664 nm gave a slope of 0.86, and Emo′ of −1.2 mV. For each mutant, at least three standard dyes were used to ensure that there was no interference from dyes to the measurement. All data are summarized in Table 1.
TABLE 1.
Steady-state kinetic parameters for the wild-type and Thr169 mutants of P2O
| Parameters | Ping-Pong |
Ternary |
||||
|---|---|---|---|---|---|---|
| WT | T169S | T169N | T169G | T169A | ||
| d-Glucose (4 °C) | kcat (s−1) | 9.7 ± 0.15 | 13.7 ± 0.2 | 5.6 ± 0.05 | 0.7 ± 0.01 | 0.06 ± 0.005 |
| KmGlc (mm) | 1.13 ± 0.07 | 1.7 ± 0.05 | 4.6 ± 0.1 | 0.9 ± 0.02 | 30 ± 1 | |
| KmO2 (mm) | 0.09 ± 0.008 | 0.99 ± 0.03 | 4.8 ± 0.1 | 4.5 ± 0.5 | 0.2 ± 0.03 | |
| kcat/KmGlc (mm−1s−1) | 8.6 | 8.1 | 1.2 | 0.8 | 0.002 | |
| kcat/KmO2 (mm−1s−1) | 110.0 | 13.8 | 1.2 | 0.2 | 0.4 | |
| d-Galactose | kcat (s−1) | 1.2 ± 0.05 | 2.5 ± 0.02 | 5.3 ± 0.1 | 1.5 ± 0.03 | 0.03 ± 0.002 |
| (25 °C) | KmGal (mm) | 15.0 ± 0.3 | 16.2 ± 0.5 | 16 ± 1 | 3.1 ± 0.08 | 50 ± 2 |
| KmO2 (mm) | 0.03 ± 0.002 | 0.07 ± 0.01 | 1.32 ± 0.1 | 3.7 ± 0.08 | 0.4 ± 0.03 | |
| kcat/KmGal (mm−1s−1) | 0.08 | 0.15 | 0.3 | 0.5 | 0.005 | |
| kcat/KmO2 (mm−1s−1) | 44.2 | 32.0 | 3.4 | 0.4 | 0.07 | |
| Reduction potential | Emo′ (mV) | −105a | −106 | −95 | −1 | −197 |
a Redox potential value is from a previous report (14).
Steady-state Kinetics
Steady-state kinetic parameters for all P2O variants were determined from initial rates of the assay reactions at various concentrations of oxygen and d-Glc or d-Gal. KmGlc denotes Km of d-Glc while KmGal represents Km of d-Gal. Double-reciprocal plots for T169N (Fig. 1, A and C), T169S, and T169G (supplemental Fig. S2) all display parallel-line patterns consistent with a Ping-Pong mechanism (Equation 3) similar to that of WT (3), whereas T169A shows an intersecting-line pattern for reactions both with d-Glc (Fig. 1B) and d-Gal (Fig. 1D), consistent with a ternary-complex mechanism (Equation 4). All steady-state kinetic parameters are summarized in Table 1.
FIGURE 1.

Steady-state kinetics of Thr169 mutants with d-Glc or d-Gal. The assay reactions were performed at 4 °C (for d-Glc) and 25 °C (for d-Gal) using the stopped-flow spectrophotometer. Double reciprocal plots of initial rates of T169N (A) with final d-Glc concentrations of 0.1, 0.2, 0.4, 1, 2, 4, 8, 16, 25, and 50 mm, and T169A (B) with final d-Glc concentrations of 8, 12.5, 25, 50, 100, 150, and 200 mm. The upper to lower lines represent, respectively, the plots of the final oxygen concentrations of 0.13, 0.26, 0.44, 0.74, and 1.09 mm. Double reciprocal plots of initial rates of T169N (C) with final d-Gal concentrations of 2, 4, 8, 16, 32, 64, and 150 mm, and T169A (D) with final d-Gal concentrations of 8, 16, 32, 64, 150, 300, and 400 mm are shown. The upper to lower lines represent, respectively, the final oxygen concentrations of 0.13, 0.26, 0.44, and 0.74 mm. The reactions of T169S and T169G with d-Glc or d-Gal showed parallel-line patterns similar to that of T169N (supplemental information).
