

Abstract
Receptors activate adenylyl cyclases through the Gαs subunit. Previous studies from our laboratory have shown in certain cell types that express adenylyl cyclase 6 (AC6), heterologous desensitization included reduction of the capability of adenylyl cyclases to be stimulated by Gαs. Here we further analyze protein kinase A (PKA) effects on adenylyl cyclases. PKA treatment of recombinant AC6 in insect cell membranes results in a selective loss of stimulation by high (>10 nM) concentrations of Gαs. Similar treatment of AC1 or AC2 did not affect Gαs stimulation. Conversion of Ser-674 in AC6 to an Ala blocks PKA phosphorylation and PKA-mediated loss of Gαs stimulation. A peptide encoding the region 660–682 of AC6 blocks stimulation of AC6 and AC2 by high concentrations of Gαs. Substitution of Ser-674 to Asp in the peptide renders the peptide ineffective, indicating that the region 660–682 of AC6 is involved in regulation of signal transfer from Gαs. This region contains a conserved motif present in most adenylyl cyclases; however, the PKA phosphorylation site is unique to members of the AC6 family. These observations suggest a mechanism of how isoform selective regulatory diversity can be obtained within conserved regions involved in signal communication.
Transmembrane signaling through the receptor–Gs–adenylyl cyclase complex has long been studied as a model for signal transduction through heterotrimeric G proteins. Receptor activation of Gs results in the dissociation of Gαs from Gβγ, and the activated Gαs stimulates adenylyl cyclase (1). Nine Gαs-regulated adenylyl cyclases have currently been cloned, and the different isoforms of adenylyl cyclases are differentially regulated (2–4). One aspect of this differential regulation is their susceptibility to participate in heterologous desensitization. Previous studies from our laboratory have shown that in addition to receptor desensitization (5), the glucagon-sensitive adenylyl cyclase can undergo desensitization at the level of Gs (6) and adenylyl cyclase (7). Desensitization at the level of adenylyl cyclase is a cAMP-dependent process and was observed in hepatocytes and S49 lymphoma cells where a decrease in Gαs-mediated adenylyl cyclase activities was seen. Cloning of adenylyl cyclase cDNAs from hepatocytes and S49 lymphoma cells indicated that adenylyl cyclase 6 (AC6) was present in both cells (7). We reasoned that the common sensitivity in hepatocytes and S49 cells of adenylyl cyclase activity to protein kinase A (PKA) could be caused by AC6. Hence we studied the effects of PKA on Gαs regulation of AC6.
MATERIALS AND METHODS
Materials.
[α-32P]ATP was from New England Nuclear. Purified catalytic subunit of PKA was purchased from Promega. Tissue culture reagents and fetal calf serum were from GIBCO. WIPTIDE and reagents for peptide synthesis were from Bachem. Anti-FLAG M2 column was from Kodak. All other reagents used were the highest grade available.
Expression of Adenylyl Cyclases.
AC1, AC2, and AC6 were tagged with the FLAG epitope at the N terminus. The FLAG tagged AC1 (8) and AC6 were constructed by using a strategy similar to that used for AC2 (9). The epitope-tagged adenylyl cyclases were expressed in Hi-5 cells by infection with recombinant baculovirus containing the required adenylyl cyclase insert. Hi-5 cells were infected with a multiplicity of 5–10, and membranes were prepared from infected cells 48 hrs after infection as described (9).
PKA Treatment.
Hi-5 cell membranes containing the recombinant adenylyl cyclase were treated for 15 min in a solution of 25 mM Tris⋅HCl, 10 mM MgCl2, 0.8 mM ATP, and protease inhibitor mixture (10) with and without 50–75 nM PKA catalytic subunit. After treatment the membranes were put on ice. Aliquots of the treated membranes were diluted 10-fold into adenylyl cyclase assay mixtures.
Adenylyl Cyclase Assays.
Adenylyl cyclase was assayed in the presence of 0.1 mM [α-32P]ATP, 2 mM MgCl2 and other reagents as described (7). Q227L-Gαs was synthesized in vitro. mRNA was transcribed by using T7 polymerase and translated in rabbit reticulocyte lysates by using the Flexi System from Promega (8). When used, the concentration of forskolin was 50 μM.
