

Abstract
Phototransduction in Limulus photoreceptors involves a G protein-mediated activation of phospholipase C (PLC) and subsequent steps involving InsP3-mediated release of intracellular Ca2+. While exploring the role of calmodulin in this cascade, we found that intracellular injection of Ca2+/calmodulin-binding peptides (CCBPs) strongly inhibited the light response. By chemically exciting the cascade at various stages, we found the primary target of this effect was not in late stages of the cascade but rather at the level of G protein and PLC. That PLCδ1 contains a calmodulin-like structure raised the possibility that PLC might be directly affected by CCBPs. To test this possibility, in vitro experiments were conducted on purified PLC. The activity of this enzyme was strongly inhibited by CCBPs and also inhibited by calmodulin itself. Our results suggest that the calmodulin-like region of PLC has an important role in regulating this enzyme.
Phototransduction in Limulus photoreceptors is a complex excitation cascade that has sufficient amplification to produce large electrical events to single photons (1). The initial stage of this cascade resembles the rhodopsin/G protein interaction found in vertebrate photoreceptors, but the enzyme activated by G protein is phosphoinositide-specific phospholipase C (PLC) rather than cGMP-phosphodiesterase. PLC, in turn, generates InsP3 (2), and the resulting InsP3-mediated Ca2+ release (3–5) leads to activation of nonspecific cation channels (2, 6), perhaps through an intermediate step involving cGMP (7, 8).
Invertebrate photoreceptors contain a high concentration of calmodulin (9). In the region of the photoreceptor specialized for phototransduction, the microvillar region, the concentration may be as high as 0.5 mM (10). Because the light-induced elevation of Ca2+ plays an obligatory role in the excitation process in Limulus (11), we suspected that a calmodulin-dependent process might play an important role in a late stage of the excitation process, perhaps coupling the InsP3-mediated elevation of Ca2+ to the opening of ion channels. If this were the case, calmodulin peptide antagonists should reduce the response to light. The experiments reported here show Ca2+/calmodulin peptide antagonists do indeed have this effect. However our results show that the primary site of this effect is not at a late stage of transduction but rather an early stage involving PLC.
PLC isoforms are found in all eukaryotic cells and are involved in signal transduction, including sensory, learning-related synaptic plasticity, and oncogenesis (for reviews see refs. 12 and 13). Recently, the crystal structure of PLCδ1 has been obtained (14). This structure revealed EF-hand domains that resemble the structure of calmodulin with Ca2+ bound (the “calmodulin-like” domain). Sequence alignment and α-helix prediction suggest the existence of similar structures in all PLC isozymes. The regulatory role of this region is unclear, but it seemed possible from our physiological results that it might be the target of calmodulin antagonists. In the second part of this study we show that purified PLC is directly inhibited by Ca2+/calmodulin-binding peptides (CCBPs).
MATERIALS AND METHODS
Electrophysiology.
Preparation of the ventral nerve and perfusion with artificial sea water were carried out as reported previously (15). Cells were impaled with an electrode containing Ca2+/calmodulin-binding or control peptide. Peptides were introduced into cells by 60–120 small pressure pulses, and the injections were monitored by an infrared video camera. Total injected volumes were estimated (16) as no more than 5% of total cell volume and usually less, i.e., the maximal intracellular peptide concentration was less than 300 μM. A second intracellular electrode was used for pressure injection of other test compounds and voltage clamp. Beam intensity was approximately 1.0 mW/cm2 and reduced by using neutral density filters.
Electrode Solutions.
The first electrode contained 6 mM Ca2+/calmodulin-binding or control peptide, 1 mM EDTA, 100 mM KCl, neutralized with Mops or Hepes, pH 7–7.5. Control peptides (LADVAEQRHLAKK and KKALHRQEAVDAL) were a gift from Leslie Griffith. Ca2+/calmodulin-dependent protein kinase II peptide 290–309 (CMKII 290–309) and vasoactive intestinal peptide (VIP) were obtained from commercial sources. Peptides were dissolved in distilled water and lyophilized to reduce organic acid content before use. For experiments using voltage clamp, the second electrode contained 3 M KCl. For excitation by Ca2+, the second electrode contained a solution of 2 mM N-(2-hydroxyethyl)ethylenedinitrilo-N,N′,N′-triacetic acid (HEDTA), 1.8 mM CaCl2, and 170 mM KCl, pH 7.6. For excitation by InsP3, the second electrode contained a solution of 100 μM InsP3, 170 KCl, and 10 mM Hepes, pH 7.2 (4).