When d-Glc was used as a substrate, the value of kcat/KmGlc for T169S was similar to that of WT (8.1 versus 8.6 mm−1s−1), whereas those of T169N and T169G were ∼7- and 11-fold lower than that of WT, respectively (Table 1). This shows that the reductive half-reaction is less affected in T169S, compared with T169N and T169G. When compared with WT, kcat/KmO2 was reduced by 8-, 92-, and 549-fold for T169S, T169N, and T169G, respectively, indicating that Thr169 is significantly important for the oxidative half-reaction. For T169A, both kcat/KmGlc and kcat/KmO2 decreased considerably, indicating that, in addition to altering the kinetic reaction pattern, replacing the side-chain hydroxyl group by a methyl group severely impairs the overall catalytic competence. It is interesting to note that with d-Glc as a substrate, T169S shows the highest kcat value (13.7 s−1), also exceeding that of the WT.
With d-Gal as a substrate, the kcat/KmGal values of T169S, T169N, and T169G were all higher (2-, 4-, and 6-fold, respectively) than that of wild-type P2O, which can be mainly attributed to the increase in kcat of these mutants (Table 1). This implies that by replacing the Thr side chain with Ser, Asn, and Gly, the overall turnover of the reaction with d-Gal has improved. When compared with WT, the kcat/KmO2 values were 1.4-, 13-, and 111-fold lower for T169S, T169N, and T169G, respectively, again confirming that alterations at the amino acid position 169 significantly affect the oxidative half-reaction.
Reduction of the Oxidized Thr169 Mutants by d-Glucose
To investigate the role of Thr169 in P2O catalysis and to identify the specific reaction steps affected by the mutations, transient kinetics were investigated. For analysis of the reductive half-reaction with d-Glc as a substrate, anaerobic enzyme solutions were mixed with buffers containing various concentrations of d-Glc, and reduction of the flavin was monitored at 458 and 395 nm. Absorbance at 458 nm gives the maximum difference between the oxidized and the reduced enzyme, and the signal at 395 nm indicates formation of an active P2O·sugar complex, as previously observed in the wild-type reaction (3).
Only the kinetic traces of the T169S and T169N reduction showed an obvious increase in absorbance at 395 nm. The first phase corresponds to a small increase in absorbance at 395 nm, concomitant with a lag at 458 nm (Fig. 2, A and B). This phase was interpreted as the formation of an active P2O·sugar complex required for flavin reduction (3). The macroscopic equilibrium dissociation constants (Kd) associated with the binding of d-Glc to the oxidized state of T169S and T169N (45 and 47 mm, respectively (Table 2)) are identical to that of WT (45 mm) (3), indicating the same binding affinity and similar nature of the P2O·Glc complex in the mutants and WT.
FIGURE 2.

Reduction of the oxidized Thr169 mutants with d-Glc. Stopped-flow traces at 458 nm are shown in dotted lines, whereas those at 395 nm are in solid lines. A, a solution of T169S (23 μm) was mixed with various concentrations of d-Glc (0.4, 0.8, 1.6, 3.2, 6.4, 15, 30, and 60 mm). All concentrations are given as after mixing. The inset a shows a plot of kobs of the first phase (absorbance increase at 395 nm) versus d-Glc concentrations, whereas the same plot of the second phase (large decrease in A458 nm) is shown in inset b. B, after storing on ice for at least 3 weeks, the third slow phase of the flavin reduction (lowest dotted line) diminished, and kinetic traces of the T169N reaction showed biphasic kinetics. C, stopped-flow experiments similar to A were performed with T169G. Traces monitored at 395 nm did not show significant absorbance increase as in the reaction of T169S in A.
Analysis of the flavin reduction phase (the decrease in absorbance at 458 nm in the second phase) is in agreement with the effects of the mutations on the first phase (Fig. 2 and Table 2). In the reactions of T169S and T169N, flavin was reduced by d-Glc with a rate constant kred (4 °C) of 13.8 ± 0.4 and 9.7 ± 0.1 s−1, respectively, which is similar to kred of the wild type (15.3 ± 0.4 s−1) (3). However, we noted that the biphasic reductive half-reaction of T169N with d-Glc (Fig. 2B) could only be observed after storing the purified enzyme on ice for 3 weeks prior to the experiment. When freshly prepared T169N was used in the experiment, the reductive half-reaction resulted in three phases; the first phase was substrate binding with the absorbance increase at 395 nm (Kd = 47 mm), and the second phase was flavin reduction (9.7 ± 0.1 s−1) as described above, while the third phase corresponded to slow flavin reduction and accounted for ∼20–30% of the total amplitude at 458 nm (Fig. 2B). The first two phases are identical to those of the stored enzyme. The observed rate constants of the third phase were hyperbolically dependent on the concentrations of d-Glc with a limiting rate constant of 0.002 s−1. Following storage on ice for at least 3 weeks, the third phase diminished, and most of the flavin reduction was now according to the fast second phase (Fig. 2B).