Mutagenesis of AC6.
Ser-674 was converted to an Ala by site-directed mutagenesis by using PCR. The 5′ oligonucleotide encoding AC6 (11) fragment from nucleotide 2,005 to 2,049 with residue T-2033 changed to a G and the 3′ oligonucleotide encoding nuclotides 2,812–2,792 were used as primers. AC6-pVL1393 cDNA was used as template. The PCR using Taq polymerase was for 28 cycles (1 min at 95°C for denaturation, 1 min at 55°C for annealing, and 2.5 min elongation). The 0.8-kb PCR product was subcloned in pCR 2.1 by using the TA cloning kit (Invitrogen) and sequenced. The fragment containing the mutated base was excised from pCR2.1 as a 0.6-kb XhoI/BlpI fragment. AC6 cDNA in pVL1393 was cut with XhoI and BlpI. The mutant fragment was then inserted into FLAG-tagged AC6 by T7 ligase. Recombinant baculovirus containing the FLAG-tagged S674A-AC6 cDNA was produced as described (10). FLAG-tagged wild-type (wt)-AC6 and S674A-AC6 were expressed in Hi-5 cells, and membranes were prepared.
PKA Phosphorylation of FLAG-Tagged wt and Mutant AC6.
Membranes containing Flag tagged wt and S674A-AC6 were used for the labeling reactions. The labeling reaction contained 40 mM Tris⋅HCl (pH 7.4), 20 mM MgCl2, 10 μM ATP, and 150 μCi [γ-32P]ATP (3,000 Ci/mmol; 1 Ci = 37 GBq); the 75 nM PKA catalytic subunit, 2 μM okadaic acid, protease inhibitor cocktail (10), and 2 mg membrane protein was present in a final volume of 150 μl. When present, the concentration of WIPTIDE was 75 μM. The reactions were incubated for 30 min at 35°C, and then 2 μl of a 1.5 mg/ml solution of DNase was added to each tube and the incubation was continued for another 15 min. The membranes were seperated from the reaction mixture by centrifugation at in a microfuge for 30 min at 4°C. The pellets were individually extracted in 1 ml of a modified RIPA (m-RIPA) buffer containing 50 mM Tris⋅HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA (pH 8.0), a phosphatase inhibitor mixture of 2 μM okadaic acid, 10 mM NaF, 10 mM β-glycerol phosphate, 1 mM Na3VO4, a protease inhibitor mixture as described above, and the following detergents: 1.5% Triton X-100, 0.1% Nonidet P-40 (NP-40), and 0.2% digitonin. The mixtures were rocked in the cold room for 4 hr, and the supernatants were collected by centrifugation at 13,000 rpm in the microfuge for 30 min. The anti-FLAG-M2 gel was equilibriated in the m-RIPA buffer containing 0.75% Triton X-100, 0.05% NP-40, and 0.1% digitonin. One milliliter of the extract was the mixed 120 μl of the packed equilibriated gel in 1 ml of the m-RIPA buffer without detergent. The suspension was mixed by rocking in the cold room for 8 hr. After mixing the gel was collected by centrifugation and washed three times with the equilibriation buffer and then eluted with 180 μl of 0.1 M glycine-HCl (pH 3.5) containing 0.05% Triton X-100 and 2 μM okadaic acid. Aliquots of the eluate were electrophoretically resolved on 6% SDS/PAGE, transferred to nitrocellulose, and blotted with the ACcomm antibody to ascertain equivalency of extraction. Samples (60 μl) were electrophoretically resolved on 6% SDS/PAGE. The gels were fixed and dried. Bands were visualized by autoradiography. The autoradiogram was scanned into a Mac-8100 computer by use of photoshop. The image was imported into canvas for labeling and printed out as a canvas file.
Peptide Synthesis.
Peptides were synthesized with an Applied Biosystems model 430A peptide synthesizer. Identity of the peptides was verified by mass spectrometry. Peptides were dissolved in water and used at a final concentration of 300 μM.
