

Abstract
Background
Reactive oxygen species (ROS) are generated by cellular metabolism as well as by exogenous agents. While ROS can promote cellular senescence, they can also act as signaling molecules for processes that do not lead to senescence. Telomere homolog oligonucleotides (T-oligos) induce adaptive DNA damage responses including increased DNA repair capacity and these effects are mediated, at least in part, through p53.
Objective
Studies were undertaken to determine whether such p53-mediated protective responses include enhanced antioxidant defenses.
Methods
Normal human fibroblasts as well as R2F fibroblasts expressing wild type or dominant negative p53 were treated with an 11-base T-oligo, a complementary control oligo or diluents alone and then examined by western blot analysis, immunofluorescence microscopy and various biochemical assays.
Results
We now report that T-oligo increases the level of the antioxidant enzymes superoxide dismutase 1 and 2 and protects cells from oxidative damage; and that telomere-based γH2AX (DNA damage) foci that form in response to T-oligos contain phosphorylated ATM and Chk2, proteins known to activate p53 and to mediate cell cycle arrest in response to oxidative stress. Further, T-oligo increases cellular ROS levels via a p53-dependent pathway, and these increases are abrogated by the NAD(P)H oxidase inhibitor diphenyliodonium chloride.
Conclusion
These results suggest the existence of innate telomere-based protective responses that act to reduce oxidative damage to cells. T-oligo treatment induces the same responses and offers a new model for studying intracellular ROS signaling and the relationships between DNA damage, ROS, oxidative stress, and cellular defense mechanisms.
Introduction
Human telomeres, tandem repeats of the sequence TTAGGG and its complement that cap the ends of chromosomes[1], play important roles in DNA damage responses[2–4] and aging[5, 6]. Telomeres exist in a loop structure that is stabilized by telomeric repeat binding factor 2 (TRF2) [7]. Disruption of the loop by a dominant negative construct (TRF2DN)2 leads to apoptosis of certain mammalian cells[8] and senescence of others[9], a process mediated at least in part through ATM and p53 activation[8], suggesting that telomere loop disruption initiates a DNA damage signal.
Interestingly, provision of telomere TTAGGG homolog oligonucleotides (T-oligos), known to rapidly accumulate in the nucleus[10–12], also stimulates DNA damage signals and adaptive responses mediated, while control oligonucleotides complementary or unrelated to the TTAGGG repeat sequence do not[10, 13–16]. Specifically, we have shown that exposure of fibroblasts to T-oligos leads to dose-dependent DNA damage responses, such as increased DNA damage repair capacity[17, 18], S-phase cell cycle arrest, apoptosis[10–12] and senescence[14, 15], mediated at least in part through ATM and p53[13–15, 19]. These cellular responses occur without affecting the cells’ own telomeres[10, 14, 19] and are independent of telomerase[15, 20]. Most recently, these T-oligo-induced responses were shown to involve formation of DNA damage foci at the telomere via WRN[19], the helicase and exonuclease mutated in the cancer-prone progeroid Werner Syndrome[21, 22]. Furthermore, p53 is known to interact with WRN both in vivo and in vitro[23–25] and fibroblasts derived from individuals with Werner Syndrome display reduced p53-mediated apoptosis, restored by introducing wild type WRN into the cells, suggesting that WRN is involved in p53 activation[24].
High levels of ROS are procarcinogenic[26, 27] and can damage cellular proteins, lipids and DNA [Reviewed in[28–30]], and a network of antioxidant enzymes has evolved to decrease ROS levels that would otherwise damage cells [Reviewed in[31–33]. Antioxidant defense mechanisms include enzymes such as glutathione peroxidase (GPX) [Reviewed in[34, 35]], glutathione reductase [Reviewed in[36, 37]], copper and zinc-dependent superoxide dismutase (SOD)1[38–40], catalase [Reviewed in[41]], and manganese-dependent SOD2[38–40] that acts preferentially in the mitochondria.
Interestingly, after UV irradiation, a DNA damaging agent that leads to the formation of DNA photoproducts and ROS, the activities of the anti-oxidant enzymes GPX, SOD1 and particularly SOD2 are induced[42], suggesting an adaptive or protective response of fibroblasts to UV-induced oxidative DNA damage. Continuous exposure to the damaging agent precipitates the fibroblast response of stress-induced premature senescence (SIPS) [43], a response similar or identical to the induction of senescence following serial cell division with critical telomere shortening[44, 45], activation of tumor supressors such as p53[46] or overexpression of Ras[47] or Raf[48] oncogenes. Oxidative stress preferentially targets guanine (G) residues, leading to formation of 8-oxo-G[49], and telomeres are particularly sensitive to oxidative stress because of their high G content. ROS exposure is well-documented to cause telomere shortening and SIPS in fibroblasts[50].
Cellular ROS can be produced by enzymatic and non-enzymatic mechanisms[51]. ROS are generated in the mitochondria through the electron transport chain and in other electron transferring cellular systems, a non-enzymatic mechanism. In contrast, ROS are also generated by the plasma membrane-associated NAD(P)H oxidase (NOX), an enzyme complex with multiple components[52–54] and thought to have a regulatory role, stimulated by growth factors and cytokines[30, 55]. Although the cellular responses mediated by NOX-generated ROS are not completely understood, it is speculated that ROS generation may enhance cell survival through upregulation of anti-oxidant defense mechanisms[56]. Thus, it has been suggested that ROS such as superoxide and hydrogen peroxide (H2O2) are utilized by cells as signaling molecules for processes that do not necessarily lead to cellular senescence or result in detectable oxidative damage[56, 57]. Rather, they propagate signaling through tyrosine phosphorylation of effector proteins, mitogen activated protein (MAP) kinase activation, DNA synthesis, and chemotaxis[58, 59]. It is speculated that lower ROS levels lead to adaptive cellular responses, while higher levels result in senescence[33, 57, 60, 61]. It has also been reported that over-expression of p53 leads to cellular ROS elevation, as well as to transcription of redox-associated genes[62].
We now report that T-oligos, known to induce a variety of DNA-protective and anti-cancer responses, also induce the level of antioxidant enzymes, specifically SOD1 and SOD2. Furthermore, T-oligos protect fibroblasts against oxidative challenge by H2O2. Finally, T-oligos upregulate ROS levels, consistent with T-oligo induced ROS signaling, a process mediated by p53 and NAD(P)H activation.