Kinetic traces of T169A and T169G do not show significant increase in absorbance at 395 nm (Fig. 2C), implying that the nature of the complex formed is different in these mutants. In addition, the reductive half-reaction in T169G and T169A showed significantly slower flavin reduction (kred of 0.7 ± 0.01 (at 4 °C) and 0.03 ± 0.007 (at 25 °C) s−1, respectively) when compared with WT (15.3 ± 0.4 s−1 (at 4 °C) and 48 ± 2 s−1 (at 25 °C)) (Table 2 and Fig. 2C).
Reduction of the Oxidized Thr169 Mutants by d-Galactose
A similar set of experiments were conducted using d-Gal as a substrate. However, due to the slow reaction rate, flavin reduction of T169A was monitored in an anaerobic cuvette. As observed with d-Glc, only kinetic traces of WT, T169S, and T169N showed obvious increases in absorbance at 395 nm (Fig. 3 and supplemental Fig. S3). Overall, the reductive half-reaction with d-Gal showed two observed phases similar to the reaction of d-Glc (Fig. 2) (3). Interestingly, in contrast to the reaction with d-Glc (Fig. 2) (3), kobs of the first substrate-binding phase is linearly dependent on [d-Gal] (insets in Fig. 3), consistent with a simple one-step binding model (Fig. 4B). The one-step binding model is also supported by agreement between (KmGal) obtained from steady-state kinetics at 4 °C (3.83 mm, data not shown), and the value calculated based on the model in Fig. 4B and kinetic parameters in Table 2 (3.88 mm). Macroscopic Kd values for T169S (5.9 ± 0.3 mm), T169N (6.6 ± 0.3 mm), and T169G (4.2 ± 0.2 mm) are all higher than that of WT (3.7 ± 0.2 mm), indicating that d-Gal binds to the mutants with lesser affinity than to WT. The second phase, flavin reduction, in T169S (0.5 ± 0.04 s−1), T169N (0.9 ± 0.01 s−1), and T169G (2.7 ± 0.02 s−1) is faster than that of WT (0.3 ± 0.03 s−1) (Table 2). Thus, the rapid kinetic results suggest that, by replacing the Thr169 side chain with Ser, Asn, or Gly, only the rate constant of the flavin reduction step is enhanced without a concomitant increase in the binding affinity of d-Gal.
FIGURE 3.

Reduction of the oxidized WT with d-Gal. Stopped-flow traces at 458 nm are shown as dotted lines while at those at 395 nm are in solid lines. A solution of the wild-type P2O (23 μm) was mixed with various concentrations of d-Gal (0.4, 0.8, 1.6, 3.2, 6.4, 12.5, and 50 mm). All concentrations are given as after mixing. Inset a shows a plot of kobs of the first phase (absorbance increase at 395 nm) versus d-Gal concentrations, whereas kobs of the second phase (large decrease in A458 nm) was plotted against d-Gal concentration in inset b. Data of the mutant are in supplemental Fig. S3.
FIGURE 4.

Kinetic mechanisms of WT and Thr169 variants. A, describes kinetic mechanisms of reactions of WT and Thr169 variants with d-Glc. The reverse rate constant (k−3) is only present in the reaction of T169A, explaining a convergent-line pattern found in this mutant. B, d-Gal binds to WT and Thr169 variants with simple one-step binding prior to the flavin reduction step.
Oxidation of the Reduced Thr169 Mutants by Oxygen
The oxidative half-reactions of the Thr169 variants were investigated using stopped-flow experiments. Kinetic traces at 395 and 458 nm for all mutants show one single phase (Fig. 5), which contrasts the reaction kinetics of WT where biphasic kinetics was observed (3, 4). In the reaction of WT, the biphasic kinetics is due to formation and decay of a C4a-hydroperoxyflavin intermediate (3, 4). All mutants show similar kinetics at all λ, indicating that only the oxidized flavin was formed, and no transient C4a-hydroperoxyflavin intermediate was stabilized. The kobs values for flavin oxidation analyzed from the traces at 395 and 458 nm were linearly dependent on [O2] and intercepts of all plots approach zero, suggesting that the reverse rate is negligible. Biomolecular rate constants for the enzyme oxidation are summarized in Table 3.
FIGURE 5.