Activation of G Proteins.
A solution of 25 μM GTP[γS], 170 KCl, and 10 mM Hepes, pH 7.2 was introduced intracellularly by using multiple small injections of GTP[γS]. Nucleotide exchange was activated by light exposure, and then the cell was dark-adapted (17).
Biochemistry.
Phospholipase C was purified (18) and assays were performed with 4 μM free Ca2+ (19) as described previously, except that calmodulin, Ca2+/calmodulin-binding peptides, and control peptide were preincubated with phospholipase C for 30 min before starting the reaction.
RESULTS
CCBPs Block Phototransduction.
We investigated the role of calmodulin by injecting the Ca2+/calmodulin-binding peptide derived from the pseudosubstrate region of calmodulin-dependent kinase II (20, 21). The peptide was injected into the cytoplasm of the photoreceptor through an intracellular microelectrode. The injection was done under visual control; the final cytoplasmic concentration of peptide was estimated to be no more than several hundred micromolar. Fig. 1A shows that the peptide rapidly reduced the photoresponse and was capable of decreasing the response to a flash by more than two orders of magnitude (Figs. 2 and 3). This inhibition slowly reversed, presumably as a result of degradation of the peptide by endogenous proteases (22). Injection of comparable and greater amounts of control peptide had no effect (Fig. 1B). To see whether the inhibition of the light response was a general feature of CCBPs we tested the peptide hormone VIP, which also binds to Ca2+/calmodulin with high affinity (23). Injection of VIP desensitized cells in a similar manner to CMKII 290–309 (Fig. 1C). We conclude that one or more steps in the transduction process are strongly inhibited by injection of CCBPs.
Figure 1.
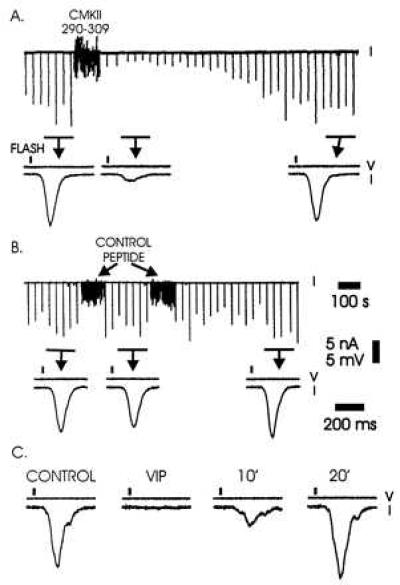
Effect of injection of Ca2+/calmodulin-binding peptides CMKII 290–309 and VIP into Limulus ventral photoreceptors. (A) Effects of intracellular pressure injection of CMKII 290–309 on the light-induced current. At the top is a continuous current trace (100-s scale bar). A dim light flash every 30 s induced inward current (downward deflection). During the period marked “CMKII 290–309,” the peptide was injected with multiple, small pressure pulses. This produced a large, but reversible decrease in the light response. Directly underneath are response averages (n = 5, 200-ms scale bar) during periods indicated by the bars and arrows. Solid rectangles represent the flash. Voltage (V) and current traces (I) are as indicated. (B) Effect of control peptide on the light-induced current. Approximately twice the number of injections of similar volume were made than in A. (C) Effect of VIP injection on the light-induced current. Single responses are shown before injection, immediately following, 10 s later, and 20 s later.
Figure 2.