Materials and Methods
Materials
Hydrogen peroxide (30% w/w, with 0.5 ppm stannate and 1 ppm phosphorus as preservatives) was obtained from Sigma (USP grade, St. Louis, MO). The stock solution was stored at 4°C and all dilutions were made in DMEM immediately before use. 2′,7′-dichlorodihydrofluorescein diacetate (DCF) from Molecular Probes, Inc., (Eugene, OR) was dissolved in DMSO to a stock concentration of 1 mg/ml and stored under nitrogen at −20°C. Propidium iodide (PI) was obtained from Sigma. Diphenyliodonium chloride (DPI) powder was obtained from A.G. Scientific, Inc. (San Diego, CA), dissolved in DMSO to a stock concentration of 5 mg/ml and frozen at −20°C until use. 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) was dissolved in dimethylformamide and added to a citric acid/NA phosphate buffer at pH 6.0 immediately before use for a final concentration of 1 mg/ml[63].
Purified 5′-phosphorylated oligonucleotides with phosphodiester linkages (Midland Certified Reagents, Inc., Midland, Texas) were obtained in lyophilized form, resuspended in sterile dH2O to generate a 2 mM stock solution and frozen in aliquots at −20°C as previously described[17]. The stock solution was further diluted to 40 μM in cell culture medium immediately before use.
Fibroblast cell culture
Normal newborn human dermal fibroblasts were cultured from foreskin specimens and maintained in DMEM supplemented with 10% calf serum (CS) as previously described[64]. In experiments using the DCF assay, cells from three different donors were combined.
R2F fibroblasts were a kind gift from Dr. J. Rheinwald (Harvard Medical School, Brigham and Women’s Hospital) The retroviral vector pL(p53DD)SN, that expresses a dominant-negative fragment of p53 (p53DN) [65, 66] was used to transfect newborn foreskin fibroblasts as described[67]. p53DN cells and isogenic wild-type p53 R2F cells were maintained in a 1:1 mixture of DMEM and Ham’s F12 medium and supplemented with 15% FBS.
Experimental Design
All telomere homolog phosphodiester-linked oligonucleotides (T-oligos) 2–20 bases in length tested to date have been demonstrated to induce genome-protective responses in human cells, with molar efficacy determined by the inter-related parameters of length, percent telomere homology, guanine (G) content and lack of cytosine (C) residues{Ohashi, 2007 #10}. Experiments reported here were performed with the 11-base 100% telomere homolog GTTAGGGTTAG (40 μM) or, in the case of the immunofluorescence microscopy studies, with a 16-base 100% homolog at half the concentration (20 μM), previously shown to be equipotent in standard T-oligo assays.
ROS induction and resistance to oxidative stress
To determine the effect of T-oligo on ROS induction, cells were stimulated once with GTTAGGGTTAG (T-oligo), the complementary control sequence CTAACCCTAAC (Cont-oligo) and/or diluent alone and were harvested at different intervals after treatment. Oligonucleotide concentrations were based on previous experiments establishing that these concentrations elicit DNA damage-like responses[17, 19, 68, 69]. To determine T-oligo protective effect, 3 × 105 fibroblasts were plated in two 100 mm dishes. Seventy-two hours after plating cells were stimulated with T-oligo (40 μM) or diluent for 72 hours. Cells were then harvested and replated at 1 × 105 cells/dish in 35 mm dishes. Twenty-four hours later, cells were treated with 25 μM fresh H2O2 or diluent for 1 hour and then were provided fresh DMEM supplemented with 10% CS. Cell numbers were determined up to 48 hours after medium change.
Dichlorofluorescein diacetate assay
DCF stock solution (1 mg/ml) was diluted in Hanks’ Balanced Salt Solution without phenol red (GIBCO Invitrogen, Carlsbad, CA) to a working concentration of 100 uM immediately before use[70] Fibroblasts were incubated for 30 minutes with 100 uM DCF solution, trypsinized and processed for FACScan analysis. The DPI stock solution was added directly to the DCF solution to achieve a final concentration of 50 μM[70]. Positive controls were fibroblasts exposed to 1 mM H2O2 for 15 minutes following the DCF incubation to rule out saturation of the DCF probe.
Western blot analysis
Total cellular proteins were harvested in RIPA buffer consisting of 0.25 M Tris HCl (pH 7.5), 0.375 M NaCl, 2.5% sodium deoxycholate, 1% Triton X-100, 25 mM MgCl2, 1 mM phenylmethyl sulfonyl fluoride, and 0.1 mg/ml aprotinin as described previously[71]. Protein concentrations were determined by the Bradford method and 35–65 ug protein/lane was separated over 10%–15% PAGE and processed for western blot analysis[71].
Membranes were probed with antibodies to SOD1 (1 ug/ml, BD Biosciences Franklin Lakes, NJ), SOD2 (1:1000 dilution, The Binding Site, Birmingham, UK); catalase (1:1000 dilution, Calbiochem, San Diego, CA) glutathione peroxidase (1:1000 dilution, Biodesign, Saco ME) and actin (1:1000 dilution, Santa Cruz Biotechnology, Santa Cruz, Ca) followed by the appropriate secondary antibodies diluted 1:2000. Antibody binding was detected with an ECL kit (NEN Life Science Products, Inc.) and exposure to XAR film (Eastman Kodak Co.).
Immunofluorescence microscopy
Normal neonatal fibroblasts were cultured on glass coverslips and always re-fed one day before treating with either 20 μM T-oligo (GTTAGGGTTAGGGTTA) or an equal volume of diluent (water). After two days, the cells were fixed and stained for γH2AX and either phosphoserine 1981-ATM or phosphothreonine 68-Chk2 using standard immunofluorescence protocols[72, 73]. γH2AX was detected using a mouse anti-γH2AX antibody (cataglog # ab18311, Abcam, Cambridge, MA) and a FITC-conjugated goat anti-mouse IgG (catalog# 115-095-146, Jackson Immunoresearch Laboratories, West Grove, PA). Phospho-ATM and phospho-Chk2 were detected using rabbit antibodies (phospho-ATM: catalog # ab2888, Abcam; phospho-Chk2: catalog # 2661, Cell Signaling Technology, Danvers, MA) and a Rhodamine Red-X-conjugated goat anti-rabbit IgG secondary antibody (catalog #111-295-144, Jackson Immunoresearch, West Grove, PA). Total nuclear DNA was stained with DAPI. The cells were examined under 1000× magnification using a Nikon Eclipse E400 microscope equipped with a RTke SPOT digital camera (Diagnostic Instruments, Inc.). FITC, TRITC and DAPI images were overlapped using the Advanced SPOT software.
Senescence-associated (SA) β-galactosidase assay
Cells were fixed with a 2% formaldehyde and 0.2% glutaraldehyde solution, washed, and stained overnight in X-gal solution[63]. The percentage of SA-β-galactosidase-positive cells was determined by counting four representative fields at 10× magnification (a minimum of 42 and up to 514 cells per field) while blinded to the identity of the treatment condition.