Oxidation of the reduced T169S at various concentrations of oxygen. A solution of reduced T169S (23 μm) was mixed with solutions containing various concentrations of oxygen: 0.13, 0.31, 0.61, and 0.96 mm. All concentrations were as after mixing, and the reactions were monitored at 395 nm (dotted lines) and 458 nm (solid lines). The trace with empty circles represents the reaction of WT with 0.13 mm oxygen monitored at 395 nm, showing formation and decay of C4a-hydroperoxyflavin (4). The inset shows kobs as a function of [O2]. A bimolecular rate constant without a reverse rate was calculated to be 1.6 ± 0.08 × 104 m−1 s−1.
TABLE 3.
Summary of bimolecular rate constants of the oxidative half-reaction of Thr169 variants at pH 7
| Variants | Bimolecular rate constant |
|---|---|
| m−1s−1 | |
| T169S | 1.6 ± 0.08 × 104 |
| T169N | 0.1 ± 0.01 × 104 |
| T169A | 1.3 ± 0.07 × 104 |
| T169G | 80 ± 4 |
Crystal Structure Analysis
We analyzed two of the four replacements at position 169, Thr → Ser and Thr → Gly, by solving the crystal structures (Fig. 6). Data collection and refinement statistics are given in supplementary Table S2. The overall structures of the P2O monomer and homotetramer are very similar to those reported earlier for wild-type and mutant T. multicolor P2O (13, 14, 23, 24). As expected, based on our previous structures, the only region in P2O that displays significant structural differences besides the point of mutation is the active-site loop (residues 451–461). Hence, only the structure of the active site will be discussed below. The two major conformational states previously observed for P2O, the open (2IGO, Fig. 7A (14)) and the closed state (1TT0, Fig. 7B (13)) serve as reference structures for structural comparisons. These two states have been discussed above.
FIGURE 6.

Active-site structure in T169S and H167A/T169G. Position 169 with Ser in T169S (A); and Gly in H167A/T169G (B).
FIGURE 7.

Comparison of active-site structures. The active site in H167A in complex with 2-deoxy-2-fluoro-d-glucose (A); wild-type with bound acetate (B); superposition of T169S (green) and H167A2FG (yellow) (C); and superposition of H167A/T169G (green) with wild-type P2O (yellow) (D). No water molecules have been drawn. H-bonds from Thr169 in the wild type to the flavin N5/O4 atoms are indicated as dashed lines.
The active-site loop in the T169S structure (Fig. 6A) folds into a slightly different conformation from the “open” state seen in other unliganded structures or the 2-deoxy-2-fluoro-d-glucose complex (14). Because the structure is unliganded, it is uncertain whether this loop conformer is relevant for the sugar-binding state (i.e. during the reductive half-reaction), or the oxidative half-reaction. The conformation is best characterized as “semi-open” (Figs. 6A and 7C), and the Ser Oγ atom assumes a position identical to that of Oγ1 in Thr169 of the sugar-bound state observed for the H167A variant in complex with 2-deoxy-2-fluoro-d-glucose (2IGO (14)) (Fig. 7, A and C).
In the case of the glycine replacement, we crystallized the double mutant, H167A/T169G, where also the flavinylation ligand (His167) had been mutated, as in the previously determined sugar-bound state (2IGO (14)). It has been shown previously that the H167A replacement does not introduce any significant change in the positioning of flavin or protein atoms in the active site beyond the absence of His167 Nϵ2-flavin C8α bond. The active-site loop in H167A/T169G is in a fully closed conformation (Fig. 6B), identical to that observed for P2O with bound acetate (1TT0 (13)) (Fig. 7, B and D), the only notable difference is at the C-terminal end of the active-site loop, in the region 457–460 that is further away from the active site. These differences may rather be due to the binding of a polyethylene glycol molecule in 1TT0 in this region, which is not present in the unliganded H167A/T169G structure shown here. In H167A/T169G, two water molecules are positioned in front of the isoalloxazine ring (Fig. 6B): one ∼3.1 Å below the flavin N5 atom and another water 2.8 Å in front of the flavin O4 oxygen. Neither of these assumes the exact position of the missing Thr side-chain hydroxyl oxygen, but the water interacting with O4 is relatively close to that position. The other water molecule, slightly “below” the flavin N5 in Fig. 6B, would not be present when sugar is bound.