All experiments were repeated at least three times with qualitatively similar results. Representative experiments are shown.
RESULTS
AC6 was expressed in Hi-5 cells by baculovirus infection, and membranes containing AC6 were treated with purified PKA and then assayed for basal, Gαs, and forskolin-stimulated activities (9). PKA treatment resulted in a 50% decrease in Gαs-stimulated adenylyl cyclase activity; there was no change in basal activity and a 30% decrease in forskolin-stimulated adenylyl cyclase activity (Fig. 1A). In contrast, when AC1 and AC2 were expressed in Hi-5 cells and membranes containing the recombinant adenylyl cyclases were treated with PKA, only small or no changes in Gαs stimulated activity was observed (Fig. 1B). Maximal effects of PKA on Gαs stimulation of AC6 activity were obtained between 50 and 75 nM PKA (data not shown). A treatment period of 15–30 min gave a maximum obtainable effect of PKA on Gαs stimulation of AC6 activity without affecting basal activity (Fig. 1C). The effect of PKA on AC6 activity was specific because the inclusion of WIPTIDE, a specific inhibitor of PKA (12), blocked the PKA-mediated effects on both Gαs and forskolin stimulation of AC6 (Fig. 1D). Because both Gαs and forskolin stimulation of AC6 activity were affected by PKA treatment, albeit to varying degrees, it was of interest to determine whether PKA treatment affected the catalytic capability of AC6. Hence we determined the effect of PKA treatment on Mn2+-stimulated AC6 activity. Mn2+-ATP is a nonphysiological substrate for adenylyl cyclase (13), and Mn2+ is thought to activate adenylyl cyclase by interaction at an allosteric site (14). Preliminary indications from the crystal structure of the catalytic core of AC2 indicate that there may be regulatory metal ion binding sites (15). Though 5–10 mM Mn2+ ions stimulated AC6 to levels seen with Gαs, PKA treatment did not affect Mn2+-stimulated AC6 activity (Fig. 1E). The native adenylyl cyclase(s) of the Hi-5 cells were not affected by PKA treatment. Gαs is not phosphorylated by PKA, and its activity is unaffected by PKA treatment (6).
Figure 1.

Effect of PKA on adenylyl cyclase activities. Adenylyl cyclases were expressed in Hi-5 cells treated without or with PKA and then assayed for indicated activities. (A) Effect of PKA treatment on basal, 20 nM Q227L-Gαs (αs*), and 50 μM forskolin-stimulated AC6 activity. (B) Effect of PKA treatment on basal and 20 nM Q227L-Gαs (αs*) stimulated AC1 and AC2 activities. (C) Effect of varying periods of treatment with PKA on αs*-stimulated AC6 activity. (D) Effect of inclusion of 75 μM WIPTIDE during PKA treatment on basal, 20 nM Q227L-Gαs (αs*), and 50 μM forskolin-stimulated AC6 activity. (E) Effect of PKA treatment on Mn2+-stimulated AC6 activity. Divalent cation concentrations are in excess over 1 mM EDTA and 0.1 mM ATP.
The data from the experiments in Fig. 1 suggest that PKA treatment results in a selective decrease in Gαs stimulation of AC6 without affecting the catalytic capability of the AC6. Hence we probed the mechanism by which PKA affected Gαs stimulation of AC6. For this we compared the effects of varying concentrations of Gαs on AC6 that had been treated in the absence and presence of PKA. Gαs stimulation of AC6 in its native membrane environment is biphasic with distinct high and low affinity components (8). The data in Fig. 2 were fitted to a two-site model described in detail elsewhere (8). In this experiment the apparent Kact for the “high affinity” site is 1.3 nM and for the “low affinity” site is 28 nM. Upon treatment of AC6-containing membranes with PKA, the high affinity stimulation by Gαs appears unaffected, but the low affinity stimulation by Gαs is inhibited by 85% (Fig. 2).
Figure 2.