Statistical Analyses
FACScan results of the DCF assay were analyzed using Cellquest software (Becton-Dickinson, CA). The position on the X-axis of the peak of each fluorescence histogram plot was determined visually (using the software). The numbers at the peak for each condition were then compared using one-way ANOVA with Scheffe or LSD post-hoc analysis (SPSS 14.0). In studies examining T-oligo protection against oxidative stress, univariate analysis of variance was used with Bonferroni post-hoc analysis.
Results
T-oligo induces intracellular ROS
To determine T-oligo effect on intracellular ROS levels, fibroblasts were stimulated with T-oligo (40 μM) or diluent as a control, and ROS levels were determined at different intervals after stimulation (Figure 1A). T-oligo induced intracellular ROS as early as 36 hours and the effect was maximal at 72 hours when the experiment was terminated. To determine the optimal T-oligo dose for ROS induction, fibroblasts were stimulated with increasing T-oligo concentrations and intracellular ROS level was determined 72 hours after stimulation (Figure 1B). Maximal ROS induction was observed with T-oligo dose of 40 μM, the dose also found to be optimal for inducing other genome protective responses[10, 12–14, 16], and no additional effect was observed at a higher dose. To assure that the result was not due to DCF fluorescence saturation, fibroblasts were stimulated with T-oligo as above and paired cultures were stimulated with H2O2 (10 mM, Figure 1C). H2O2 induced a detectably higher ROS level than T-oligo, confirming that the assay was not saturated (Figure 1C). To assure that the T-oligo effect was specific, fibroblasts were stimulated with T-oligo (40 μM), diluent, or a complementary oligonucleotide (Cont-oligo, 40 μM) as an additional control (Figure 1D). Only T-oligo induced intracellular ROS levels (p<0.02, T-oligo vs Cont-oligo).
Figure 1. T-oligos stimulate intracellular ROS levels.




(A) Normal newborn human fibroblasts were stimulated with T-oligo or diluent and ROS levels were determined using the DCF assay[70, 74, 114, 115]. Within 36 hours intracellular ROS levels were induced only in T-oligo-treated cultures and the levels increased up to 72 hours when the experiment was terminated. One of five representative experiments is shown. (B) Fibroblasts were stimulated as above with increasing T-oligo concentrations and ROS levels were determined 72 hours after stimulation. Maximal induction of ROS was observed at T-oligo concentrations of ≥ 40 μM. One of two reproducible experiments is shown. (C) Fibroblasts were stimulated with T-oligo as above or with 1 mM H2O2 for 15 minutes to examine the possibility of DCF probe saturation. H2O2 increased fluorescent intensity beyond that of T-oligo-induced fluorescence, confirming that DCF fluorescence was not saturated. One of three reproducible experiments is shown. (D) Fibroblasts were stimulated with T-oligo (40 μM), Cont-oligo (40 μM) or diluent as above and ROS level was determined 72 hours after treatment. Only T-oligo induced intracellular ROS levels. One of five experiments with comparable results shown.
T-oligo-stimulated ROS production is p53-dependent
Because p53 mediates many T-oligo effects and can induce ROS accumulation[62] we investigated whether p53 is causally related to T-oligo-induced ROS increase in fibroblasts. p53DN R2F fibroblasts that lack p53 activity and normal p53 wild type (p53WT) isogenic R2F cells (positive control) were stimulated with T-oligo as above. Increased intracellular ROS levels were observed only in p53WT cells, but not in p53DN cells (Figure 2), strongly suggesting that ROS production in response to T-oligo is p53-dependent.
Figure 2. T-oligo induced ROS levels are p53-mediated.

p53DN mouse fibroblasts and isogenic p53 wild type cells were stimulated with T-oligo or diluent for 3 days and ROS levels were examined. Only p53WT cells displayed increased ROS levels, confirming that functional p53 is required for T-oligo-mediated ROS induction. (One of two representative experiments is shown.)
T-oligo stimulated ROS induction is NAD(P)H dependent
To determine if p53-dependent ROS production in fibroblasts is mediated, at least in part, by NAD(P)H oxidase activity, a specific NAD(P)H oxidase inhibitor, DPI[74, 75], was added to fibroblasts stimulated with T-oligo and ROS levels were determined. DPI treatment consistently abrogated the increase in ROS stimulated by T-oligo (Figure 3), suggesting that T-oligo-induced ROS production is mediated by NAD(P)H oxidase and further confirming the role of p53 in T-oligo-mediated ROS increases.
Figure 3. T-oligo-induced ROS production is NAD(P)H-dependent.
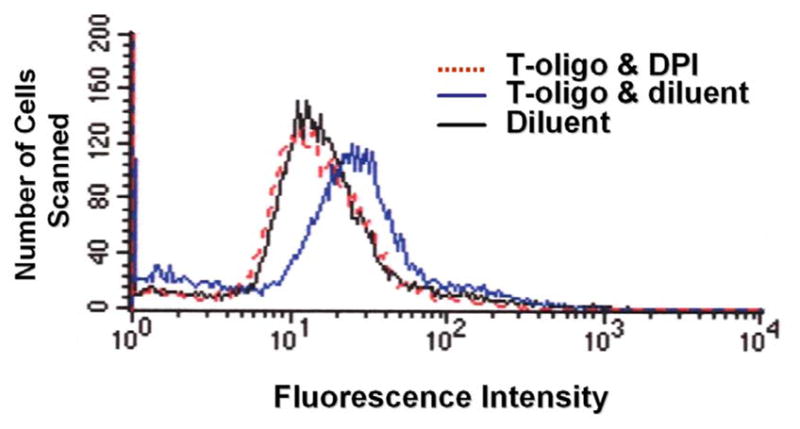
Fibroblasts were treated with T-oligo in the presence or absence of the specific NAD(P)H inhibitor DPI for 3 days as per Material and Methods and DCF fluorescence was examined. DPI, added to DCF assay medium, completely abrogated T-oligo mediated ROS induction, verifying that T-oligo induced ROS is NAD(P)H-dependent. (One of six experiments with comparable results is shown.)
T-oligo induction of ROS is not accompanied by fibroblast senescence
We have reported that treating fibroblasts with T-oligo (40uM) for 7 days induces senescence as indicated by positive staining for SA β-galactosidase in >60% of the cells, comparable to cultures rendered senescent by serial passage[14]. To determine the effect of briefer T-oligo exposure, sufficient to induce ROS production, fibroblasts were stimulated with T-oligo and the SA β-galactosidase assay was performed 72 hours after stimulation. Within this time frame, < 1 % of the cells were blue, and their number did not differ between T-oligo and diluent-treated dishes (data not shown), demonstrating that within the 72 hours required for ROS generation T-oligo does not induce fibroblast senescence.