DISCUSSION
Based on our kinetic data presented here and its strategic position in the P2O active site (14, 13), it is clear that Thr169 is an intriguing and important amino acid in catalysis by P2O. Our findings indicate that a side chain at position 169 capable of forming H-bonds near the productive P2O·sugar complex is needed for efficient flavin reduction and identify mutants that can help improving application of P2O in d-tagatose synthesis. Stabilization of C4a-hydroperoxyflavin in P2O also requires the presence of threonine at position 169. Thr169 has been observed to assume two rotamer conformations in P2O: a rotamer with the Oγ pointing away from the flavin and toward amino acids in the dynamic active-site loop, as seen when the loop is open to allow sugar binding (Fig. 7A), and another rotamer where Oγ is pointing toward the flavin and offers H-bond(s) to the N5/O4 locus when the substrate loop is closed (Fig. 7B) (14). Based on available crystal structures, the Oγ atom of Thr169 in P2O assumes a position equivalent to that of Thr266 in electron transfer flavoprotein from Methylophilus methylotrophus (25); Ser254 in electron transfer flavoprotein from the methylotrophic bacterium W3A1 (26); Ser101 in choline oxidase (27); Ser106 in alditol oxidase (28); Asp170 in vanillyl alcohol oxidase (29); the backbone amide group of Gly120 in Streptomyces or Brevibacterium cholesterol oxidase (30). The observed distances between atoms of the proteins and the flavin-N5/O4 locus indicate that they are mostly within H-bonding distance. Hydrogen-bonding interactions in the vicinity of the flavin N5/O4 locus are recurrent features in flavoenzymes (17). Despite the commonality of these interactions, little is known about the precise functional roles and mechanistic implications. In electron transfer flavoprotein from methylotrophic bacterium W3A1, the H-bond to N5 has been suggested to increase Emo′ of the enzyme (26). Clearly, more detailed studies on other flavoproteins are needed to determine whether the mechanistic implications of Thr169 in P2O, especially in flavin reduction, can be generalized to other flavoenzymes.
Our results suggest that a protein side chain capable of forming H-bonds near the productive P2O·sugar complex is required for efficient flavin reduction. Results with d-Glc as a substrate indicate that the replacement of Thr169 with Asn and Ser has a negligible effect on the reductive half-reaction. The Kd values for the binding of d-Glc in WT, T169S, and T169N are almost identical. In addition, T169S and T169N all form an active P2O·Glc complex characterized by an absorbance increase at 395 nm (3). The Emo′ values (Table 1) and the rates of flavin reduction (kred, Table 2) of WT, T169S, and T169N are also in the same range, indicating that the oxidative power of these mutants is preserved. Interestingly, although the Emo′ of T169G was significantly higher than the other mutant enzymes (−1 mV versus −105 mV or lower), the flavin reduction by d-Glc was affected inversely to the thermodynamic changes, i.e. >10-fold decrease in kred (Table 2). It has been shown for the flavoenzyme 2-methyl-3-hydroxypyridine-5-carboxylic monooxygenase that, despite similar Emo′ values, flavin reduction rates can differ up to 1600-fold due to changes in the precise geometry of the reactants (31, 32). This suggests that the binding mode of d-Glc in T169G is different from that in WT, T169S, and T169N, thus resulting in lower efficiency of the hydride transfer. In T169A, the flavin reduction by d-Glc is significantly impaired (Table 2). The small absorbance increase at 395 nm accompanying the binding of d-Glc to T169G and T169A also supports the idea that substrate binding is different than in WT, T169N, and T169S. We may conclude that efficient hydride transfer from the sugar C2 to flavin N5 during the reductive half-reaction requires an enzyme conformational state that is compatible with Thr, Ser, and Asn at the position 169.
Stopped-flow experiments of WT-P2O with d-Gal show that, unlike the binding of d-Glc, P2O binds d-Gal in one step. Replacement of Thr169 by Ser, Asn, and Gly increased flavin reduction rates when compared with that of WT without increasing the binding affinity. This result can be interpreted as d-Gal binds to the mutants with the geometry allowing the reactants to carry out the hydride-transfer reaction more optimal than in the WT. This is likely due to the relief of steric hindrance close to the axial C4 hydroxyl group of d-Gal, as predicted by our previous structural analysis (14). In T169A, flavin reduction by d-Gal is very slow, resulting in an almost inactive enzyme. As for the reaction with d-Glc discussed above, these data suggest that, by substituting Thr169 with Ala, the reductive half-reaction is significantly impaired.
