

Abstract
Crystallization of human membrane proteins in lipidic cubic phase often results in very small but highly ordered crystals. Advent of the sub-10 µm minibeam at the APS GM/CA CAT has enabled the collection of high quality diffraction data from such microcrystals. Herein we describe the challenges and solutions related to growing, manipulating and collecting data from optically invisible microcrystals embedded in an opaque frozen in meso material. Of critical importance is the use of the intense and small synchrotron beam to raster through and locate the crystal sample in an efficient and reliable manner. The resulting diffraction patterns have a significant reduction in background, with strong intensity and improvement in diffraction resolution compared with larger beam sizes. Three high-resolution structures of human G protein-coupled receptors serve as evidence of the utility of these techniques that will likely be useful for future structural determination efforts. We anticipate that further innovations of the technologies applied to microcrystallography will enable the solving of structures of ever more challenging targets.
Keywords: lipidic cubic phase, G protein-coupled receptor, minibeam, microcrystallography
1. Introduction
One of the bottlenecks in structural studies of membrane proteins is obtaining diffraction quality crystals. Perhaps even more challenging are proteins with low ratio of hydrophilic to hydrophobic surface area such as G protein-coupled receptors (GPCRs). Crystallization in lipidic cubic phase (LCP; Landau & Rosenbusch 1996) is advantageous for promoting crystal packing contacts between hydrophilic as well as between hydrophobic protein surfaces (Caffrey 2009). Crystals grown in LCP, also referred as in meso crystallization (Caffrey 2003), are in general well ordered probably because of the increase in available crystal packing area and diffusion driven mechanism; however, on average, these crystals have a smaller size than the typical crystals grown in aqueous solution. In certain cases, it has not been possible to obtain crystals larger than approximately 1000 µm3, a volume that would contain approximately 109 unit cells. Despite surprisingly good diffraction limits, such crystals present a significant challenge for standard crystallographic techniques in that detection of the crystals in the drop, harvesting and collecting high quality diffraction data become non-trivial and a feat nearly impossible without the development of microcrystallography techniques.
Microcrystallography emerged and gained popularity more than a decade ago with the appearance of high brilliance beams at the third generation synchrotron sources. Much of the pioneering work was done at ID-13 at the ESRF (Pebay-Peyroula et al. 1997). The development of a micro-diffractometer at the EMBL-Grenoble (Perrakis et al. 1999) paved the way for routine microcrystallography experiments. Small X-ray beams with diameters less than 10 µm have been used to collect data from thin needles (Rasmussen et al. 2007) and small microcrystals (Coulibaly et al. 2007), as well as to improve diffraction from inhomogeneous (Sanishvili et al. 2008) or mosaic (Xiao et al. 2003) crystals.
The General Medical Sciences and Cancer Institute Collaborative Access Team (GM/CA CAT) at the Advance Photon Source, Argonne, IL, USA, has developed a dedicated protein microcrystallography minibeam apparatus (Fischetti et al. 2009). A systematic feasibility study using this apparatus for data collection from test crystals measuring less than 10 × 10 × 10 µm3 has been published (Sanishvili et al. 2008). Here we report the results of a collaboration between the Cherezov–Kuhn–Stevens groups at The Scripps Research Institute (TSRI) and APS GM/CA CAT to develop advanced methods to centre optically invisible crystals and collect high quality diffraction data. The group at Argonne developed the automated software to raster search a user defined sample volume using the GM/CA minibeam. The groups at TSRI developed improved methods for growing and harvesting in meso grown GPCR microcrystals, conducted the manual rastering of optically invisible crystals, provided valuable feedback on the automated rastering capability and developed data collection strategies to mitigate the effects of radiation damage.
2. Material and methods
2.1. Protein expression and purification
Human β2-adrenergic GPCR (β2AR) was engineered by replacing part of the intracellular loop 3 with lysozyme from T4 phage (β2AR-T4L) in order to improve the receptor's stability and increase its chances for successful crystallization (Rosenbaum et al. 2007). Initial purified β2AR-T4L protein, in which residues Gln231–Ser262 were replaced with residues 2–161 of T4 lysozyme, the C-terminus was truncated at Gly365 and one glycosylation site was eliminated through Asn187 to Glu mutation, was obtained from B. Kobilka (Stanford University). Thermally stabilized β2AR-T4L construct (β2AR(E122W)-T4L) and human adenosine A2A receptor T4L fusion construct (A2AR-T4L) were designed, cloned, expressed and purified in house (Hanson et al. 2008; Jaakola et al. 2008). In β2AR(E122W)-T4L, residues Gln231–Ser262 were replaced with residues 2–161 of T4 lysozyme, the C-terminus was truncated at Lys348, one glycosylation site was eliminated through Asn187 to Glu mutation and Glu122Trp mutation (Roth et al. 2008) was introduced to increase the stability. In A2AR-T4L, residues Leu209–Ala221 were replaced with residues 2–161 of T4 lysozyme and the C-terminus was truncated at Ala316.
The constructs were generated using standard cloning techniques and recombinantly expressed in baculovirus infected sf9 insect cells as described in detail in Rosenbaum et al. (2007), Hanson et al. (2008) and Jaakola et al. (2008). The cells were harvested 48 h after infection and disrupted by a nitrogen cavitation bomb. The cell membranes were isolated and washed using repeated centrifugation followed by resuspension and homogenization. The protein was solubilized from the membranes using 0.5 per cent (w/v) n-dodecyl-β-d-maltopyranoside/0.01 per cent (w/v) cholesterol hemisuccinate (CHS) and purified with Talon (Clonetech) and Ni Sepharose (GE LifeScience) IMAC columns. PNGase F (New England Biolabs) was used on the Ni Sepharose column to de-glycosylate the protein. After elution from the Ni Sepharose column, the protein was concentrated to 30–70 mg ml−1 using a 100 kDa cutoff Vivaspin concentrator (Sartorius).
The final protein samples were of high purity (>95% by SDS–PAGE) and displayed a single monomer peak on an analytical size exclusion chromatography column.
2.2. Robotic in meso crystallization setup
Protein solution at 30–70 mg ml−1 concentration was mixed with a lipid (typically monoolein or monoolein supplemented with 5–10 wt% of cholesterol or other lipids) in the ratio of two parts of protein solution to three parts of lipid using a syringe lipid mixer (Cheng et al. 1998). After a clear homogeneous LCP was formed, it was transferred into a 100 µl gas-tight syringe (Hamilton Company). A short 10 mm removable needle (gauge 26) was attached to the syringe and the syringe was mounted on an in meso crystallization robot (Cherezov et al. 2004). The robot dispensed 25–50 nl of protein-laden cubic phase and 0.8 µl of precipitant solution per well of a 96-well glass sandwich plate (Cherezov et al. 2004). The wells were sealed with a cover glass using a brayer. All manipulations on setting up crystallization trials were performed at room temperature (21–23°C). After the plates were sealed, they were transferred into an incubator/imager (RockImager 1000, Formulatrix), where they were stored at 20°C and imaged according to a specified schedule (typically on 0, 1, 3, 5, 7, 10, 14, 21 and 31 days after setup).