T-oligo-induced γH2AX foci contain phosphorylated ATM and Chk2 proteins
ATM and its effector protein Chk2 play a role in the phosphorylation of p53 (serine 15) and activation of p53 in response to oxidative stress [76] and also in cell cycle arrest in response to oxidative stress[77, 78]. Because these proteins are recruited to γH2AX foci in cells experiencing DNA damage, for example in response to ionizing radiation (IR), we asked whether the γH2AX foci induced by T-oligo treatment also contained phosphorylated (activated) ATM and Chk2. Within 48 hours, fibroblasts treated once with T-oligo and then processed for immunofluorescent staining showed co-localization of phospho-ATM and phosphor-Chk2 with γH2AX (Figure 4). These data are consistent with WRN-T-oligo interactions[19] initiating a signaling casade that involves ATM, Chk2 and p53 and that serves to protect cells from oxidative stress.
Figure 4. T-oligo-induced γH2AX foci contain phospho-ATM and phospho-Chk2.

Normal neonatal fibroblasts were grown on glass coverslips and treated for 2 days with 20 μM T-oligo (GTTAGGGTTAGGGTTA). Cells were then fixed in paraformaldehyde and prepared for immunostaining as described in Experimental Procedures. Representative cells are shown. Top row: γH2AX. Middle row: phospho-ATM (left) or phospho Chk2 (right). Bottom row: merged images with DAPI nuclear stain.
T-oligo induces the level of antioxidant enzymes
Because we and others have shown that antioxidant enzymes are induced after oxidative DNA damage due to various agents[42, 79], we investigated whether T-oligo treatment also induces antioxidant enzyme levels.
Compared to diluent and control oligo, T-oligo induced the levels of SOD1 and SOD2 as most strikingly observed 16 hours after stimulation, in light of superimposed “feeding effects” following addition of fresh medium at time 0 and 24 hours before the 96 and 168 hour timepoints; GPX and catalase were not modulated (Figure 5).
Figure 5. T-oligo induces the levels of SOD1 and SOD2 in fibroblasts.

Newborn fibroblasts were plated and processed as per Materials and Methods. Total cellular proteins were harvested 16, 24, 48, 72, 96 and 168 hr after oligonucleotide stimulation at time 0. Cultures were provided fresh medium lacking oligonucleotides immediately after the 72 hour timepoint and after 144 hours. Proteins were processed for western blot analysis and the blot was reacted with the following antibodies: SOD1, SOD2, Catalase, GPX and actin, followed by appropriate secondary antibodies. Compared to diluent (D) and control oligo (C), within 16 hours T-oligo (T) induced the levels of SOD1 and SOD2 and the induction persisted through 96 hours. GPX and catalase were not modulated. Differences in SOD1 and SOD2 band intensity between control oligo and diluent appear largely to reflect loading differences as demonstrated by the actin bands.
T-oligo protects fibroblasts from oxidative damage
Because T-oligo induced the levels of SOD1 and SOD2, we investigated its capacity to protect against oxidative damage. Fibroblasts were processed as per Materials and Methods, and cell yields following an H2O2 challenge were determined (Figure 6). H2O2 (25 μM) reduced fibroblast yield in all cultures, as expected. However, 48 hours after H2O2 challenge, cell yields in T-oligo pretreated cultures were significantly higher than in diluent pretreated cultures (p<0.007, Figure 6), 75 ± 3% vs 52 ± 1% of their respective diluent-challenged controls, demonstrating that T-oligo pretreatment protects against oxidative stress.
Figure 6. T-oligo protects fibroblasts from oxidative damage.

Newborn fibroblasts were processed as per Materials and Methods, and cells were then treated with 25 μM fresh H2O2 or diluent for one hour and then provided fresh medium without T-oligos. Cell yields were determined 8, 16, 24 and 48 hours later. Fibroblasts pretreated with T-oligo and then diluent proliferated more slowly than diluent pretreated cells (p< 0.0001), as expected, given the initial T-oligo induced S-phase arrest[13], but both showed exponential growth after the first 24 hours. Interestingly, fibroblasts pretreated with T-oligo and then challenged with H2O2 proliferated significantly better than diluent pretreated H2O2-challenged fibroblasts (p< 0.006). Data points are the mean ± SEM for 3 separate experiments with different donor cells.
Discussion
Our data demonstrate substantial effects of T-oligo on redox status and resistance against oxidative stress in normal human fibroblasts. We found that T-oligo induces ROS levels in fibroblasts and that the effect is dependent on the tumor suppressor and transcription factor p53. Using a colorectal cell line transfected with a p53 expression vector, Polyak et al identified 14 transcripts that were markedly increased in p53-expressing cells. Interestingly, several of these p53-induced genes (PIGs) encoded proteins that generate ROS or function as antioxidants[62].
The authors concluded that p53 transcriptionally induces redox-related genes, followed by upregulation of ROS in the cells. Our data are consistent with the above observation, as T-oligo supplementation induces and activates p53 in fibroblasts within 24 hours[14], approximately 12 hours prior to ROS elevation. Furthermore, in fibroblasts with inactive p53, we show that ROS are not generated in response to T-oligo, demonstrating the requirement for this transcription factor in T-oligo-stimulated ROS upregulation.
We have shown in both normal and malignant cells of several lineages that T-oligos induce DNA damage-like responses[10–17, 19]. In fibroblasts, T-oligo-induced responses include formation of DNA damage foci at the telomere without associated loss of the single-stranded telomere overhang, followed by p53 induction and phosphorylation[19]. These respones require the presence of WRN protein, a 3′ → 5′ exonuclease and helicase mutated in Werner syndrome[19]. In the present study, we expand this observation by documenting the presence of phosphorylated (activated) ATM and its effector protein Chk2 in these foci, strongly suggesting that the T-oligo signal is transduced at these sites to p53. This would provide a mechanism for the presumed role of ATM in protecting against oxidative stress[80].
ROS are continuously generated by normal cellular metabolism and appear to contribute to the development of senescence[81–83]. However, recent evidence suggests that ROS can also mediate cellular signaling by growth factors such as PDGF and EGF[58, 59]. In this context ROS, specifically H2O2, was shown to induce protein phosphorylation, MAP kinase activation, DNA synthesis, and chemotoxis of cells[58, 59], suggesting that ROS can mediate outcomes other than senescence.