The crystal structures of T169S and T169G mutants also support the idea that the H-bonding ability of a side chain at position 169 is likely to be required for efficient flavin reduction. The structure of T169S shows that the Oγ of Ser assumes the same position as Oγ of Thr169 in the sugar-bound state of H167A P2O where the Thr169 side chain is pointing away from the flavin N5/O4 locus (Figs. 6A and 7C). Thus, by allowing identical positioning of the Thr/Ser Oγ, the reductive half-reaction remains unperturbed. In T169G, which lacks the possibility of forming a H-bond, two water molecules are found near the flavin N5/O4, offering the possibility of H-bonds. In T169G, the rate of flavin reduction by d-Glc decreased, but the rate with d-Gal increased when compared with that of the WT (Table 2). This mutant is thus quite competent in the reductive half-reaction, maybe due to the possibility of solvent-mediated H-bonds to the productive P2O·Gal complex. Although the structure of T169N is not available, the Asn side chain should also be able to H-bond to important groups in the productive ES complex. Hence, by maintaining the H-bonding ability at position 169 (as in T169S, T169N, and T169G), efficient flavin reduction can be maintained.
Thr169 is important for formation of a C4a-hydroperoxy-FAD intermediate. Our previous study with the WT has shown that P2O is thus far unique among flavoprotein oxidases for its ability to stabilize the C4a-hydroperoxy-FAD during the oxidative half-reaction (4). It was suggested that the cavity at the active site of this enzyme can provide the required space for accommodating a peroxide group at the flavin C4a position (4). In other flavoprotein oxidases, the active sites may be too restricted to allow formation of the intermediate (4, 28, 33). We could not detect the C4a-hydroperoxy-FAD intermediate in any of the Thr169 variants. All mutants reacted with oxygen in a second-order fashion without a reverse rate constant, similar to other flavoprotein oxidases (34, 35). The closed loop conformation observed in the wild-type P2O-acetate complex (1TT0 (13)) shows Thr169 Oγ1 interacting with the flavin N5/O4 locus, offering the possibility of H-bonds to N5 and O4 (Fig. 7B). In the structure of T169S, although the position of Ser169 Oγ (Fig. 6) is different from the position of Thr169 Oγ1 in the closed loop structure of WT (1TT0 (13)), a similar H-bond as in WT may be possible if loop movement occurs during the oxidative half-reaction. However, the presence of a group capable of making a H-bond to the flavin N5/O4 is unlikely to be the main factor for stabilizing the intermediate, because in T169S, no intermediate was detected. A possible explanation may be that, in the closed conformational state, the position of Thr169 provides an occluded reaction chamber that helps to control precisely the reactivity of the reduced flavin and oxygen to ensure the formation of the flavin-C4a intermediate prior to the subsequent H2O2 elimination.
The steady-state kinetic data are in agreement with the kinetic model interpreted from stopped-flow data and suggest that T169S and T169N can be considered to be good biocatalysts. With d-Glc as a substrate, kcat of T169S is higher than the kcat of WT and other mutants. In all mutants, the effect on kcat followed the trend seen for kred (T169S > T169N > T169G > T169A). In WT, the overall turnover with d-Glc is governed by two steps: flavin reduction (kred) and C4a-hydroperoxyflavin decay (3). Because in all of the mutants the oxidative half-reaction is second-order without an intermediate (Fig. 5), the reaction is limited only by flavin reduction (kred). Therefore, kcat of T169S with d-Glc is higher than the value of WT (Table 1). These data also imply that, if the reaction is equilibrated with high concentration of oxygen, T169S can be employed as a better catalyst than WT when using d-Glc as a substrate. When using d-Gal as a substrate, kcat of T169S and T169N are 2- and 4-fold higher than the value of WT, due to the increase in kred (rate-limiting step) of the mutants (Fig. 3 and Tables 1 and 2). These two mutants would also be better biocatalysts than the WT in d-Gal oxidation for producing d-tagatose. Effects of the mutations based on steady-state parameters reported in Table 1 are different from kcat and KmGlc,Gal values reported before (15, 21) where the values were from steady-state assay under air saturation. Deviations of the values reported in previous studies (15, 21) from the actual kinetic parameters in Table 1 are due to high values of KmO2, especially for the mutants T169N and T169G; the concentration of oxygen under air-saturation (0.26 mm) is below the KmO2 values of all the mutants (Table 1).