2.3. Crystal harvesting
Crystals were harvested directly from the glass sandwich plates. All operations were performed under a variable zoom stereo microscope (Nikon SMZ1500). To open an individual well, the cover glass was scored using a sharp corner of a glass capillary cutting stone (Hampton Research, cat# HR4-334) with four short strokes forming a square centred inside the well boundaries. Strong sharp point tweezers (Ted Pella, cat# 510) were used to press around the scored perimeter to propagate the scratch through the thickness of the cover glass. Then two holes were punched at two opposite corners of the square, followed by an injection of 2–3 µl of precipitant solution through one of the holes inside the well to reduce dehydration during the well opening manipulations. A sharp angled needle probe (Ted Pella, cat# 13650) was used to free the glass square and carefully lift it up. An extra 5 µl of precipitant solution was added on top of the opened well. The crystals were harvested directly from the LCP using MicroMounts (MiTeGen) with the diameter matching the crystal size (typically 30 µm). A polarizer and analyser attached to the microscope were used at close to cross at 90° orientation to facilitate visualization of birefringent crystals while still providing enough light to see the MicroMount approaching the crystal. The amount of the lipid phase surrounding the harvested crystal was minimized as much as possible. If the crystal was sitting deep inside the LCP bolus, a MicroMount was used to remove the top LCP layer and expose the crystal at the surface, after which another clean MicroMount was used to harvest the crystal. The harvested crystal was immediately plunged into liquid nitrogen. As the precipitant contained at least 25 per cent PEG400, it protected the crystal during the freezing and no additional cryoprotectant was used.
2.4. Loop rastering and crystal centring
Optically invisible microcrystals were located and centred in the minibeam using diffraction from a highly attenuated X-ray beam to minimize radiation damage. A manual centring procedure was developed initially and later it was replaced by an automatic raster scanning. Both these approaches are described in this section.
2.4.1. Manual centring
As the loop size was in general significantly larger than the size of the minibeam, the whole loop area was initially scanned using a beam with a cross section of 69 × 21 µm2 (full width at half maximum, FWHM) at the sample position. The beam size was defined by slits that were located 230 mm upstream of the sample position and were set to 75 × 25 µm2 defining the beam that was focused at the sample position. The beam was attenuated 20-fold to minimize radiation damage. When diffraction was observed the corresponding area was coarsely scanned with a 20-fold attenuated minibeam using 10 µm steps and consequently fine tuned using 2 µm steps with a 10-fold attenuated minibeam. During the fine tuning, the integrated intensity within a strongly diffracting spot served as feedback for selecting the best position. The direction of the crystal motion during the fine tuning was determined by the gradient descent method, which allowed a minimum number of steps but could also potentially lead to a local minima trap, especially in the case of larger crystals. After the crystal was centred in one orientation, the loop was rotated 90° and the coarse 10 and fine 2 µm scans were repeated in the direction perpendicular to the rotation axis. The alignment of each crystal took 30–60 min and exposed the crystal to a total X-ray dose of <1 MGy, which was equivalent to <1 s exposure with unattenuated beam.
2.4.2. Automatic rastering
Automatic rastering of samples was developed as a tool invoked within the GM/CA CAT adaptation of Blu-Ice (McPhillips et al. 2002; Stepanov et al. 2006). A similar tool was developed in the original Blu-Ice for automated centring (Song et al. 2007) of larger crystals with larger X-ray beams. The rastering tool developed at GM/CA CAT uses the minibeam to centre optically invisible crystals in the X-ray beam and to map better ordered regions of large crystals. The user can choose to collect data from the best part of the crystal using the full focused beam (or that defined by the upstream slits) as was suggested earlier (Sanishvili et al. 2008) or to collect partial datasets by walking along the crystal with the minibeam. At present, automated rastering follows manual centring of the loop on the crosshairs where the X-ray beam is also centred independently. To minimize the number of exposures during rastering of optically independent crystals, it is recommended to rotate the sample mount so that its smallest profile is rastered first.
Automated rastering starts with larger steps and the beam defined by slits, as described above for manual rastering. The initial grid parameters are entered in the raster tab of Blu-Ice-EPICS, and the resulting grid is displayed over the sample image. On clicking start, diffraction images are taken for each cell in the grid. No search optimization is currently done.
Diffraction images are analysed with DISTL (Zhang et al. 2006) on the fly and/or by a user manually with image viewing software. DISTL produces per-image quality indicators including spot total and ice rings. The indicators are displayed in sortable columns in the results table. Processing with DISTL is done in the background on a SAN-connected workstation and produces results at the same pace as rastering. With a 0.5 s exposure time this rate is 15 frames min−1.
The user can select the grid point with the best diffraction as determined by DISTL by double clicking on the image identifier in the list prepared by the software. The centre of the cell of the corresponding grid point is then automatically centred.
For rastering with finer steps (≤10 µm), the user can select the 5 or 10 µm beam from a pull-down menu in the GM/CA CAT version of Blu-Ice. Once the sample is centred in the given plane, it is rotated by 90° and a single vertical scan is carried out with the minimum step size used in the previous scan. Exposure times and attenuation are selected as a compromise to achieve detectable diffraction while minimizing X-ray induced radiation damage to the sample. The total estimated dose absorbed by the crystal during a typical automatic rastering is less than 0.5 MGy.
2.5. Data collection and processing
X-ray data were collected on beamline 23ID-B at GM/CA CAT at the Advanced Photon Source, Argonne, IL, using a minibeam with a wavelength of 1.033 Å (12.000 keV) passing through a 10 µm collimator (Sanishvili et al. 2008; Fischetti et al. 2009) and a MarMosaic 300 CCD detector (Rayonix, Evanston, IL; formerly Mar USA). The minibeam in the earlier experiments before February 2008 had an oval shape with a size of 7.8 × 6.3 µm2 FWHM at the sample position as measured by a knife-edge scan, and with a total flux of 1 × 1011 photons s−1 (100 mA)−1, measured with an ion chamber (Sanishvili et al. 2008). Since February 2008, a new 10 µm collimator was used producing a 10.6 × 11.6 µm2 beam with an intensity of 2.2 × 1011 photons s−1 (100 mA)−1 at the sample position (Fischetti et al. 2009).