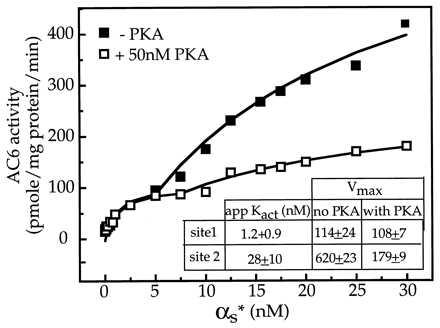
Effect of PKA treatment on stimulation of AC6 by varying concentrations of Gαs. AC6 containing Hi-5 cell membranes were treated in the presence and absence of PKA and then assayed in the presence of indicated concentrations of activated Gαs (αs*). Data were analyzed on a Sun Work station by using the program prophet. The plots of the data points and the fitted curves were generated by prophet. The data best fit a two-site model (8), and the indicated constants were obtained from the two site fit. The printed plots were exported to the canvas program in a Mac 8100 by use of an optical scanner. The plots were labeled within canvas and printed as canvas files.
There is a single predicted PKA phosphorylation site at Ser-674 in AC6 (11). To determine whether Ser-674 is phosphorylated by PKA, we converted the Ser-674 to an Ala by site-directed mutagenesis. Because prior experience with AC2 suggested that we might need to purify the recombinant enzyme expressed in insect cells to be able to visualize the phosphorylated enzyme (9), we introduced the FLAG epitope at the N terminus of the wt and S674A mutant of AC6. The epitope-tagged enzymes were expressed in Hi-5 cells by baculovirus infection. To determine whether PKA phosphorylated AC6, and whether mutation of S674 had any effect on the phosphorylation, membranes containing the wt and mutant adenylyl cyclase were incubated with [γ-32P]ATP in the absence and presence of PKA and with PKA plus the inhibitor peptide WIPTIDE. The membrane proteins were then extracted, and the FLAG-tagged adenylyl cyclases were adsorbed on to the anti FLAG-M2 affinity gel. The gels were washed and the enzymes eluted at low pH and resolved electrophoretically on SDS/polyacrylamide and then visualized by autoradiography. When AC6-containing Hi-5 cell membranes were exposed to PKA, a single labeled band of ≈135 kDa was observed. Inclusion of WIPTIDE blocked PKA-dependent labeling of the 135-kDa band (Fig. 3A, lane 3). In contrast, when the S674A-AC6 was expressed in Hi-5 cells and the cell membranes were treated with PKA, no labeled band was observed in the absence or presence of WIPTIDE (Fig. 3A, three right lanes). These observations indicate that there is a single PKA phosphorylation site at Ser-674 in AC6. We next determined the effects of phosphorylation of Ser-674 on AC6 activity. For this we compared the effect of PKA treatment on Gαs stimulation of wt and S674A-AC6. Conversion of Ser-674 to Ala results in a small (10–20%) increase in basal activity but otherwise did not affect AC6 activities. Gαs extensively stimulated both wt and S674A-AC6. PKA treatment of AC6 resulted in a 50% decrease in stimulation by 20 nM Gαs, but PKA treatment of S674A-AC6 did not affect Gαs stimulation (Fig. 3B). These results suggest that the region around Ser-674 of AC6 may be involved in regulation of stimulation by Gαs.
Figure 3.
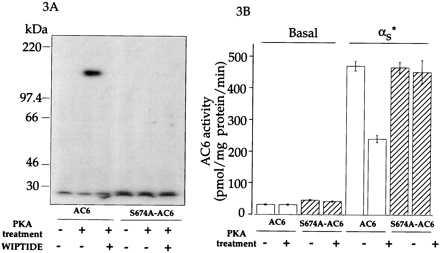
Effect of mutating AC6 Ser-674 to Ala on PKA effects on AC6. (A) Protein kinase-mediated phosphorylation of Hi-5 cell membranes expressing wt and S674A forms of AC6. Treatment was in the absence or presence of 50 nM PKA or PKA and 75 μM WIPTIDE. After treatment the membranes were extracted and the FLAG-tagged AC6 and S674A-AC6 were isolated on anti FLAG-M2 columns. The column eluates were resolved by SDS/PAGE and visualized by autoradiography. For detailed procedures, see Materials and Methods. (B) Effect of PKA treatment of Hi-5 cell membranes expressing wt and S674A forms of AC6 on basal and activated Gαs (αs*)-stimulated activities. Treatment was in the absence or presence of PKA as described (7). Concentration of αs* in the assay was 20 nM.