All variants except T169A showed parallel-line patterns when d-Glc or d-Gal were used as substrates. This is similar to the reaction of WT with d-Glc as a substrate (3). The kinetic mechanism of WT at pH 7.0 is Ping-Pong where the 2-keto-d-sugar product is released prior to the oxygen reaction. The parallel-line pattern can also be explained by a model in which the sugar product remains bound during the reaction of molecular oxygen, but the reduction step is essentially irreversible (k3 ≫ k−3) (3). Interestingly, the steady-state kinetics of T169A with d-Glc or d-Gal as substrates show an intersecting-line pattern instead of a parallel-line pattern, suggesting that, for T169A, regardless of the sugar used, the reaction of O2 with the reduced enzyme may occur while the 2-keto-d-sugar remains bound to the enzyme. These data also imply that the reverse rate constant (k−3 in Fig. 4A) of T169A is significant, in agreement with its low Emo′. The ternary-complex pattern indicates that a 2-keto-sugar product resulting from the reductive half-reaction fails to dissociate and remains bound to the active site during the reaction with O2 in the oxidative half-reaction. One possible explanation is that space allocation at the re-side of the isoalloxazine ring is increased in T169A compared with the WT and other mutants investigated. A shift in the reaction mechanism from the typical Ping-Pong type to a ternary-complex mechanism was also observed in the reaction of P2O at pH 8 or above (36).
In conclusion, our results suggest that the residue at position 169 is important for both half-reactions in P2O catalysis. Efficient hydride transfer from the sugar C2 to flavin N5 during the reductive half-reaction requires a residue at this position that can maintain a H-bonding within the ES complex, i.e. Thr (WT), Ser, Asn, and Gly (H-bond possibly maintained by solvent). When compared with the WT, the overall turnover of T169S and T169N with d-Gal was faster due to higher rate constants of the flavin reduction. In addition, the side chain of Thr169 is crucial for stabilizing the C4a-hydroperoxy-FAD during the oxidative half-reaction and cannot be substituted by other residues.
Supplementary Material
Acknowledgments
We thank Janewit Wongratana for constructing plasmids for expression of T169S, T169N, and T169A and the beam-line staff scientists at MAX-lab (Lund, Sweden) for support during data collection. We thank Bruce Palfey for critical reading of the manuscript.
This work was supported in part by the Thailand Research Fund through Grants BRG5180002 (to P. C.), MRG4980117 (to J. S.), and PHD/0151/2547 of the Royal Golden Jubilee Ph.D. program (to M. P.) and by the Faculty of Science, Mahidol University (to P. C.) and from the Faculty of Dentistry Chulalongkorn University (to J. S.).
The atomic coordinates and structure factors (codes 3K4B and 3K4C) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Tables S1 and S2, and Figs. S1–S3.
- P2O
- pyranose 2-oxidase
- ABTS
- 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt
- λ
- wavelength
- WT
- wild type
- Mes
- 4-morpholineethanesulfonic acid.
REFERENCES
- 1.Halada P., Leitner C., Sedmera P., Haltrich D., Volc J. (2003) Anal. Biochem. 314, 235–242 [DOI] [PubMed] [Google Scholar]
- 2.Leitner C., Volc J., Haltrich D. (2001) Appl. Environ. Microbiol. 67, 3636–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prongjit M., Sucharitakul J., Wongnate T., Haltrich D., Chaiyen P. (2009) Biochemistry 48, 4170–4180 [DOI] [PubMed] [Google Scholar]
- 4.Sucharitakul J., Prongjit M., Haltrich D., Chaiyen P. (2008) Biochemistry 47, 8485–8490 [DOI] [PubMed] [Google Scholar]
- 5.Palfey B. A., Massey V. (1998) in Comprehensive Biological Catalysis, Vol. III (Michael S. ed) pp. 83–153, San Diego, Academic Press [Google Scholar]
- 6.Ballou D. P., Entsch B., Cole L. J. (2005) Biochem. Biophys. Res. Commun. 338, 590–598 [DOI] [PubMed] [Google Scholar]