Data were integrated, scaled and merged using XDS (Kabsch 1993). Initial phase information was obtained by molecular replacement with Phaser (McCoy 2007) using the coordinates of T4 lysozyme and a polyalanine model of the rhodopsin 7 transmembrane (TM) helical bundle. Refinement was performed iteratively with Refmac5 (Murshudov et al. 1997), Phenix (Adams et al. 2002) and manual rebuilding in Coot (Emsley & Cowtan 2004).
3. Results and discussion
3.1. In meso crystallization
All GPCR crystals for this work were obtained using automated nanolitre in meso crystallization (Cherezov et al. 2004). Robotic setup allowed for extensive screening and optimization while consuming minimal amounts of protein. As little as 10 µl of concentrated protein solution (the minimal volume achievable with commercial centrifugation-based concentrators) was sufficient to perform 700–800 trials.
Unoptimized conditions generally produce crystals in meso appearing as showers of extremely small microcrystals (figure 1a). Detection of such tiny colourless protein crystals from within the LCP background is challenging. However, the glass sandwich plates in which the crystallization trials are set up in Cherezov et al. (2004) provide a superior optical environment for obtaining high-quality crystal images using a microscope with both bright-field illumination and cross-polarization capability (figures 1–3). Trace labelling proteins with a fluorescent dye are also useful for initial crystal detection (figure 2c). Alternatively a UV-fluorescence microscope can be used to both detect microcrystals in the low background LCP and confirm that they contain protein (figure 3c).
Figure 1.

Improvements in size and shape of β2AR-T4L crystals as the result of optimizations of crystallization conditions. (a) Initial crystals appearing as showers of microneedles with largest dimension of a few micrometres. (b) Crystals achieved after optimization of concentrations of the main precipitant (PEG 400), salt (sodium sulphate) and pH of the buffer. (c) Crystals obtained after optimization of the lipid additives. (d) Final crystals diffracting to high resolution. Scale bars, 25 µm (a,b); 50 µm (c,d).
Figure 2.

Imaging of small A2AR-T4L crystals in LCP. (a) Image recorded with a Zeiss AxioImager microscope using a 10× objective and a bright-field illumination. LCP is very homogeneous and crystals are easily detected (scale bar, 25 µm). To confirm that the crystals are made of protein, the protein was labelled with a dye and fluorescence image was taken in (c). (b) Image recorded with a Zeiss AxioImager microscope using a 10× objective and crossed polarizers. Crystals show good birefringency, which also depends on their orientation in LCP. (c) Image recorded with a Zeiss AxioImager microscope using an epi-illumination with an excitation filter centred at 543 nm with a bandwidth of 22 nm and an emission filter with transmission between 575 and 640 nm. The protein was labelled with Cy3 succinimidyl ester at a trace ratio of below 1%. The fluorescence image provides a very good contrast even for very small, micrometre-size, crystals, confirming that the crystals contain protein.
Figure 3.

Imaging of small β2AR-T4L crystals in LCP. (a) Image recorded with RockImager 1000 (Formulatrix) using a bright-field illumination. LCP contains many defects and irregularities scattering light and obscuring crystal detection. Scale bar, 50 µm. (b) Image recorded with RockImager 1000 using crossed polarizers. Crystals in this case display very low birefringency, which depends on their orientation, making them difficult to be detected. (c) UV-fluorescence image recorded with Korima UV microscope (excitation 280 nm, emission 350 nm). Slight protein precipitation and several protein crystals are clearly seen in this image.
Optimization of in meso crystallization conditions involves adjustment of many of the same parameters as one would explore in a typical crystallization experiment in an aqueous solution (Benvenuti & Mangani 2007). In addition, the identity of the host lipid and the identity and concentration of lipid-like or hydrophobic additives may be explored to great effect. These additives can be mixed directly with the host lipid before combining them with the protein solution to form the LCP matrix. We found that cholesterol, an important component of biological membranes in mammalian cells, dramatically improves the size and the quality of β2AR-T4L and A2AR-T4L crystals grown in LCP (Cherezov et al. 2007; Hanson et al. 2008; Jaakola et al. 2008). To increase the crystal size, it was necessary to concentrate protein between approximately 50 and 70 mg ml−1. At higher protein concentration, the detergent content was too high, causing the LCP to transform into a lamellar phase (Misquitta & Caffrey 2003), which was detrimental to the crystal growth. Another important optimization parameter was the volume of the lipidic mesophase bolus, which reproducibly improved the final crystal size for β2AR-T4L when decreased from 50 to 20 nl. This phenomenon is likely related to the differences in equilibration times after adding precipitant to the lipidic mesophase, which can range from hours to days depending on the volume of the mesophase bolus and the size of the precipitant molecules. Even after extensive optimization, the in meso crystals were still rather small by typical crystallography standards (figure 1d), which could be explained by the relatively low stability of GPCRs. Obtaining the most diffraction information from such small crystals required using a high-intensity X-ray beam with a diameter less than 10 µm.
For crystallographic data collection the microcrystals must first be harvested from the LCP. The glass sandwich plates were initially developed for high throughput screening and optimization (Cherezov et al. 2004). The wells in these plates were tightly sealed with two glass slides complicating the opening of individual wells for harvesting. Conditions optimized in the glass sandwich plates have been successfully translated into commercial plastic minibatch or vapour diffusion plates to grow harvestable crystals of light harvesting complex II (Cherezov et al. 2006a), outer membrane vitamin B12 transporter BtuB (Cherezov et al. 2006b) and outer membrane adhesion OpcA (Cherezov et al. 2008). However, after performing extensive GPCR crystallization trials in plastic trays, we were not able to obtain similar size and quality crystals as those grown in the glass sandwich plates. The inability to translate the crystal growth conditions into a more traditional plate format triggered the development of a procedure for harvesting crystals directly from the glass sandwich screening plates. Each well containing crystals was accessed by first scoring and removing a small square piece of the cover glass followed by harvesting of the crystals directly from the 20 nl cubic phase bolus. Thus, the utility of the glass sandwich plates was extended and currently all crystals are directly harvested from these plates without the need to translate conditions saving time and protein resources. It has previously been demonstrated that crystals can be released from the LCP gel for harvesting using an enzymatic lipid digestion (Nollert & Landau 1998), detergent solutions (Luecke et al. 1999), sponge inducing additives (Cherezov et al. 2006a) or mineral oils. We observed that in the case of GPCR crystals any of these perturbations decreased the diffraction quality of the harvested crystals. Moreover, it was easier to harvest colourless microcrystals when they were embedded in the LCP gel, rather than free floating in solution. Another benefit of harvesting directly from the LCP was that lipids surrounding the crystal provide additional cryoprotection during the freezing in liquid nitrogen. One major drawback of this approach is that upon freezing, the lipidic mesophase turns opaque rendering the embedded crystals invisible, which complicates data collection (see §3.3). Furthermore, the lipids surrounding the crystal contribute to the background scattering, thus minimizing the amount of mesophase harvested with the crystal is essential for optimal diffraction. We prefer to use MiTeGen MicroMounts (www.mitegen.com) for harvesting as they are thin but sufficiently rigid to easily penetrate the LCP allowing a minimal amount of extraneous lipid when the size of the MicroMount appropriately matches the size of the crystal (figure 4b).