Although sustained (7 day) exposure to T-oligo induces senescence in more than 60% of fibroblasts as determined by SA β-galactosidase activity, induction of senescence-associated proteins and failure to resume growth following serum stimulation[14, 15], at the time of maximal T-oligo-induced cellular ROS (72 hours) there were very few cells positive for SA β-galactosidase activity and the number of occasional SA β-galactosidase positive cells did not differ between T-oligo-treated and diluent-treated cultures, demonstrating that within 72 hours T-oligo does not irrevocably commit fibroblasts to senescence and suggesting that this ROS elevation may mediate other cellular processes.
Several studies show that exposure to DNA damaging agents in sublethal doses induces adaptive DNA damage responses in the cells[84–89], and reviewed in[90, 91]. It is tantalizing to speculate that T-oligos, known to concentrate in the nucleus[10, 12], induce a telomere-based DNA damage signal and lead to upregulation of antioxidant enzymes, as well as DNA repair enzymes[18], to reduce DNA damage from future insults[90]. Of note, the enzymes upregulated by T-oligo, SOD1 and SOD2, catalyze the conversion of O2•− to H2O2, shown to be the key signaling molecule among all ROS[58, 59], suggesting that T-oligo induces H2O2-mediated signaling.
Our studies show that T-oligo-induced ROS elevation is p53-mediated. This is consistent with studies by Macip et al. [92], who showed that over-expression of the p53-inducible protein p21, also induced by T-oligos[14, 18, 93], in normal human fibroblasts increases ROS levels in the cells.
DNA damage responses include increased DNA repair mediated by p53 itself, as well as by p53-upregulated repair proteins[94–102], and in the case of overwhelming damage, upregulation of pro-apoptotic proteins and eventual elimination of the cell through apoptosis[97, 103–105]. The same responses of increased DNA repair and apoptosis are induced by T-oligo[10–12, 16, 18, 84], acting through p53 or in the absence of p53 through its homolog p73[10]. At least in fibroblasts, activation of p53 by T-oligo also induces antioxidant enzymes, but in a time course expected to transiently downregulate potentially damaging ROS. At low levels, these ROS appear to serve as additional signal transducers[58, 59], although the precise downstream events are as yet unclear; while at high levels, the ROS and particularly H2O2 can drive cells toward senescence[50], again removing them from the proliferative pool, an outcome understood to be, like apoptosis, an anti-cancer defense mechanism[106].
The three major enzymes that catalyze the formation of cellular ROS are mitochondrial oxidases, primarily NADH oxidase[107], the cell surface-associated p53-regulated NAD(P)H oxidase[108] and 5-lipoxygenase that catalyzes ROS production from arachidonic acid[108–111]. In this study we show that both p53DN and the specific inhibitor of NAD(P)H oxidase DPI[74, 75] abrogate T-oligo-mediated ROS induction, demonstrating that T-oligo stimulates ROS production through the activation of p53-dependent membrane-associated NAD(P)H oxidase.
Our results demonstrate the existence of a p53-dependent NAD(P)H-mediated redox response to telomere homolog oligonucleotides that mimic the effect of telomere disruption in cells[12, 14], suggesting an innate telomere-based genome-protective response. These data provide the strongest demonstration of T-oligo-induced antioxidant defenses complementing the observation that chronic topical T-oligo treatment reduces the level of 8-oxoG in chronically irradiated mouse skin[112]. The inducible antioxidant responses complement the previously demonstrated cell cycle arrest, adaptive differentiation, enhanced repair of DNA photoproducts and chemical adducts, and reduced mutation frequency[13, 17, 18, 84, 93] that together reduce carcinogenesis in the face of repeated carcinogenic insults[113]. The data also imply that T-oligo signaling involves generation of ROS. Our data suggest that cells have evolved to respond to DNA damage by upregulating proteins that protect against possible future damage in a manner at least functionally analogous to the bacterial SOS response[90]. Because telomere homologs presumptively mimic the DNA damage signal selectively, in contrast to UV irradiation or oxidative stress that are expected to also damage cell membranes and other cellular constituents, they provide a unique tool for analyzing such protective mechanisms. Finally, by inducing protective response in the absence of initial damage, T-oligos may provide a novel approach to the prevention and treatment of diseases due to oxidative stress.
Acknowledgments
This work was supported by NIH grant 1 R01 CA 105156 (BAG), and NIH training grants T32 AR07562-16 and T32 AG00115-17 (MSL-B., trainee; MY, preceptor; BAG, director).
Footnotes
Conflict of Interest Statement
Portions of the work reported in this article pertain to a patent application for which M.S. L-B, M.Y., M.S.E. and B.A.G. are co-inventors and, if awarded, will be assigned to the Trustees of Boston University (their employer) and then licensed to SemaCo, Inc., a for-profit company created to commercialize intellectual property arising out of their laboratory. M.Y., M.S.E. and B.A.G. all hold equity in SemaCo, and B.A.G. is SemaCo’s Chief Scientific Officer.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.IJAS, Greider CW. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Molecular biology of the cell. 2003;14:987–1001. doi: 10.1091/mbc.02-04-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shay JW, Wright WE. Telomeres are double-strand DNA breaks hidden from DNA damage responses. Molecular cell. 2004;14:420–421. doi: 10.1016/s1097-2765(04)00269-2. [DOI] [PubMed] [Google Scholar]
- 4.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 5.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science (New York, NY) 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 6.Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science (New York, NY) 2003;299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 7.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 8.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science (New York, NY) 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 9.van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 10.Eller MS, Puri N, Hadshiew IM, Venna SS, Gilchrest BA. Induction of apoptosis by telomere 3′ overhang-specific DNA. Experimental cell research. 2002;276:185–193. doi: 10.1006/excr.2002.5531. [DOI] [PubMed] [Google Scholar]
- 11.Ohashi N, Yaar M, Eller MS, Truzzi F, Gilchrest BA. Features that determine telomere homolog oligonucleotide-induced therapeutic DNA damage-like responses in cancer cells. Journal of cellular physiology. 2007;210:582–595. doi: 10.1002/jcp.20848. [DOI] [PubMed] [Google Scholar]