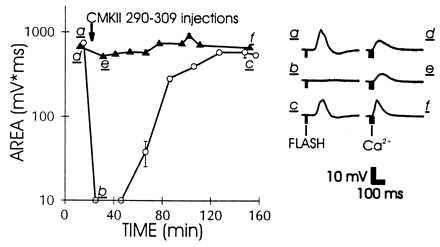
Comparison of the inhibitory effects of CMKII 290–309 on the excitation (depolarization) produced by light (open circles) or Ca2+ injection (solid triangles). a–f match averaged (n = 4–5) areas (Left) to time course (Right) before a d, after b e, and following recovery from c f peptide injection. The two light response data points immediately after peptide injection represent upper limits on the area.
Figure 3.
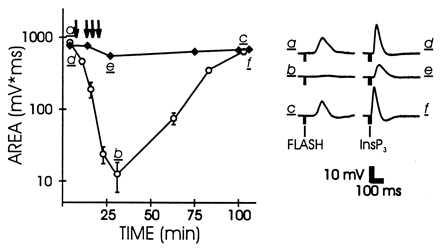
Comparison of the effects of CMKII 290–309 on the excitation produced by light (open circles) and InsP3 (solid diamonds). For details see Fig. 2.
CCBPs Block Either G Protein or PLC.
We initially suspected that CCBPs might act at a late stage of the cascade and involve the processes by which InsP3-mediated Ca2+ release affects the opening of ion channels. If this was the case, the peptide should strongly reduce the excitation caused by intracellular Ca2+ injection, just as peptide reduces the response to light. Previous work showed that brief injections of Ca2+ into the transducing lobe of the photoreceptor excite this photoreceptor. The conductance opened in this way appears to be the same as that opened by light (11, 24).
Ca2+ injection from electrodes containing high concentrations of Ca2+ are notoriously difficult because the electrodes usually become blocked within minutes, presumably as a result of Ca2+ precipitates forming in the electrode tip. We developed a new method for injecting Ca2+ that involves the Ca2+ buffer HEDTA (25). Injection of a solution containing 2 mM HEDTA (in the microelectrode) with no added Ca2+ failed to excite cells (Fig. 4A). However, if 1.8 mM Ca2+ was added to 2 mM HEDTA, pH 7.6, the free Ca2+ in the solution would be ≈8.5 μM. Injection of this solution gave highly reproducible excitation to consecutive injections (Fig. 4A). An initial “use effect” often occurred with responses increasing to a stable level during the first set of injections (Fig. 4B). The response amplitude and duration depended on the duration of the pressure pulse (Fig. 4C). As duration increased, the response amplitude saturated, but the response area continued to increase. An important rationale for use of HEDTA is that it does not buffer intracellular Ca2+ effectively. Ca2+ buffers have been shown to greatly reduce the response to light (11, 26, 27). HEDTA fails as an intracellular Ca2+ buffer because of poor selectivity for Ca2+ relative to Mg2+ (only about 16-fold) (25). Once injected into cells HEDTA should equilibrate as a Mg2+ salt (estimated at 97%), because the intracellular Mg2+ concentration (mM) is orders of magnitude higher than resting Ca2+ concentration (sub-μM) (28, 29). HEDTA fails to deplete cell Mg2+ levels as each injection is rapidly diluted >100-fold into the total cell volume. Intracellular Mg2+ levels presumably are replenished from the bath solution (48 mM Mg2+). Indeed, normal responses to light (Fig. 4D), including unchanged responses to single photons (Fig. 4E), were observed after >100 injections of either 2 mM HEDTA, as in Fig. 4, or the Ca2+/HEDTA.
Figure 4.