Figure 4.

Images of loops with harvested β2AR-T4L crystals under a cryo-stream recorded through the inline optics at the GM/CA beamline at APS. (a) A nylon loop with several optically invisible β2AR-T4L crystals embedded into a frozen lipidic mesophase. Scanning the loop with the minibeam revealed a distinct protein diffraction at locations marked with the black circles. (b) A MiTeGen MicroMount containing a small amount of lipidic mesophase with a single β2AR-T4L crystal. The crystal is optically invisible but can be easily located using the automated rastering procedure implemented at the GM/CA beamlines. Scale bars, 50 µm.
3.2. Minibeam development
Approaches to developing microbeams for macromolecular crystallography fall in two major categories. Beamline ID23-2 of the European Synchrotron Source uses short focal length (approx. 1 m) mirrors arranged in Kirkpatrick–Beaz (KB) geometry positioned close to the sample to focus the beam to the desired size (approx. 7 µm). Any movement of the source or mirrors can result in the beam position drifting at the sample position.
The GM/CA CAT beamlines at the APS use long focal length mirrors (approx. 5–8 m) also arranged in a KB geometry to focus the beam to 20–25 × 65–120 µm2, depending on the particular beamline. A minibeam collimator positions a pin-hole aperture 30 mm upstream of the sample position which defines a 5 or 10 µm beam at the sample position. Experiments prior to February 2008 used single minibeam collimators with either 5 or 10 µm apertures. Some data reported in this paper used a 10 µm pin hole that provided a 7.8 × 6.3 µm2 (FWHM) beam with an intensity of 1 × 1011 photons s−1 (100 mA)−1 (Sanishvili et al. 2008). Later experiments used a triple minibeam collimator to position any one of three, user selectable, beam defining, pin-hole apertures (Fischetti et al. 2009). The collimator houses 5 and 10 µm beam defining apertures and a 300 µm scatter guard. The 10 µm pin hole provided a 10.6 × 11.6 µm2 beam with an intensity of 2.2 × 1011 photons s−1 (100 mA)−1. This later pin hole had a slightly larger diameter than the previous one accounting for the increased beam size and intensity. The minibeams at GM/CA have Gaussian profiles and virtually no tails. The mirrors intercept the full cone of emission from the undulator source on both beamlines providing a high-intensity focused beam at the sample position. A high level of positional and intensity stability of the beam is achieved because of the focused beam being significantly larger than the small pin holes (Fischetti et al. 2009). The long focal length mirrors provide a beam with low convergence, which is further reduced by the pin-hole apertures.
Beamline X06SA of the Swiss Light Source combines the two approaches such that the beam is focused to the desired size in the vertical direction and defined in the horizontal direction using the beam shaper of the MD-2 micro-diffractometer (Perrakis et al. 1999; Cipriani et al. 2007).
3.3. Loop rastering, crystal location and centring
GPCR microcrystals, harvested directly from the LCP and frozen in liquid nitrogen, were invisible through the beamline optics (figure 4). To locate the crystals in the loops and precisely align them with the 10 µm minibeam, we have developed an approach using diffraction from a highly attenuated beam of variable size as a probe (see §2.4). Applying this procedure manually was rather tedious taking 30–60 min to align each crystal. This inconvenience prompted development and implementation of an automatic rastering utility, which was fully integrated in the beamline controlling software (Blu-Ice; figure 5). With this routine, the crystal alignment time decreased to approximately 10 min, significantly increasing the throughput of screening for optimal diffraction.
Figure 5.

A screenshot of the automated rastering interface window in Blu-Ice. After centring the loop at the crosshair, the user defines a grid for rastering, selects exposure parameters at each cell of the grid and starts the scan. Collected diffraction frames are processed in real time using the program DISTL (Zhang et al. 2006) and the results are displayed in the window at the lower part of the screen.
3.4. Radiation damage
Collecting high-resolution data from small microcrystals requires exposing them to a higher X-ray dose, inevitably leading to increased radiation damage. It was suggested that a twofold decrease in diffraction intensity during data collection owing to the radiation damage is tolerable (Henderson 1990; Owen et al. 2006; Holton 2009). Such decay can be partially compensated for computationally (Diederichs 2006) and typically will not introduce significant errors in the electron density. Henderson estimated the limiting X-ray dose that destroys half of the diffraction intensity in any protein crystal as 20 MGy (Henderson 1990). Recently, more comprehensive studies on seven approximately 200 µm crystals extended this number to 43 MGy and suggested an upper dose limit of 30 MGy for data collection (Owen et al. 2006). These estimates should, of course, be followed with caution as different crystals may have different sensitivities to the damage (Holton 2009). In the case of micrometre-size crystals, theoretical calculations predict that the radiation damage could be significantly lower owing to an escape of the secondary electrons from the crystal (Nave & Hill 2005); a systematic experimental study to test these predictions is currently under way (R. F. Fischetti and R. Sanishvili 2009, unpublished data).
We measured the decrease in the diffracted intensity in different resolution shells owing to radiation damage for a small microcrystal of A2AR-T4L (40 × 15 × 3 µm3; figure 6). As expected, the higher resolution reflections decay faster, making it increasingly more difficult to collect higher resolution data from microcrystals. Several authors estimated the minimum size of the crystal, from which it is possible to collect a full dataset, as approximately 20–30 µm in all three dimensions (Glaeser et al. 2000; Teng & Moffat 2000; Sliz et al. 2003). As all our crystals were on average of smaller size, we had to use multiple crystals to collect a complete dataset. While working with the in meso grown β2AR-T4L microcrystals of typical size 25 × 8 × 4 µm3, we empirically established the following correlations between the beam intensity, the amount of collected data and the achieved maximal resolution: using 1 s exposures with 20-fold attenuated beam and 1° oscillation allowed one to collect approximately 200° of data with resolution of approximately 5–6 Å, corresponding to a total absorbed X-ray dose of approximately 10 MGy (calculated with the RADDOSE program; Murray et al. 2004); using 1 s exposures with fivefold attenuation and 1° oscillation allowed one to collect 40° with resolution approximately 3–3.5 Å (total absorbed X-ray dose of approximately 8 MGy); using 1 s exposures without attenuation and 1° oscillation allowed one to collect 7° with resolution approximately 2.5 Å (total absorbed X-ray dose approx. 7 MGy); and, finally, using 5 s exposures without attenuation and 1° oscillation allowed one to collect just one frame with resolution approximately 2.2 Å (total absorbed X-ray dose approx. 5 MGy). The actual resolution would of course vary from crystal to crystal and the final determination for useable resolution and amount of data was ultimately based on scaling statistics after incorporation of each wedge of data into the main dataset.