- 12.Yaar M, Eller MS, Panova I, Kubera J, Wee LH, Cowan KH, Gilchrest BA. Telomeric DNA induces apoptosis and senescence of human breast carcinoma cells. Breast Cancer Res. 2007;9:R13. doi: 10.1186/bcr1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eller MS, Li GZ, Firoozabadi R, Puri N, Gilchrest BA. Induction of a p95/Nbs1-mediated S phase checkpoint by telomere 3′ overhang specific DNA. Faseb J. 2003;17:152–162. doi: 10.1096/fj.02-0197com. [DOI] [PubMed] [Google Scholar]
- 14.Li GZ, Eller MS, Firoozabadi R, Gilchrest BA. Evidence that exposure of the telomere 3′ overhang sequence induces senescence. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:527–531. doi: 10.1073/pnas.0235444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li GZ, Eller MS, Hanna K, Gilchrest BA. Signaling pathway requirements for induction of senescence by telomere homolog oligonucleotides. Experimental cell research. 2004;301:189–200. doi: 10.1016/j.yexcr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 16.Puri N, Eller MS, Byers HR, Dykstra S, Kubera J, Gilchrest BA. Telomere-based DNA damage responses: a new approach to melanoma. Faseb J. 2004;18:1373–1381. doi: 10.1096/fj.04-1774com. [DOI] [PubMed] [Google Scholar]
- 17.Eller MS, Maeda T, Magnoni C, Atwal D, Gilchrest BA. Enhancement of DNA repair in human skin cells by thymidine dinucleotides: evidence for a p53-mediated mammalian SOS response. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:12627–12632. doi: 10.1073/pnas.94.23.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goukassian DA, Eller MS, Yaar M, Gilchrest BA. Thymidine dinucleotide mimics the effect of solar simulated irradiation on p53 and p53-regulated proteins. The Journal of investigative dermatology. 1999;112:25–31. doi: 10.1046/j.1523-1747.1999.00468.x. [DOI] [PubMed] [Google Scholar]
- 19.Eller MS, Liao X, Liu S, Hanna K, Backvall H, Opresko PL, Bohr VA, Gilchrest BA. A role for WRN in telomere-based DNA damage responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15073–15078. doi: 10.1073/pnas.0607332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilchrest BA. Using DNA damage responses to prevent and treat skin cancers. The Journal of dermatology. 2004;31:862–877. doi: 10.1111/j.1346-8138.2004.tb00619.x. [DOI] [PubMed] [Google Scholar]
- 21.Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA. The Werner syndrome protein is a DNA helicase. Nature genetics. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- 22.Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner’s syndrome gene. Science (New York, NY) 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 23.Blander G, Kipnis J, Leal JF, Yu CE, Schellenberg GD, Oren M. Physical and functional interaction between p53 and the Werner’s syndrome protein. The Journal of biological chemistry. 1999;274:29463–29469. doi: 10.1074/jbc.274.41.29463. [DOI] [PubMed] [Google Scholar]
- 24.Spillare EA, Robles AI, Wang XW, Shen JC, Yu CE, Schellenberg GD, Harris CC. p53-mediated apoptosis is attenuated in Werner syndrome cells. Genes & development. 1999;13:1355–1360. doi: 10.1101/gad.13.11.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang XW, Tseng A, Ellis NA, Spillare EA, Linke SP, Robles AI, Seker H, Yang Q, Hu P, Beresten S, Bemmels NA, Garfield S, Harris CC. Functional interaction of p53 and BLM DNA helicase in apoptosis. The Journal of biological chemistry. 2001;276:32948–32955. doi: 10.1074/jbc.M103298200. [DOI] [PubMed] [Google Scholar]
- 26.Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer letters. 2008;266:6–11. doi: 10.1016/j.canlet.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 27.Mates JM, Segura JA, Alonso FJ, Marquez J. Intracellular redox status and oxidative stress: implications for cell proliferation, apoptosis, and carcinogenesis. Archives of toxicology. 2008;82:273–299. doi: 10.1007/s00204-008-0304-z. [DOI] [PubMed] [Google Scholar]
- 28.Camhi SL, Lee P, Choi AM. The oxidative stress response. New horizons (Baltimore, Md) 1995;3:170–182. [PubMed] [Google Scholar]
- 29.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. Journal of cellular physiology. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 30.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. American journal of physiology. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 31.Karihtala P, Soini Y. Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. Apmis. 2007;115:81–103. doi: 10.1111/j.1600-0463.2007.apm_514.x. [DOI] [PubMed] [Google Scholar]
- 32.Mates JM, Sanchez-Jimenez F. Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci. 1999;4:D339–345. doi: 10.2741/mates. [DOI] [PubMed] [Google Scholar]
- 33.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. The international journal of biochemistry & cell biology. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Brigelius-Flohe R. Glutathione peroxidases and redox-regulated transcription factors. Biological chemistry. 2006;387:1329–1335. doi: 10.1515/BC.2006.166. [DOI] [PubMed] [Google Scholar]
- 35.Lei XG, Cheng WH, McClung JP. Metabolic regulation and function of glutathione peroxidase-1. Annual review of nutrition. 2007;27:41–61. doi: 10.1146/annurev.nutr.27.061406.093716. [DOI] [PubMed] [Google Scholar]
- 36.Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxidants & redox signaling. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 37.Hirt RP, Muller S, Embley TM, Coombs GH. The diversity and evolution of thioredoxin reductase: new perspectives. Trends in parasitology. 2002;18:302–308. doi: 10.1016/s1471-4922(02)02293-6. [DOI] [PubMed] [Google Scholar]
- 38.Culotta VC, Yang M, O’Halloran TV. Activation of superoxide dismutases: putting the metal to the pedal. Biochimica et biophysica acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson F, Giulivi C. Superoxide dismutases and their impact upon human health. Molecular aspects of medicine. 2005;26:340–352. doi: 10.1016/j.mam.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free radical biology & medicine. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 41.Kirkman HN, Gaetani GF. Mammalian catalase: a venerable enzyme with new mysteries. Trends in biochemical sciences. 2007;32:44–50. doi: 10.1016/j.tibs.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Leccia MT, Yaar M, Allen N, Gleason M, Gilchrest BA. Solar simulated irradiation modulates gene expression and activity of antioxidant enzymes in cultured human dermal fibroblasts. Experimental dermatology. 2001;10:272–279. doi: 10.1034/j.1600-0625.2001.100407.x. [DOI] [PubMed] [Google Scholar]
- 43.Toussaint O, Medrano EE, von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Experimental gerontology. 2000;35:927–945. doi: 10.1016/s0531-5565(00)00180-7. [DOI] [PubMed] [Google Scholar]
- 44.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Experimental cell research. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 45.Stewart SA, Weinberg RA. Telomerase and human tumorigenesis. Seminars in cancer biology. 2000;10:399–406. doi: 10.1006/scbi.2000.0339. [DOI] [PubMed] [Google Scholar]