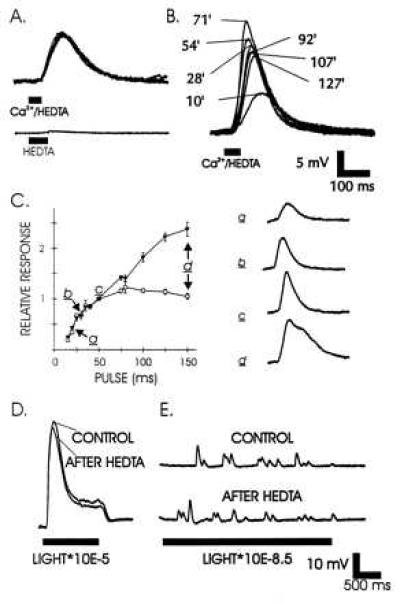
Characterization of the excitation produced by intracellular pressure injection of 1.8 mM Ca2+/2 mM HEDTA, pH 7.6 solution. (A) Superposed depolarizations produced by five consecutive injections of Ca2+/HEDTA solution (40-ms pressure pulses spaced 1 min apart). Below is the effect of 50 ms pressure pulse injection of 2 mM HEDTA only in a control experiment. (B) Superposed responses to Ca2+/HEDTA injections by 50-ms pressure pulses at times indicated over more than 2 h. (C) Relationship between pressure pulse length and response peak amplitude (open circles) and areas (solid triangles). a–d match amplitude and pulse duration (Left) to voltage trace (Right). Bars represent the standard error of 3–10 measurements over the 2-h experiment. Responses were normalized by dividing response amplitude and area by those of concurrent 50-ms pressure pulse responses. (D) Lack of effect of HEDTA buffer on the light response after >100 injections in the same control experiment. (E) Lack of effect of HEDTA buffer on the response to single photons after >100 injections in the same control experiment. The stimulating light beams were attenuated as indicated.
By using the reproducible responses to Ca2+ injection, we tested whether these responses were reduced by CCBPs. Fig. 2 shows that the response to Ca2+ injection was not strongly inhibited by CMKII 290–309, whereas the excitation by light in the same cell was strongly inhibited. To exclude the possibility that the lack of effect was because of saturation of the response to Ca2+ at the site of injection, we used a pressure pulse producing only 50–70% of maximal response amplitude (as in Fig. 4C). The minor effects of CCBP on the response to Ca2+ injection produced under these conditions indicate that the primary effect of the Ca2+/calmodulin-binding peptide on excitation is upstream from the excitatory role of Ca2+ in the cascade.
If CCBPs affected the InsP3 receptor, thereby blocking InsP3-mediated Ca2+ release, the peptide should have strong effects on the excitation produced by InsP3 injection. This form of excitation also was relatively insensitive to CMKII 290–309 (Fig. 3). This indicates that the primary action of Ca2+/calmodulin-binding peptide is upstream from the production of InsP3 in the cascade.
We next examined the excitation produced at an early step of the transduction by using the G protein activator GTP[γS] (30). After injection of this activator into the cell and stimulation of guanine nucleotide exchange by light there was a sustained increase in the frequency of discrete events in the dark, as previously reported for this and other G protein activators (17, 31, 32). Fig. 5 shows that the frequency of these events was greatly reduced by CCBP, in parallel with the reduction in the response to light. The reduction in event rate because of CMKII 290–309 was observed in three additional experiments. We lack sufficient data to characterize the rare GTP[γS]-induced events observed during maximal desensitization to light after peptide injection, but they appeared normal in shape and normal, or slightly reduced, in size. We interpret the pattern of change as follows: GTP[γS] directly activates G proteins, bypassing the need for activation by rhodopsin. Active G protein subunits bind and activate PLCβ (33–35). Although the stoichiometry has not been measured, the discrete events likely reflect the activation of individual PLCβ molecules. If Ca2+/calmodulin-binding peptide eliminated the activity of some fraction of G protein or PLCβ, the result would be a reduction in the frequency of discrete events, as observed. However, those activated PLCβ molecules that are not inhibited by CCBP would generate normal-size discrete waves, as observed. We conclude that the CCBP primarily acts downstream from rhodopsin and upstream from InsP3; the results can be explained by an effect of peptide on either G protein or PLCβ.
Figure 5.