Figure 6.
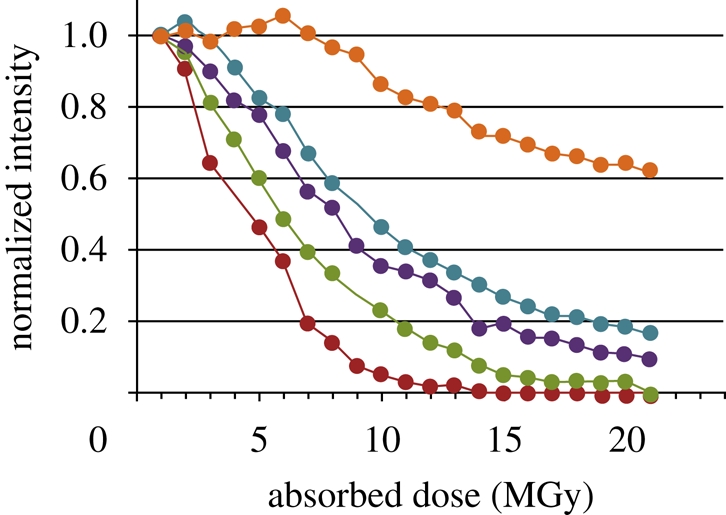
Effect of radiation damage on the intensity of diffraction spots collected in different resolution shells (identified as different colours) from a crystal of A2AR-T4L. The crystal was exposed multiple times at the same orientation with the unattenuated minibeam using the 1° oscillation and 1 s exposure per frame. Each frame corresponds to an estimated radiation dose of 1 MGy absorbed by the crystal (the dose was calculated using the program RADDOSE; Murray et al. 2004). The crystal lost half of its diffraction intensity at 3 Å resolution after absorbing approximately 5 MGy. Red line, 3–3.1 Å; green line, 4–4.2 Å; violet line, 6–6.5 Å; blue line, 7.5–8 Å; orange line, 9–12 Å.
3.5. Data collection strategies
Because radiation damage limits the collection of full datasets from small crystals, one has to carefully design the data collection strategy. Several strategies have been recently applied to mitigate the effect of radiation damage on data collected from crystals with small volumes (Coulibaly et al. 2007; Moukhametzianov et al. 2008). The strategy applied here combined a low-resolution (4–5 Å) full dataset recorded from a single crystal with high-resolution (2.2–2.8 Å) wedges collected from multiple crystals. The low-resolution data served as a reference set for scaling the small wedges of high-resolution data. When enough wedges were collected to assemble a complete dataset, the low-resolution set was removed from the final data.
To optimize the collection of high-resolution data, we first estimated the maximum obtainable diffraction limit. Although the best crystals diffracted to about 2.0 Å or better using 5–10 s exposure with unattenuated beam, the majority of them fell in the 2.0–2.5 Å resolution range. Therefore, we took a conservative approach of taking 1 s exposures per frame, which allowed us to collect 10–15 usable frames with 1° oscillation from each crystal. Merging data from multiple crystals relies on the crystal isomorphism. The in meso grown GPCR crystals did provide such isomorphism in contrast to often highly non-isomorphic crystals of membrane proteins grown in detergent solutions (Carpenter et al. 2008). While it is possible to merge just single frames from hundreds of crystals (Grimes et al. 1998), the data quality certainly suffers as the number of frames collected from each crystal decreases. We have found empirically that 10–15 frames per crystal provided the best compromise between data quality and resolution for the type and size of crystals we were using. Initially, the high-resolution data wedges were collected starting from the same loop orientation assuming random orientation of crystals in the loop. This approach worked well with β2AR-T4L crystals. However, in the case of A2AR-T4L this resulted in systematic omission of reflections at certain orientations. Using the strategy to select the starting angles for the high-resolution wedges as implemented in XDS (Kabsch 1993) helped to collect a complete dataset. In order to minimize the radiation damage from the X-ray exposure during this procedure, we attenuated the beam 10-fold and based our prediction on diffraction data at a maximal resolution of 4–5 Å. We found that collecting an initial wedge of 5–10 frames resulted in a more reliable indexing of the low-resolution diffraction images allowing an assignment of the orientation matrix and prediction of the best rotation angle to add completeness to the high-resolution dataset.
The data collected from multiple crystals were processed and scaled with XDS (Kabsch 1993). XDS can account for the radiation damage (Diederichs 2006) and compensate for the movement of the crystal out of the beam due to small deviations of the rotation axis from the crystal centre. The final data collection and scaling statistics are shown in table 1. Data from 27 crystals were merged together to obtain the final dataset for β2AR-T4L/carazolol structure. For β2AR(E122W)-T4L/timolol and A2AR-T4L/ZM241385, 11 and 13 crystals were used, respectively. Fewer crystals were required in the last two cases because of the higher symmetry space group and lower resolution. In practice, at least two times more data were collected in each case. The high-resolution data wedges that did not scale well with the low-resolution set, representing less than 20 per cent of all crystals, were discarded. The best combinations of the high-resolution data providing the lowest Rmerge and the highest completeness were selected to represent the final merged data. The quality of the final data was very good with the total Rmerge ranging between 10 and 15 per cent.
Table 1.
Data collection statistics (data for the highest resolution shell are shown in parentheses).
| protein | β2AR-T4L | β2AR(E122W)-T4L | A2AR-T4L |
| ligand | carazolol | timolol | ZM241385 |
| PDB ID | 2RH1 | 3D4S | 3EML |
| space group | C2 | P212121 | P21 |
| number of crystals | 27 | 11 | 13 |
| cell dimensions | |||
| a, b, c (Å) | 106.3, 169.2, 40.2 | 40.0, 75.7, 172.7 | 47.7, 76.9, 86.6 |
| α, β, γ (°) | 90, 105.6, 90 | 90, 90, 90 | 90, 101.3, 90 |
| total reflections | 245 571 | 54 405 | 64 526 |
| unique reflections | 26 574 | 12 782 | 18 465 |
| resolution (Å) | 50−2.4 (2.5−2.4) | 50−2.8 (3.0−2.8) | 20−2.6 (2.8−2.6) |
| Rmerge | 12.7 (67.8) | 14.3 (56.9) | 9.8 (38.9) |
| I/σ | 9.6 (2.2) | 6.9 (1.9) | 7.0 (2.3) |
| redundancy | 9.4 (4.8) | 4.2 (2.9) | 3.5 (2.3) |
| completeness (%) | 99.5 (99.1) | 94.0 (91.0) | 96.8 (93.9) |
3.6. Summary of G protein-coupled receptor structure results
Structure of β2AR-T4L bound to diffusible ligand carazolol was solved by molecular replacement (Cherezov et al. 2007). Initial solution was found by Phaser (McCoy 2007) using a polyalanine model of seven TM helices of rhodopsin (PDB ID 1U19) together with a model of T4 lysozyme. Subsequent structures of β2AR(E122W)-T4L/timolol (Hanson et al. 2008) and A2AR-T4L/ZM241385 (Jaakola et al. 2008) were also solved by molecular replacement using separated structures of β2AR and T4L from the first β2AR-T4L/carazolol structure (PDB ID 2RH1) as the search models.