To explicitly test whether the region around Ser-674 was involved in regulation by Gαs, we synthesized a peptide encoding region 660–682 of AC6 and tested whether this peptide (FLLT peptide) affected Gαs stimulation of AC6. The location of the region utilized for the synthesis of the peptide is shown within the predicted topographic structure of a mammalian adenylyl cyclase in Fig. 4A. Also shown is the sequence of residues for other adenylyl cyclases in this region. We tested the effect of the peptide on stimulation of AC6 by varying concentrations of Gαs. In the absence of the FLLT peptide, a typical biphasic stimulation of AC6 by Gαs was observed. The FLLT peptide did not significantly affect the high affinity stimulation by Gαs but substantially (≈88%) reduced the low affinity stimulation by Gαs (Fig. 4B). The Gαs stimulation in the presence of the FLLT peptide is very similar to that seen after PKA treatment (Compare Figs. 4B and 2). We next tested whether the inhibition of the Gαs stimulation by the FLLT peptide and inhibition by PKA treatment of AC6 was nonadditive. The effect of the FLLT peptide was tested on AC6 treated in the absence and presence of PKA. Both the FLLT peptide and PKA treatment individually inhibited Gαs stimulation, and their inhibition was nonadditive (Fig. 4C). To test the specificity of the FLLT peptide, we converted the Ser corresponding to Ser-674 to an Asp. Asp is often used to mimic a phosphorylated Ser or Thr. Because phosphorylation at Ser-674 blocks Gαs interactions with AC6 (Fig. 3B), a negative charge at this position should render the FLLT peptide incapable of interfering with Gαs stimulation of AC6. This assumption was tested in the experiment shown in Fig. 4D. Although the FLLT peptide inhibited Gαs stimulation by 55%, the S674D-FLLT peptide did not affect Gαs stimulation of AC6 (Fig. 4D). We next tested the effect of varying concentrations of FLLT peptide on Gαs stimulation and found that the peptide inhibited Gαs stimulation with an IC50 of 40 μM (Fig. 4E). The FLLT peptide (300 μM) does not affect Mn2+-stimulated activity but reduced forskolin-stimulated activity by 20–30% (data not shown). We then tested whether the FLLT peptide blocked Gαs stimulation of AC2 We found that the peptide inhibited stimulation of AC2 by high concentrations of Gαs (Fig. 4F).
Figure 4.

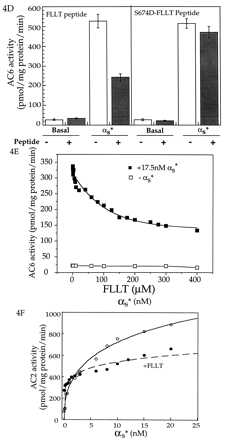
Effects of the peptide encoding region 660–682 of AC6 on Gαs regulation of adenylyl cyclases. (A) The region of AC6 containing the predicted PKA site used for the synthesis of the peptide is shown as part of the presumed topographical arrangement of mammalian adenylyl cyclases. Sequences of this region are compared between the different adenylyl cyclases from several species. Alignment of sequences was obtained by using the Genetics Computer Group program in a VAX computer. Fully conserved residues are shown in red. Mostly conserved charged residues are shown in blue, and mostly conserved hydrophobic residues are in green. The existence of a motif conserved amongst the different adenylyl cyclases is visually obvious. (B) AC6 containing Hi-5 cell membranes were assayed in the presence of indicated concentrations of activated Gαs (αs*) with or without the FLLT peptide. The data best fit a two-site model (8). Indicated constants were obtained from the two-site fit. (C) AC6 containing Hi-5 cell membranes that had been treated in the absence and presence of PKA were assayed for basal and 20 nM αs*-stimulated activities in the absence and presence of 300 μM FLLT peptide. (D) Comparison of the effect of the FLLT peptide and a modified peptide (S674D-FLLT peptide) where the Ser corresponding to Ser-674 was substituted with an Asp (D) on AC6 containing Hi-5 cell membrane basal and 20 nM αs*-stimulated adenylyl cyclase activities. Concentrations of both peptides were 300 μM. (E) Effect of varying concentrations of FLLT peptide on stimulation of AC6 by 17.5 nM αs*. (F) AC2 containing Hi-5 cell membranes assayed in the presence of indicated concentrations of αs* in the absence and presence of 300 μM FLLT peptide.