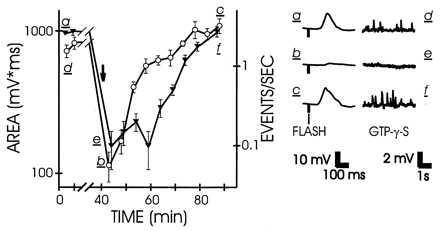
Comparison of the effects of CMKII 290–309 on the excitation produced by light (open circles, left axis) and by GTP[γS] (solid inverted triangles, right axis). The GTP[γS]-induced activity is quantified by the rate of discrete events. For details see Fig. 2.
CCBPs Inhibit PLC in Vitro.
The occurrence of a calmodulin-like domain in PLCδ1 (14) and our results (Figs. 1, 2, 3 and 5) suggested the possibility that CCBPs might interact directly with PLC. Therefore, we tested whether the activity of pure PLC isoforms are affected by the CCBPs. Both PLCδ and γ isozymes were strongly inhibited by the CCBPs, VIP and CMKII 290–309 (Fig. 6) that inhibited the light response in our physiological experiments (Figs. 1, 2, 3 and 5). The inhibition occurred in the range of 1–100 μM peptide. Control peptide (100 μM) had no effect on PLC activities. PLCβ isozyme, including the norpA PLC involved in invertebrate phototransduction (36, 37), was not available in pure form.
Figure 6.
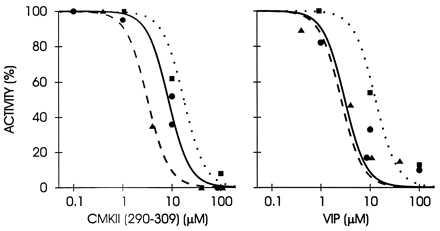
Inhibitory effects of Ca2+/calmodulin-binding peptides CMKII 290–309 and VIP on phospholipases C δ1 (circles), δ3 (triangles), and γ1 (squares). Data points represent the mean of duplicate assays normalized to controls lacking peptide.
Essen et al. (14) noted that sequence similarity occurs between representatives of known PLC isozymes in the calmodulin-like domain and showed that the program PHD predicts α-helical structure consistent with EF-hands in β and γ isoforms. This suggests that the calmodulin-like structure has been conserved in all PLC isoforms.
One interesting possibility is that the calmodulin-like structure in PLC binds to some other region of the enzyme that resembles a calmodulin-binding peptide. If this linkage exists, it may be necessary for enzyme activity, because deletion of the calmodulin-like domain abolishes activity (38–40). This type of an interaction might be broken by calmodulin as well as by Ca2+/calmodulin-binding peptides. Consistent with this prediction, addition of 10 μM calmodulin to in vitro assays decreased PLCδ1, δ3, and γ1 isoform activities greater than 30%.
DISCUSSION
Our central finding is that the light response of Limulus ventral photoreceptors can be desensitized more than two orders of magnitude by CCBPs. At the low light intensities used in this study, there was no evidence of a component of the response that was insensitive to this inhibition. We used two different Ca2+/calmodulin-binding peptides, and both were potent inhibitors of the light response. A third CCBP, M5 (41), has been reported to decrease the light response in Drosophila (42, 43).
Our physiological results indicate that the primary site of inhibition by CCBPs is at the G protein/PLC stage of the transduction cascade. Our in vitro results by using three pure isoforms of PLC show that CCBPs can directly inhibit PLC activity. Therefore, the in vivo effects of CCBPs as calmodulin antagonists appear less specific than previously thought. The use of CCBPs in living cells as probes for calmodulin-dependent processes must be carefully examined.
Given the abundant calmodulin found in invertebrate photoreceptors (9, 10), it is somewhat surprising that CCBPs are effective at inhibiting the light response through their interaction with PLC. However, in the cases of Drosophila photoreceptors, brush border epithelia cells, and the brain, the bulk of calmodulin appears to be bound to Ca2+-independent high affinity sites (10, 44, 45). In addition, CCBPs have much lower affinity for calmodulin (μM) compared with Ca2+/calmodulin (nM) (21, 46). Thus, at low levels of Ca2+ in dark-adapted cells, the free concentration of any form of calmodulin may be too low to prevent inhibition of PLC by CCBPs. Consistent with this interpretation are the findings by Arnon et al. (42, 43) that the CCBP M5 did not inhibit the light response in wild-type Drosophila photoreceptors exposed to bright lights in the presence of bath Ca2+ (when Ca2+ would be high). However, when bath Ca2+ or calmodulin was reduced (presumably lowering Ca2+/calmodulin), an inhibition of the light response by M5 was observed.