All three structures are composed of a seven TM bundle and a short helix 8 running along the membrane at the cytoplasmic side (figure 7). Structural alignments of the rhodopsin, β2AR and A2AR backbones reveal slight shifts, rotations and twists between the corresponding helices, resulting in mutual displacements ranging between 1 and 7 Å depending on the helix and on the position of the residue in the helix. Rotations and twists occur mostly around conserved proline residues. The most divergent part of the seven TM bundle is at the extracellular side, around the ligand binding site. In contrast to rhodopsin, in which the N-terminus together with the second extracellular loop (ECL2) form a four-stranded beta-sheet, closing access to the ligand from the extracellular side, in both β2AR-T4L and A2AR-T4L the N-terminus is disordered and the ECL2 is shaped with disulphide bonds (two disulphides in β2AR-T4L and four disulphides in A2AR-T4L) so that the passage to the ligand binding site is open for diffusible ligand. The ligand binding sites and interactions of ligands with receptors' residues are well defined in all solved structures. Binding modes of β2AR antagonists carazolol and timolol are similar to each other, and their orientations and locations with respect to the receptors resemble those of retinal in rhodopsin. The A2AR antagonist ZM241385, however, binds in an unexpected, orthogonal to the membrane plane orientation. In both β2AR and A2AR, the ligands substantially interact with residues on ECL2. Loop conformations and thus these interactions are difficult to predict.
Figure 7.
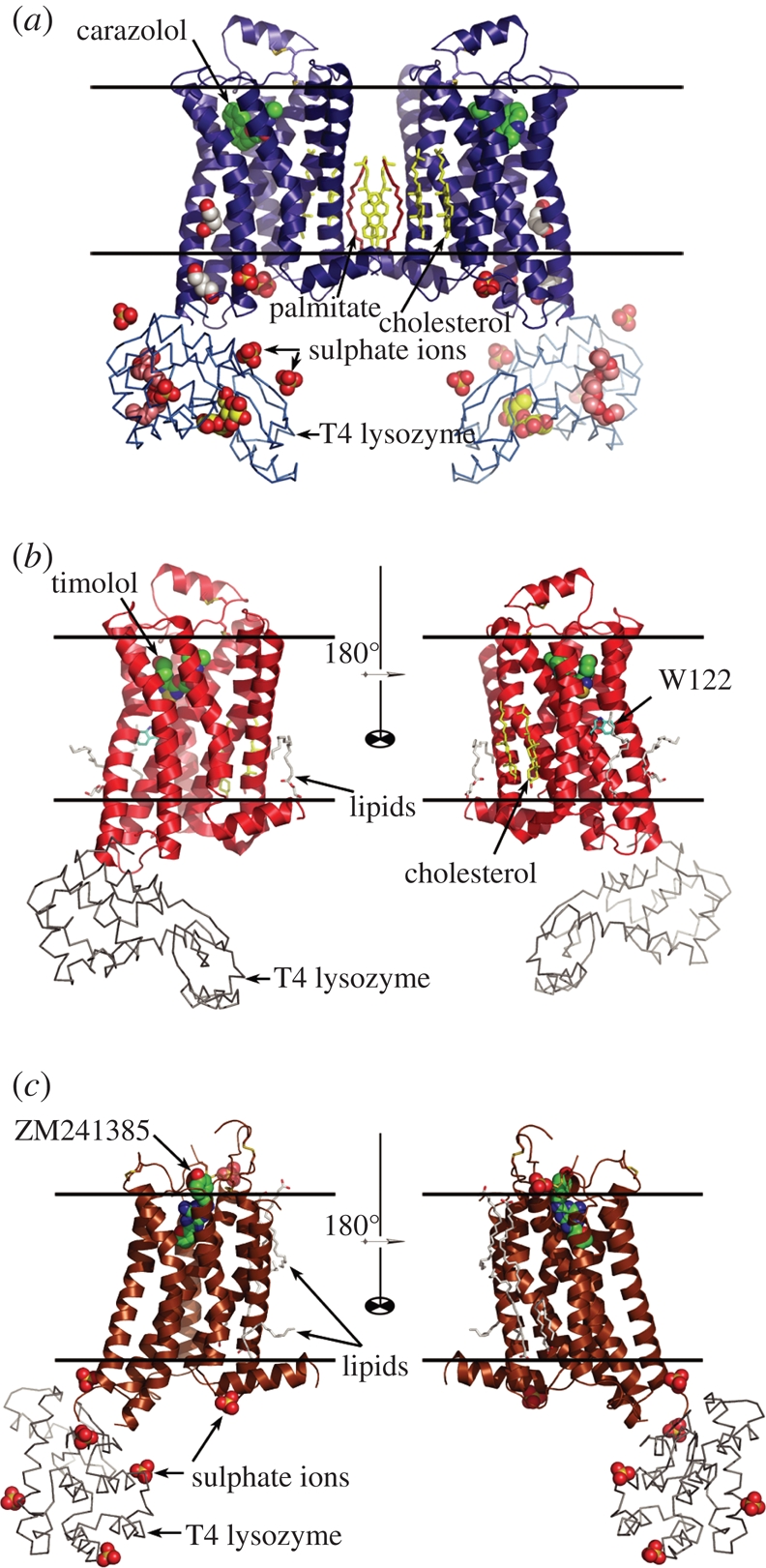
Crystal structures of (a) β2AR-T4L bound to carazolol (PDB ID 2RH1; Cherezov et al. 2007), (b) β2AR(E122W)-T4L bound to timolol (PDB ID 3D4S; Hanson et al. 2008) and (c) A2AR-T4L bound to ZM241385 (PDB ID 3EML; Jaakola et al. 2008). β2AR-T4L/carazolol crystallized as a parallel crystallographic dimer as shown in (a). Both β2AR(E122W)-T4L/timolol and A2AR-T4L/ZM241385 crystallized as monomers, and, therefore, two monomers rotated 180° around the vertical axis are shown in (b) and (c). Approximate locations of the lipid membrane boundaries are shown with two parallel black lines. Receptors are oriented with their extracellular side facing the top of the figure.