- 46.Sugrue MM, Shin DY, Lee SW, Aaronson SA. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9648–9653. doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 48.Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes & development. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helbock HJ, Beckman KB, Shigenaga MK, Walter PB, Woodall AA, Yeo HC, Ames BN. DNA oxidation matters: the HPLC-electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:288–293. doi: 10.1073/pnas.95.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Zglinicki T. Role of oxidative stress in telomere length regulation and replicative senescence. Annals of the New York Academy of Sciences. 2000;908:99–110. doi: 10.1111/j.1749-6632.2000.tb06639.x. [DOI] [PubMed] [Google Scholar]
- 51.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Laboratory investigation; a journal of technical methods and pathology. 1982;47:412–426. [PubMed] [Google Scholar]
- 52.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 53.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Archives of biochemistry and biophysics. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 54.Bokoch GM, Knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends in biochemical sciences. 2003;28:502–508. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 55.Thannickal VJ, Aldweib KD, Fanburg BL. Tyrosine phosphorylation regulates H2O2 production in lung fibroblasts stimulated by transforming growth factor beta1. The Journal of biological chemistry. 1998;273:23611–23615. doi: 10.1074/jbc.273.36.23611. [DOI] [PubMed] [Google Scholar]
- 56.Thannickal VJ. The paradox of reactive oxygen species: injury, signaling, or both? American journal of physiology. 2003;284:L24–25. doi: 10.1152/ajplung.00279.2002. [DOI] [PubMed] [Google Scholar]
- 57.Droge W. Free radicals in the physiological control of cell function. Physiological reviews. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 58.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. The Journal of biological chemistry. 1997;272:217–221. [PubMed] [Google Scholar]
- 59.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science (New York, NY) 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 60.Davies KJ. The broad spectrum of responses to oxidants in proliferating cells: a new paradigm for oxidative stress. IUBMB life. 1999;48:41–47. doi: 10.1080/713803463. [DOI] [PubMed] [Google Scholar]
- 61.Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJ. Free radical biology and medicine: it’s a gas, man! Am J Physiol Regul Integr Comp Physiol. 2006;291:R491–511. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- 62.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 63.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Plisko A, Gilchrest BA. Growth factor responsiveness of cultured human fibroblasts declines with age. Journal of gerontology. 1983;38:513–518. doi: 10.1093/geronj/38.5.513. [DOI] [PubMed] [Google Scholar]
- 65.Gottlieb E, Haffner R, von Ruden T, Wagner EF, Oren M. Down-regulation of wild-type p53 activity interferes with apoptosis of IL-3-dependent hematopoietic cells following IL-3 withdrawal. The EMBO journal. 1994;13:1368–1374. doi: 10.1002/j.1460-2075.1994.tb06390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shaulian E, Zauberman A, Ginsberg D, Oren M. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Molecular and cellular biology. 1992;12:5581–5592. doi: 10.1128/mcb.12.12.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rheinwald JG, Hahn WC, Ramsey MR, Wu JY, Guo Z, Tsao H, De Luca M, Catricala C, O’Toole KM. A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Molecular and cellular biology. 2002;22:5157–5172. doi: 10.1128/MCB.22.14.5157-5172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eller MS, Yaar M, Gilchrest BA. DNA damage and melanogenesis. Nature. 1994;372:413–414. doi: 10.1038/372413a0. [DOI] [PubMed] [Google Scholar]
- 69.Marwaha V, Chen YH, Helms E, Arad S, Inoue H, Bord E, Kishore R, Sarkissian RD, Gilchrest BA, Goukassian DA. T-oligo treatment decreases constitutive and UVB-induced COX-2 levels through p53- and NFkappaB-dependent repression of the COX-2 promoter. The Journal of biological chemistry. 2005;280:32379–32388. doi: 10.1074/jbc.M503245200. [DOI] [PubMed] [Google Scholar]
- 70.Narayanan PK, Goodwin EH, Lehnert BE. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer research. 1997;57:3963–3971. [PubMed] [Google Scholar]
- 71.Verdier-Sevrain S, Yaar M, Cantatore J, Traish A, Gilchrest BA. Estradiol induces proliferation of keratinocytes via a receptor mediated mechanism. Faseb J. 2004;18:1252–1254. doi: 10.1096/fj.03-1088fje. [DOI] [PubMed] [Google Scholar]
- 72.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 73.Pichierri P, Rosselli F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. The EMBO journal. 2004;23:1178–1187. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chakraborty S, Massey V. Reaction of reduced flavins and flavoproteins with diphenyliodonium chloride. The Journal of biological chemistry. 2002;277:41507–41516. doi: 10.1074/jbc.M205432200. [DOI] [PubMed] [Google Scholar]
- 75.Cross AR, Jones OT. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. The Biochemical journal. 1986;237:111–116. doi: 10.1042/bj2370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xie S, Wang Q, Wu H, Cogswell J, Lu L, Jhanwar-Uniyal M, Dai W. Reactive oxygen species-induced phosphorylation of p53 on serine 20 is mediated in part by polo-like kinase-3. The Journal of biological chemistry. 2001;276:36194–36199. doi: 10.1074/jbc.M104157200. [DOI] [PubMed] [Google Scholar]
- 77.Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS. The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. The Journal of biological chemistry. 2001;276:21951–21959. doi: 10.1074/jbc.M011303200. [DOI] [PubMed] [Google Scholar]
- 78.Takao N, Li Y, Yamamoto K. Protective roles for ATM in cellular response to oxidative stress. FEBS letters. 2000;472:133–136. doi: 10.1016/s0014-5793(00)01422-8. [DOI] [PubMed] [Google Scholar]
- 79.Kinscherf R, Deigner HP, Usinger C, Pill J, Wagner M, Kamencic H, Hou D, Chen M, Schmiedt W, Schrader M, Kovacs G, Kato K, Metz J. Induction of mitochondrial manganese superoxide dismutase in macrophages by oxidized LDL: its relevance in atherosclerosis of humans and heritable hyperlipidemic rabbits. Faseb J. 1997;11:1317–1328. doi: 10.1096/fasebj.11.14.9409551. [DOI] [PubMed] [Google Scholar]
- 80.Rotman G, Shiloh Y. The ATM gene and protein: possible roles in genome surveillance, checkpoint controls and cellular defence against oxidative stress. Cancer surveys. 1997;29:285–304. [PubMed] [Google Scholar]
- 81.Chen QM, Bartholomew JC, Campisi J, Acosta M, Reagan JD, Ames BN. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. The Biochemical journal. 1998;332(Pt 1):43–50. doi: 10.1042/bj3320043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, Mazarati JB, Eliaers F, Remacle J, Toussaint O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free radical biology & medicine. 2000;28:361–373. doi: 10.1016/s0891-5849(99)00249-x. [DOI] [PubMed] [Google Scholar]