Although the primary site of action of CCBPs appears to be at PLC, our results suggest that there may be a second site of action late in the transduction cascade. The evidence for a second site is the observation that CCBP produced a small (2-fold) but consistent reduction in the response to InsP3 or Ca2+ injection. This result may reflect the depression of free calmodulin levels by CCBPs, consistent with a role for calmodulin in the late stages of transduction. Further work will be needed to understand the mechanism of this effect.
Because the amino-terminal domain of PLC contains a region that strongly resembles the three-dimensional structure of Ca2+/calmodulin, it seems likely that CCBPs are binding to this domain and that this domain has a role in regulating enzyme activity. We have also found that pure PLC is inhibited by calmodulin, indicating that there is a region of the enzyme with calmodulin-binding properties. Together these results suggest a possible model of PLC function in which the endogenous calmodulin-like domain binds to the endogenous calmodulin-binding domain; disruption of this linkage by either calmodulin or CCBPs leads to inhibition of enzyme activity.
Our finding that PLC activity can be inhibited by Ca2+/calmodulin suggests a new site for the control of PLC transduction cascades. Negative feedback by Ca2+ is important for reducing transduction gain when photoreceptors become light-adapted (15, 26, 46). Roles for calmodulin in this process have been inferred from the effects on the Limulus photoresponse of inhibiting Ca2+/calmodulin-dependent protein kinase II and calcineurin (47) and electrophysiology of Drosophila ninaC mutants (48, 49). Work on head membrane extracts from Drosophila has found that high Ca2+ exerts an inhibitory effect on PLC activity (50, 51). Our results suggest that this may be a direct effect of Ca2+/calmodulin on PLC. Because PLC activity can lead to rapid Ca2+ release in photoreceptors and other cell types (for reviews see refs. 12 and 13), the inhibition of PLC by Ca2+/calmodulin could provide negative feedback needed to limit further Ca2+ release.
Acknowledgments
Drs. Carolyn Cohen, Leslie Griffith, and Anne Houdousse participated in helpful discussions during the course of this work. Support was provided by the W. M. Keck Foundation and grants from the National Institutes of Health (EY01496).
ABBREVIATIONS
- CCBP
Ca2+/calmodulin-binding peptide
- CMKII 290–309
Ca2+/calmodulin-dependent protein kinase II peptides 290–309
- HEDTA
N-(2-hydroxyethyl)ethylenedinitrilo-N,N′,N′-triacetic acid
- InsP3
inositol 1,4,-trisphosphate
- PLC
phospholipase C
- VIP
vasoactive intestinal peptide
References
- 1.Borsellino A, Fuortes M G F. J Physiol. 1968;196:507–539. doi: 10.1113/jphysiol.1968.sp008521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown J E, Rubin L J, Ghalayini A J, Tarver A P, Irvine R F, Berridge M J, Anderson R E. Nature (London) 1984;311:160–163. doi: 10.1038/311160a0. [DOI] [PubMed] [Google Scholar]
- 3.Brown J E, Rubin L J. Biochem Biophys Res Commun. 1984;125:1137–1142. doi: 10.1016/0006-291x(84)91402-5. [DOI] [PubMed] [Google Scholar]