In addition to establishing high-resolution snapshots of two GPCRs bound to diffusible antagonists, the new structures are invaluable in providing high-quality templates for generating models of closely related receptors suitable for virtual ligand screening (Katritch et al. 2009; Reynolds et al. 2009). The work on expanding the coverage of the diverse GPCR structural space, as well as pursuit towards the activated receptor state, is in progress.
4. Conclusions
Technology developments in membrane protein crystallization and synchrotron microcrystallography combined with advancements in protein stabilization and purification have enabled structural determination of two members of the pharmaceutically important and highly challenging family of GPCRs. Highly ordered, yet small (<1000 µm3), microcrystals of engineered human β2 adrenergic and adenosine A2A receptors bound to diffusible ligands were obtained through utilization of high-throughput robotic crystallization techniques in lipidic mesophases. Development of the minibeam instrumentation and rastering software by the GM/CA CAT at the Advance Photon Source allowed for high-quality crystallographic data collection from multiple GPCR microcrystals. Using the 10 µm minibeam substantially improved the signal-to-noise ratio of collected reflections and extended the resolution, when compared with conventional beams defined by slits. Data collection from microcrystals with the minibeam was limited by the radiation damage. Obtaining maximum structural information from available microcrystals at a minimal damage level required careful planning and optimization of data collection strategies. Further decrease in the synchrotron beam size may provide intriguing opportunities for improving data quality and mitigating radiation damage.
Acknowledgements
This work was supported in part by the NIH Roadmap Initiative grant P50 GM073197 and Protein Structure Initiative grant U54 GM074961. The authors acknowledge D. Rosenbaum and B. Kobilka from Stanford University for providing initial β2AR-T4L samples, crystals of which were used to develop some of the approaches described in this paper, as well as Y. Zheng, Ohio State University, and M. Caffrey, University of Limerick, for the generous loan of the in meso robot (built with support from the NIH (GM075915), the NSF (IIS0308078) and Science Foundation Ireland (02-IN1-B266)). The GM/CA CAT beamline (23-ID) is supported by the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104).
Footnotes
One contribution of 13 to a Theme Supplement ‘Biological physics at large facilities’.
References
- Adams P. D., et al. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954. ( 10.1107/S0907444902016657) [DOI] [PubMed] [Google Scholar]
- Benvenuti M., Mangani S. 2007. Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat. Protoc. 2, 1633–1651. ( 10.1038/nprot.2007.198) [DOI] [PubMed] [Google Scholar]
- Caffrey M. 2003. Membrane protein crystallization. J. Struct. Biol. 142, 108–132. ( 10.1016/S1047-8477(03)00043-1) [DOI] [PubMed] [Google Scholar]
- Caffrey M. 2009. Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Annu. Rev. Biophys. 38, 29–51. ( 10.1146/annurev.biophys.050708.133655) [DOI] [PubMed] [Google Scholar]
- Carpenter E. P., Beis K., Cameron A. D., Iwata S. 2008. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 18, 581–586. ( 10.1016/j.sbi.2008.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A., Hummel B., Qiu H., Caffrey M. 1998. A simple mechanical mixer for small viscous lipid-containing samples. Chem. Phys. Lipids 95, 11–21. ( 10.1016/S0009-3084(98)00060-7) [DOI] [PubMed] [Google Scholar]
- Cherezov V., Peddi A., Muthusubramaniam L., Zheng Y. F., Caffrey M. 2004. A robotic system for crystallizing membrane and soluble proteins in lipidic mesophases. Acta Crystallogr. D Biol. Crystallogr. 60, 1795–1807. ( 10.1107/S0907444904019109) [DOI] [PubMed] [Google Scholar]
- Cherezov V., Clogston J., Papiz M. Z., Caffrey M. 2006a. Room to move: crystallizing membrane proteins in swollen lipidic mesophases. J. Mol. Biol. 357, 1605–1618. ( 10.1016/j.jmb.2006.01.049) [DOI] [PubMed] [Google Scholar]
- Cherezov V., Yamashita E., Liu W., Zhalnina M., Cramer W. A., Caffrey M. 2006b. In meso structure of the cobalamin transporter, BtuB, at 1.95 Å resolution. J. Mol. Biol. 364, 716–734. ( 10.1016/j.jmb.2006.09.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V., et al. 2007. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265. ( 10.1126/science.1150577) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V., Liu W., Derrick J. P., Luan B., Aksimentiev A., Katritch V., Caffrey M. 2008. In meso crystal structure and docking simulations suggest an alternative proteoglycan binding site in the OpcA outer membrane adhesion. Proteins 71, 24–34. ( 10.1002/prot.21841) [DOI] [PubMed] [Google Scholar]
- Cipriani F., Felisaz F., Lavault B., Brockhauser S., Ravelli R., Launer L., Leonard G., Renier M. 2007. 9th Int. Conf. Synchrotron Radiation Instrumentation (eds Choi J.-Y., Rah S.), pp. 1928–1931. New York, NY: American Institute of Physics. [Google Scholar]
- Coulibaly F., Chiu E., Ikeda K., Gutmann S., Haebel P. W., Schulze-Briese C., Mori H., Metcalf P. 2007. The molecular organization of cypovirus polyhedral. Nature 446, 97–101. ( 10.1038/nature05628) [DOI] [PubMed] [Google Scholar]
- Diederichs K. 2006. Some aspects of quantitative analysis and correction of radiation damage. Acta Crystallogr. D Biol. Crystallogr. 62, 96–101. ( 10.1107/S0907444905031537) [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. ( 10.1107/S0907444904019158) [DOI] [PubMed] [Google Scholar]
- Fischetti R. F., et al. 2009. Mini-beam collimator enables microcrystallography experiments on standard beamlines. J. Synchrotron Radiat. 16, 217–225. ( 10.1107/S0909049508040612) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser R., Facciotti M., Walian P., Rouhani S., Holton J., MacDowell A., Celestre R., Cambie D., Padmore H. 2000. Characterization of conditions required for X-ray diffraction experiments with protein microcrystals. Biophys. J. 78, 3178–3185. ( 10.1016/S0006-3495(00)76854-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes J. M., Burroughs J. N., Gouet P., Diprose J. M., Malby R., Zientara S., Mertens P. P., Stuart D. I. 1998. The atomic structure of the bluetongue virus core. Nature 395, 470–478. ( 10.1038/26694) [DOI] [PubMed] [Google Scholar]
- Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V. P., Chien E. Y., Velasquez J., Kuhn P., Stevens R. C. 2008. A specific cholesterol binding site is established by the 2.8 Å structure of the human beta(2)-adrenergic receptor. Structure 16, 897–905. ( 10.1016/j.str.2008.05.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R. 1990. Cryo-protection of protein crystals against radiation damage in electron and X-ray diffraction. Proc. R. Soc. Lond. B 241, 6–8. ( 10.1098/rspb.1990.0057) [DOI] [Google Scholar]
- Holton J. M. 2009. A beginner's guide to radiation damage. J. Synchrotron Radiat. 16, 133–142. ( 10.1107/S0909049509004361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V. P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y., Lane J. R., Ijzerman A. P., Stevens R. C. 2008. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217. ( 10.1126/science.1164772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26, 795–800. ( 10.1107/S0021889893005588) [DOI] [Google Scholar]
- Katritch V., Cherezov V., Hanson M. A., Roth R. B., Yeager M., Abagyan R. 2009. Analysis of full and partial agonists binding to beta(2)-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J. Mol. Recognit. 22, 307–318. ( 10.1002/jmr.949) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau E. M., Rosenbusch J. P. 1996. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl Acad. Sci. USA 93, 14 532–14 535. ( 10.1073/pnas.93.25.14532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke H., Schobert B., Richter H. T., Cartailler J. P., Lanyi J. K. 1999. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291, 899–911. ( 10.1006/jmbi.1999.3027) [DOI] [PubMed] [Google Scholar]
- McCoy A. J. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41. ( 10.1107/S0907444906045975) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips T. M., et al. 2002. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406. ( 10.1107/S0909049502015170) [DOI] [PubMed] [Google Scholar]
- Misquitta Y., Caffrey M. 2003. Detergents destabilize the cubic phase of monoolein: implications for membrane protein crystallization. Biophys. J. 85, 3084–3096. ( 10.1016/S0006-3495(03)74727-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moukhametzianov R., Burghammer M., Edwards P. C., Petitdemange S., Popov D., Fransen M., McMullan G., Schertler G. F., Riekel C. 2008. Protein crystallography with a micrometre-sized synchrotron-radiation beam. Acta Crystallogr. D Biol. Crystallogr. 64, 158–166. ( 10.1107/S090744490705812X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. W., Garman E. F., Ravelli R. B. G. 2004. X-ray absorption by macromolecular crystals: the effects of wavelength and crystal composition on absorbed dose. J. Appl. Cryst. 37, 513–522. ( 10.1107/S0021889804010660) [DOI] [Google Scholar]
- Murshudov G. N., Vagin A. A., Dodson E. J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255. ( 10.1107/S0907444996012255) [DOI] [PubMed] [Google Scholar]
- Nave C., Hill M. A. 2005. Will reduced radiation damage occur with very small crystals? J. Synchrotron Radiat. 12, 299–303. ( 10.1107/S0909049505003274) [DOI] [PubMed] [Google Scholar]
- Nollert P., Landau E. M. 1998. Enzymic release of crystals from lipidic cubic phases. Biochem. Soc. Trans. 26, 709–713. [DOI] [PubMed] [Google Scholar]
- Owen R. L., Rudino-Pinera E., Garman E. F. 2006. Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc. Natl Acad. Sci. USA 103, 4912–4917. ( 10.1073/pnas.0600973103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebay-Peyroula E., Rummel G., Rosenbusch J. P., Landau E. M. 1997. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 277, 1676–1681. ( 10.1126/science.277.5332.1676) [DOI] [PubMed] [Google Scholar]
- Perrakis A., Cipriani F., Castagna J.-C., Claustre L., Burghammer M., Riekel C., Cusack S. 1999. Protein microcrystals and the design of a microdiffractometer: current experience and plans at EMBL and ESRF/ID13. Acta Crystallogr. D Biol. Crystallogr. 55, 1765–1770. ( 10.1107/S0907444999009348) [DOI] [PubMed] [Google Scholar]
- Rasmussen S. G., et al. 2007. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383–387. ( 10.1038/nature06325) [DOI] [PubMed] [Google Scholar]
- Reynolds K. A., Katritch V., Abagyan R. 2009. Identifying conformational changes of the beta(2) adrenoceptor that enable accurate prediction of ligand/receptor interactions and screening for GPCR modulators. J. Comput. Aided Mol. Des. 23, 273–288. ( 10.1007/s10822-008-9257-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M., et al. 2007. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273. ( 10.1126/science.1150609) [DOI] [PubMed] [Google Scholar]
- Roth C. B., Hanson M. A., Stevens R. C. 2008. Stabilization of the human beta2-adrenergic receptor TM4–TM3–TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. J. Mol. Biol. 376, 1305–1319. ( 10.1016/j.jmb.2007.12.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanishvili R., Nagarajan V., Yoder D., Becker M., Xu S., Corcoran S., Akey D. L., Smith J. L., Fischetti R. F. 2008. A 7 microm mini-beam improves diffraction data from small or imperfect crystals of macromolecules. Acta Crystallogr. D Biol. Crystallogr. 64, 425–435. ( 10.1107/S0907444908001741) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliz P., Harrison S. C., Rosenbaum G. 2003. How does radiation damage in protein crystals depend on X-ray dose? Structure 11, 13–19. ( 10.1016/S0969-2126(02)00910-3) [DOI] [PubMed] [Google Scholar]
- Song J., Mathew D., Jacob S. A., Corbett L., Moorhead P., Soltis S. M. 2007. Diffraction-based automated crystal centering. J. Synchrotron Radiat. 14, 191–195. ( 10.1107/S0909049507004803) [DOI] [PubMed] [Google Scholar]
- Stepanov S., Makarov O., Urakhchin A., Devarapalli S., Yoder D., Fischetti R. 2006. GM/CA CAT beamline control system for protein crystallography at the APS. In NOBUGS-2006 Conf. Program & Abstracts, LBL, Berkeley, CA, 2–4 October 2006, pp. 31–32. [Google Scholar]
- Teng T. Y., Moffat K. 2000. Primary radiation damage of protein crystals by an intense synchrotron X-ray beam. J. Synchrotron Radiat. 7, 313–317. ( 10.1107/S0909049500008694) [DOI] [PubMed] [Google Scholar]
- Xiao B., et al. 2003. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc. Natl Acad. Sci. USA 100, 2363–2368. ( 10.1073/pnas.0436813100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Sauter N. K., van den Bedem H., Snell G., Deacon A. M. 2006. Automated diffraction image analysis and spot searching for high-throughput crystal screening. J. Appl. Cryst. 39, 112–119. ( 10.1107/S0021889805040677) [DOI] [Google Scholar]