- 7.van Berkel W. J., Kamerbeek N. M., Fraaije M. W. (2006) J. Biotechnol. 124, 670–689 [DOI] [PubMed] [Google Scholar]
- 8.Orville A. M., Lountos G. T., Finnegan S., Gadda G., Prabhakar R. (2009) Biochemistry 48, 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Héroux A., Bozinovski D. M., Valley M. P., Fitzpatrick P. F., Orville A. M. (2009) Biochemistry 48, 3407–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mallett T. C., Claiborne A. (1998) Biochemistry 37, 8790–8802 [DOI] [PubMed] [Google Scholar]
- 11.Ghisla S., Massey V. (1989) Eur. J. Biochem. 181, 1–17 [DOI] [PubMed] [Google Scholar]
- 12.Albrecht M., Lengauer T. (2003) Bioinformatics 19, 1216–1220 [DOI] [PubMed] [Google Scholar]
- 13.Hallberg B. M., Leitner C., Haltrich D., Divne C. (2004) J. Mol. Biol. 341, 781–796 [DOI] [PubMed] [Google Scholar]
- 14.Kujawa M., Ebner H., Leitner C., Hallberg B. M., Prongjit M., Sucharitakul J., Ludwig R., Rudsander U., Peterbauer C., Chaiyen P., Haltrich D., Divne C. (2006) J. Biol. Chem. 281, 35104–35115 [DOI] [PubMed] [Google Scholar]
- 15.Salaheddin C., Spadiut O., Ludwig R., Tan T. C., Divne C., Haltrich D., Peterbauer C. (2009) Biotechnol. J. 4, 535–543 [DOI] [PubMed] [Google Scholar]
- 16.Bannwarth M., Heckmann-Pohl D., Bastian S., Giffhorn F., Schulz G. E. (2006) Biochemistry 45, 6587–6595 [DOI] [PubMed] [Google Scholar]
- 17.Fraaije M. W., Mattevi A. (2000) Trends Biochem. Sci. 25, 126–132 [DOI] [PubMed] [Google Scholar]
- 18.Haltrich D., Leitner C., Neuhauser W., Nidetzky B., Kulbe K. D., Volc J. (1998) Ann. N.Y. Acad. Sci. 864, 295–299 [DOI] [PubMed] [Google Scholar]
- 19.Massey V. (1991) in: Flavins and Flavoproteins (Curti B., Ronchi S., Zanetti G. eds) pp. 59–66, Walter de Gruyter, Berlin [Google Scholar]
- 20.Dalziel K. (1957) Acta Chem. Scand. 11, 1706–1723 [Google Scholar]
- 21.Spadiut O., Leitner C., Tan T. C., Ludwig R., Divne C., Haltrich D. (2008) Biocatal. Biotran. 26, 120–127 [Google Scholar]
- 22.Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 23.Spadiut O., Leitner C., Salaheddin C., Varga B., Vertessy B. G., Tan T. C., Divne C., Haltrich D. (2009) FEBS J. 276, 776–792 [DOI] [PubMed] [Google Scholar]
- 24.Spadiut O., Radakovits K., Pisanelli I., Salaheddin C., Yamabhai M., Tan T. C., Divne C., Haltrich D. (2009) Biotechnol. J. 4, 525–534 [DOI] [PubMed] [Google Scholar]
- 25.Roberts D. L., Frerman F. E., Kim J. J. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 14355–14360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang K. Y., Swenson R. P. (2007) Biochemistry 46, 2289–2297 [DOI] [PubMed] [Google Scholar]
- 27.Quaye O., Lountos G. T., Fan F., Orville A. M., Gadda G. (2008) Biochemistry 47, 243–256 [DOI] [PubMed] [Google Scholar]
- 28.Forneris F., Heuts D. P., Delvecchio M., Rovida S., Fraaije M. W., Mattevi A. (2008) Biochemistry 47, 978–985 [DOI] [PubMed] [Google Scholar]
- 29.van den Heuvel R. H., Fraaije M. W., Mattevi A., van Berkel W. J. (2000) J. Biol. Chem. 275, 14799–14808 [DOI] [PubMed] [Google Scholar]
- 30.Yue Q. K., Kass I. J., Sampson N. S., Vrielink A. (1999) Biochemistry 38, 4277–4286 [DOI] [PubMed] [Google Scholar]
- 31.Chaiyen P., Brissette P., Ballou D. P., Massey V. (1997) Biochemistry 36, 2612–2621 [DOI] [PubMed] [Google Scholar]
- 32.Chaiyen P. (2010) Arch. Biochem. Biophys. 493, 62–70 [DOI] [PubMed] [Google Scholar]
- 33.Alfieri A., Fersini F., Ruangchan N., Prongjit M., Chaiyen P., Mattevi A. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 1177–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattevi A. (2006) Trends Biochem. Sci. 31, 276–283 [DOI] [PubMed] [Google Scholar]
- 35.Gibson Q. H., Swoboda B. E., Massey V. (1964) J. Biol. Chem. 239, 3927–3934 [PubMed] [Google Scholar]
- 36.Rungsrisuriyachai K., Gadda G. (2009) Arch. Biochem. Biophys. 483, 10–15 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.