- 4.Payne R, Corson D W, Fein A, Berridge M J. J Gen Physiol. 1986;88:127–142. doi: 10.1085/jgp.88.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Payne R, Fein A. J Cell Biol. 1987;104:933–937. doi: 10.1083/jcb.104.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fein A, Payne R, Corson D W, Berridge M J, Irvine R F. Nature (London) 1984;311:157–160. doi: 10.1038/311157a0. [DOI] [PubMed] [Google Scholar]
- 7.Bacigalupo J, Johnson E C, Vergara C, Lisman J E. Proc Natl Acad Sci USA. 1991;88:7938–7942. doi: 10.1073/pnas.88.18.7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson E C, Robinson P R, Lisman J E. Nature (London) 1986;324:468–470. doi: 10.1038/324468a0. [DOI] [PubMed] [Google Scholar]
- 9.de Couet H G, Jablonski P P, Perkin J L. Cell Tissue Res. 1989;244:315–319. [Google Scholar]
- 10.Porter J A, Yu M, Doberstein S K, Pollard T D, Montell C. Science. 1993;262:1038–1042. doi: 10.1126/science.8235618. [DOI] [PubMed] [Google Scholar]
- 11.Shin J, Richard E A, Lisman J E. Neuron. 1993;11:845–855. doi: 10.1016/0896-6273(93)90114-7. [DOI] [PubMed] [Google Scholar]
- 12.Minke B, Selinger Z. In: Sensory Transduction. Corey D P, Roper SD, editors. New York: Rockefeller University Press; 1992. pp. 201–217. [Google Scholar]
- 13.Noh D Y, Shin S H, Rhee S G. Biochim Biophys Acta. 1995;1242:99–113. doi: 10.1016/0304-419x(95)00006-0. [DOI] [PubMed] [Google Scholar]
- 14.Essen L O, Perisic O, Cheung R, Katan M, Williams R L. Nature (London) 1996;380:595–602. doi: 10.1038/380595a0. [DOI] [PubMed] [Google Scholar]
- 15.Lisman J E, Brown J E. J Gen Physiol. 1972;59:701–719. doi: 10.1085/jgp.59.6.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corson D W, Fein A. Biophys J. 1983;44:299–309. doi: 10.1016/S0006-3495(83)84303-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolsover S R, Brown J E. J Physiol. 1982;332:325–342. doi: 10.1113/jphysiol.1982.sp014416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh S, Lowenstein J M. Gene. 1996;176:249–255. doi: 10.1016/0378-1119(96)00260-0. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh S, Pawelczyk T, Lowenstein J M. Protein Expression Purif. 1997;9:262–278. doi: 10.1006/prep.1996.0682. [DOI] [PubMed] [Google Scholar]
- 20.Payne M E, Fong Y L, Ono T, Colbran R J, Kemp B E, Soderling T R, Means A R. J Biol Chem. 1988;263:7190–7195. [PubMed] [Google Scholar]
- 21.Hanley R M, Means A R, Ono T, Kemp B E, Burgin K E, Waxham N, Kelly P T. Science. 1987;237:293–297. doi: 10.1126/science.3037704. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez A, Mery J, Vandromme M, Basset M, Cavadore J C, Lamb N J. Exp Cell Res. 1991;195:468–477. doi: 10.1016/0014-4827(91)90398-e. [DOI] [PubMed] [Google Scholar]
- 23.Stallwood D, Brugger C H, Baggenstoss B A, Stemmer P M, Shiraga H, Landers D F, Paul S. J Biol Chem. 1992;267:19617–19621. [PubMed] [Google Scholar]
- 24.Payne R, Corson D W, Fein A. J Gen Physiol. 1986;88:107–126. doi: 10.1085/jgp.88.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martell A E, Smith R M. Critical Stability Constants: Amino Acids. Vol. 1. New York: Plenum; 1974. [Google Scholar]
- 26.Lisman J E, Brown J E. J Gen Physiol. 1975;66:489–506. doi: 10.1085/jgp.66.4.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank T M, Fein A. J Gen Physiol. 1991;97:697–723. doi: 10.1085/jgp.97.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ukhanov K Y, Flores T M, Hsiao H S, Mohapatra P, Pitts C H, Payne R. J Gen Physiol. 1995;105:95–116. doi: 10.1085/jgp.105.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Day P M, Gray-Keller M P. J Gen Physiol. 1989;93:473–494. doi: 10.1085/jgp.93.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeuffer T, Helmreich E J M. J Biol Chem. 1975;250:867–876. [PubMed] [Google Scholar]