DISCUSSION
These results indicate that we have identified a region of AC6 that is involved in PKA regulation. Mutational analysis of AC6, as well as a peptide derived from AC6, support this conclusion. Although on an overall basis this region is not highly conserved among the different adenylyl cyclases, as can be seen from the sequence comparison in Fig. 4A, this region does contains a motif conserved among the various adenylyl cyclases. The presence of a Ser in this region that is capable of being phosphorylated by PKA is, however, unique to AC6 and AC5, and hence Gαs interactions with AC6 are selectively regulated by PKA. Forskolin and Gαs stimulation of AC5, which is very closely related to AC6, has also been shown to be inhibited by PKA (16). AC1 and AC2 do not contain a predicted phosphorylation site in this region, and our data indicate that they are not susceptible to PKA regulation under our assay conditions. The 660–682 region of AC6 appears to be involved in regulating Gαs stimulation because the FLLT peptide inhibits Gαs stimulation of AC2 as well. For both AC6 and AC2, the FLLT peptide specifically blocks stimulation by higher concentrations of Gαs but does not appear to affect the high affinity Gαs stimulation that occurs with an apparent Kact of 1–2 nM. These observations imply that in the membrane environment stimulation of adenylyl cyclases may involve high and low affinity interactions with Gαs. This does not necessarily mean that there are two Gαs binding sites per adenylyl cyclase. Crosslinking studies with the olfactory adenylyl cyclase in the membrane indicate a 1:1 stoichiometery (17), and recently a 1:1 stoichiometery has also been clearly demonstrated for Gαs interactions with a chimeric functional enzyme consisting of soluble fragments from AC5 and AC2 (18). If one assumes a 1:1 binding, then our data would be compatible with a model where adenylyl cyclase function as dimers in the membrane environment. Each adenylyl cyclase monomer would bind on Gαs, and the kinetic data suggest cooperativity between the two sites (8). In such a model the 660–682 region could be involved in either in an intra- or intermolecular interactions with the cytoplasmic tail resulting in a functional Gαs binding site. Phosphorylation of Ser-674 would block this interaction and thus disrupt Gαs interactions. Data from analysis of soluble AC5 support such a model (19). Alternatively this region could be involved in direct interactions with Gαs and phosphorylation disrupts this interaction. Further experiments are needed with both soluble and full-length membrane bound forms to obtain a better understanding of how phosphorylation regulates Gαs stimulation. Irrespective of the detailed mechanism by which PKA phosphorylation regulates Gαs stimulation, such feedback regulation contributes to desensitization of this transmembrane signaling system. In this context it is noteworthy that Gαs stimulation of AC6, a widely expressed adenylyl cyclase, is selectively but tightly regulated by feedback inhibition by PKA. Such regulation ensures that hormone-stimulated cAMP production is transitory and does not result in the pathophysiologies associated with prolonged elevation of cAMP levels (20).
Acknowledgments
We thank Dr. Patel for a preprint of his paper. This research was supported by National Institutes of Health Grants DK38761 and GM 54508. Y.C. was a predoctoral trainee supported by Molecular Endocrinology Training Grant DK-0745. A.H. was supported by National Research Service Award Predoctoral Fellowship GM15599. M.J.S. was supported by a fellowship from the Netherlands Organization for Scientific Research. A.H. was supported by National Research Service Award Predoctoral Fellowship GM15599. G.W. is an Aaron Diamond Fellow,
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: AC1, 2, etc., adenylyl cyclase 1, 2, etc.; wt, wild type; PKA, protein kinase A; FLAG epitope, DYKDDDDK; WIPTIDE, PKA inhibitory peptide.