- 83.Reichelt J, Schachtschabel DO. Energetic stress induces premature aging of diploid human fibroblasts (Wi-38) in vitro. Archives of gerontology and geriatrics. 2001;32:219–231. doi: 10.1016/s0167-4943(01)00102-9. [DOI] [PubMed] [Google Scholar]
- 84.Arad S, Konnikov N, Goukassian DA, Gilchrest BA. T-oligos augment UV-induced protective responses in human skin. Faseb J. 2006;20:1895–1897. doi: 10.1096/fj.06-5964fje. [DOI] [PubMed] [Google Scholar]
- 85.de Toledo SM, Asaad N, Venkatachalam P, Li L, Howell RW, Spitz DR, Azzam EI. Adaptive responses to low-dose/low-dose-rate gamma rays in normal human fibroblasts: the role of growth architecture and oxidative metabolism. Radiation research. 2006;166:849–857. doi: 10.1667/RR0640.1. [DOI] [PubMed] [Google Scholar]
- 86.Smith DM, Raaphorst GP. Adaptive responses in human glioma cells assessed by clonogenic survival and DNA strand break analysis. International journal of radiation biology. 2003;79:333–339. doi: 10.1080/0955300032000093137. [DOI] [PubMed] [Google Scholar]
- 87.Xu Y. DNA damage: a trigger of innate immunity but a requirement for adaptive immune homeostasis. Nature reviews. 2006;6:261–270. doi: 10.1038/nri1804. [DOI] [PubMed] [Google Scholar]
- 88.Jeeves WP, Rainbow AJ. Gamma-ray-enhanced reactivation of irradiated adenovirus in Xeroderma pigmentosum and Cockayne syndrome fibroblasts. Radiation research. 1983;94:480–498. [PubMed] [Google Scholar]
- 89.Jeeves WP, Rainbow AJ. U.V. enhanced reactivation of U.V.- and gamma-irradiated adenovirus in normal human fibroblasts. International journal of radiation biology and related studies in physics, chemistry, and medicine. 1983;43:599–623. doi: 10.1080/09553008314550721. [DOI] [PubMed] [Google Scholar]
- 90.Gilchrest BA, Eller MS. The tale of the telomere: implications for prevention and treatment of skin cancers. The journal of investigative dermatology Symposium proceedings/the Society for Investigative Dermatology, Inc. 2005;10:124–130. doi: 10.1111/j.1087-0024.2005.200406.x. [DOI] [PubMed] [Google Scholar]
- 91.Preston RJ. Radiation biology: concepts for radiation protection. Health physics. 2005;88:545–556. doi: 10.1097/00004032-200506000-00003. [DOI] [PubMed] [Google Scholar]
- 92.Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, Aaronson SA. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. The EMBO journal. 2002;21:2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goukassian DA, Bagheri S, el-Keeb L, Eller MS, Gilchrest BA. DNA oligonucleotide treatment corrects the age-associated decline in DNA repair capacity. Faseb J. 2002;16:754–756. doi: 10.1096/fj.01-0829fje. [DOI] [PubMed] [Google Scholar]
- 94.Adams MM, Carpenter PB. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell division. 2006;1:19. doi: 10.1186/1747-1028-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bargonetti J, Manfredi JJ. Multiple roles of the tumor suppressor p53. Current opinion in oncology. 2002;14:86–91. doi: 10.1097/00001622-200201000-00015. [DOI] [PubMed] [Google Scholar]
- 96.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkp oints. Journal of cellular physiology. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 97.Gomez-Lazaro M, Fernandez-Gomez FJ, Jordan J. p53: twenty five years understanding the mechanism of genome protection. Journal of physiology and biochemistry. 2004;60:287–307. doi: 10.1007/BF03167075. [DOI] [PubMed] [Google Scholar]
- 98.Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med. 2006;7:165–172. doi: 10.1016/j.carrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 99.Livneh Z. Keeping mammalian mutation load in check: regulation of the activity of error-prone DNA polymerases by p53 and p21. Cell cycle (Georgetown, Tex. 2006;5:1918–1922. doi: 10.4161/cc.5.17.3193. [DOI] [PubMed] [Google Scholar]
- 100.Loffler H, Lukas J, Bartek J, Kramer A. Structure meets function--centrosomes, genome maintenance and the DNA damage response. Experimental cell research. 2006;312:2633–2640. doi: 10.1016/j.yexcr.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 101.Lukas J, Bohr VA, Halazonetis TD. Cellular responses to DNA damage: current state of the field and review of the 52nd Benzon Symposium. DNA repair. 2006;5:591–601. doi: 10.1016/j.dnarep.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 102.Vousden KH. Activation of the p53 tumor suppressor protein. Biochimica et biophysica acta. 2002;1602:47–59. doi: 10.1016/s0304-419x(02)00035-5. [DOI] [PubMed] [Google Scholar]
- 103.Hoglund P. DNA damage and tumor surveillance: one trigger for two pathways. Sci STKE. 2006;2006:pe2. doi: 10.1126/stke.3172006pe2. [DOI] [PubMed] [Google Scholar]
- 104.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends in molecular medicine. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 105.Yee KS, Vousden KH. Complicating the complexity of p53. Carcinogenesis. 2005;26:1317–1322. doi: 10.1093/carcin/bgi122. [DOI] [PubMed] [Google Scholar]
- 106.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 107.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. The Journal of biological chemistry. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 108.Meier B, Cross AR, Hancock JT, Kaup FJ, Jones OT. Identification of a superoxide-generating NADPH oxidase system in human fibroblasts. The Biochemical journal. 1991;275(Pt 1):241–245. doi: 10.1042/bj2750241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 110.Catalano A, Rodilossi S, Caprari P, Coppola VA. Procopio, 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. The EMBO journal. 2005;24:170–179. doi: 10.1038/sj.emboj.7600502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. American journal of respiratory and critical care medicine. 2002;166:S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 112.Arad S, Zattra E, Hebert J, Epstein EH, Jr, Goukassian DA, Gilchrest BA. Topical thymidine dinucleotide treatment reduces development of ultraviolet-induced basal cell carcinoma in Ptch-1+/− mice. The American journal of pathology. 2008;172:1248–1255. doi: 10.2353/ajpath.2008.071117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goukassian DA, Helms E, van Steeg H, van Oostrom C, Bhawan J, Gilchrest BA. Topical DNA oligonucleotide therapy reduces UV-induced mutations and photocarcinogenesis in hairless mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3933–3938. doi: 10.1073/pnas.0306389101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holland PC, Clark MG, Bloxham DP, Lardy HA. Mechanism of action of the hypoglycemic agent diphenyleneiodonium. The Journal of biological chemistry. 1973;248:6050–6056. [PubMed] [Google Scholar]
- 115.Majander A, Finel M, Wikstrom M. Diphenyleneiodonium inhibits reduction of iron-sulfur clusters in the mitochondrial NADH-ubiquinone oxidoreductase (Complex I) The Journal of biological chemistry. 1994;269:21037–21042. [PubMed] [Google Scholar]