- 31.Fein A, Corson D W. Science. 1981;212:555–557. doi: 10.1126/science.6782676. [DOI] [PubMed] [Google Scholar]
- 32.Erickson M A, Robinson P, Lisman J. Science. 1992;257:1255–1258. doi: 10.1126/science.1519062. [DOI] [PubMed] [Google Scholar]
- 33.Devary O, Heichal O, Blumenfeld A, Cassel D, Suss E, Barash S, Rubinstein C T, Minke B, Selinger Z. Proc Natl Acad Sci USA. 1987;84:6939–6943. doi: 10.1073/pnas.84.19.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hepler J R, Kozasa T, Smrcka A V, Simon M I, Rhee S G, Sternweis P C, Gilman A G. J Biol Chem. 1993;268:14367–14375. [PubMed] [Google Scholar]
- 35.Smrcka A V, Sternweis P C. J Biol Chem. 1993;268:9667–9674. [PubMed] [Google Scholar]
- 36.Bloomquist B T, Shortridge R D, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak W L. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- 37.Pearn M T, Randall L L, Shortridge R D, Burg M G, Pak W L. J Biol Chem. 1996;271:4937–4945. doi: 10.1074/jbc.271.9.4937. [DOI] [PubMed] [Google Scholar]
- 38.Bristol A, Hall S M, Kriz R W, Stahl M L, Fan Y S, Byers M G, Eddy R L, Shows T B, Knopf J L. Cold Spring Harbor Symp Quant Biol. 1988;53:915–920. doi: 10.1101/sqb.1988.053.01.105. [DOI] [PubMed] [Google Scholar]
- 39.Ellis M V, Carne A, Katan M. Eur J Biochem. 1993;213:339–347. doi: 10.1111/j.1432-1033.1993.tb17767.x. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh S. Ph.D. thesis. Waltham, MA: Brandeis University; 1996. [Google Scholar]
- 41.Kennelly P J, Edelman A M, Blumenthal D K, Krebs E G. J Biol Chem. 1987;262:11958–11963. [PubMed] [Google Scholar]
- 42.Arnon A, Cook B, Montell C, Selinger Z, Minke B. Science. 1997;275:1119–1121. doi: 10.1126/science.275.5303.1119. [DOI] [PubMed] [Google Scholar]
- 43.Arnon A, Cook B, Gillo B, Montell C, Selinger Z, Minke B. Proc Natl Acad Sci USA. 1997;94:5894–5899. doi: 10.1073/pnas.94.11.5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alexander K A, Cimler B M, Meier K E, Storm D R. J Biol Chem. 1987;262:6108–6113. [PubMed] [Google Scholar]
- 45.Glenney J R J, Bretscher A, Weber K. Proc Natl Acad Sci USA. 1980;77:6458–6462. doi: 10.1073/pnas.77.11.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payne R, Walz B, Levy S, Fein A. Philos Trans R Soc London B. 1988;320:359–379. doi: 10.1098/rstb.1988.0082. [DOI] [PubMed] [Google Scholar]
- 47.Kass L, Bray W O. J Photochem Photobiol B. 1996;35:105–113. doi: 10.1016/1011-1344(96)07301-0. [DOI] [PubMed] [Google Scholar]
- 48.Hofstee C A, Henderson S, Hardie R C, Stavenga D G. Visual Neurosci. 1996;13:897–906. doi: 10.1017/s0952523800009147. [DOI] [PubMed] [Google Scholar]
- 49.Porter J A, Minke B, Montell C. EMBO J. 1995;14:4450–4459. doi: 10.1002/j.1460-2075.1995.tb00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inoue H, Yoshioka T, Hotta Y. J Biochem (Tokyo) 1988;103:91–94. doi: 10.1093/oxfordjournals.jbchem.a122246. [DOI] [PubMed] [Google Scholar]
- 51.Running Deer J L, Hurley J B, Yarfitz S L. J Biol Chem. 1995;270:12623–12628. doi: 10.1074/jbc.270.21.12623. [DOI] [PubMed] [Google Scholar]