References
- 1.Gilman A G. Annu Rev Biochem. 1987;56:615–693. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 2.Iyengar R. FASEB J. 1993;7:768–775. doi: 10.1096/fasebj.7.9.8330684. [DOI] [PubMed] [Google Scholar]
- 3.Sunahara R K, Dessauer C W, Gilman A G. Annu Rev Pharmacol Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 4.Premont R T, Matsuoka I, Mattei M-G, Pouille Y, Defer N, Hanoune J. J Biol Chem. 1996;217:13900–13907. doi: 10.1074/jbc.271.23.13900. [DOI] [PubMed] [Google Scholar]
- 5.Premont R T, Iyengar R. J Biol Chem. 1988;263:16087–16095. [PubMed] [Google Scholar]
- 6.Premont R T, Iyengar R. Endocrinology. 1989;125:1151–1160. doi: 10.1210/endo-125-3-1151. [DOI] [PubMed] [Google Scholar]
- 7.Premont R T, Jacobowitz O, Iyengar R. Endocrinology. 1992;131:2774–2784. doi: 10.1210/endo.131.6.1332848. [DOI] [PubMed] [Google Scholar]
- 8.Harry A, Chen Y-b, Magnusson R, Iyengar R, Weng G. J Biol Chem. 1997;272:19017–19021. doi: 10.1074/jbc.272.30.19017. [DOI] [PubMed] [Google Scholar]
- 9.Jacobowitz O, Iyengar R. Proc Natl Acad Sci USA. 1994;91:10630–10634. doi: 10.1073/pnas.91.22.10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pieroni J P, Harry A, Chen J-Q, Jacobowitz O, Magnusson P R, Iyengar R. J Biol Chem. 1995;270:21368–21373. doi: 10.1074/jbc.270.36.21368. [DOI] [PubMed] [Google Scholar]
- 11.Premont R T, Chen J, Ma H-W, Ponnapalli M, Iyengar R. Proc Natl Acad Sci USA. 1992;89:9808–9813. doi: 10.1073/pnas.89.20.9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng H-C, Kemp B E, Pearson R B, Smith A J, Misconi L, Vanpatten S M, Walsh D A. J Biol Chem. 1986;261:989–992. [PubMed] [Google Scholar]
- 13.Ross E M, Howlett A W, Ferguson K M, Gilman A G. J Biol Chem. 1978;253:6401–6412. [PubMed] [Google Scholar]
- 14.Somkuti S G, Hildebrandt J D, Herberg J T, Iyengar R. J Biol Chem. 1982;257:6387–6393. [PubMed] [Google Scholar]
- 15.Zhang G, Liu Y, Ruoho A E, Hurley J H. Nature (London) 1997;386:247–253. doi: 10.1038/386247a0. [DOI] [PubMed] [Google Scholar]
- 16.Iwami G, Kawabe J, Ebina T, Cannon P J, Homcy C J, Ishikawa Y. J Biol Chem. 1995;270:12481–12484. doi: 10.1074/jbc.270.21.12481. [DOI] [PubMed] [Google Scholar]
- 17.Pfeuffer E, Mollner S, Lancent D, Pfeuffer T. J Biol Chem. 1989;264:18803–18807. [PubMed] [Google Scholar]
- 18.Sunahara R K, Dessauer C W, Whisnant R E, Kleuss C, Gilman A G. J Biol Chem. 1997;272:22265–22271. doi: 10.1074/jbc.272.35.22265. [DOI] [PubMed] [Google Scholar]
- 19.Scholich K, Wittpoth C, Barbier A J, Mullinex J B, Patel T B. Proc Natl Acad Sci USA. 1997;94:9602–9607. doi: 10.1073/pnas.94.18.9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinstein L S, Shenker A, Gejman P V, Merrino M, Friedman E, Speigel A M. N Eng J Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]