

Abstract
Aggregation of α-synuclein (α-syn), a process that generates oligomeric intermediates, is a common pathological feature of several neurodegenerative disorders. Despite the potential importance of the oligomeric α-syn intermediates in neuron function, their biochemical properties and pathobiological functions in vivo remain vastly unknown. Here we used two-dimensional analytical separation and an array of biochemical and cell-based assays to characterize α-syn oligomers that are present in the nervous system of A53T α-syn transgenic mice. The most prominent species identified were 53 Å detergent-soluble oligomers, which preceded neurological symptom onset, and were found at equivalent amounts in regions containing α-syn inclusions as well as histologically unaffected regions. These oligomers were resistant to SDS, heat, and urea but were sensitive to proteinase-K digestion. Although the oligomers shared similar basic biochemical properties, those obtained from inclusion-bearing regions were prominently reactive to antibodies that recognize oxidized α-syn oligomers, significantly accelerated aggregation of α-syn in vitro, and caused primary cortical neuron degeneration. In contrast, oligomers obtained from non-inclusion-bearing regions were not toxic and delayed the in vitro formation of α-syn fibrils. These data indicate that specific conformations of α-syn oligomers are present in distinct brain regions of A53T α-syn transgenic mice. The contribution of these oligomers to the development of neuron dysfunction appears to be independent of their absolute quantities and basic biochemical properties but is dictated by the composition and conformation of the intermediates as well as unrecognized brain-region-specific intrinsic factors.
Introduction
α-Synuclein (α-syn) is a soluble, acidic protein that typically assumes a random coil structure, but it acquires α-helical conformation during binding to anionic phospholipids (Davidson et al., 1998; Ulmer et al., 2005). Although the exact function(s) of α-syn remain uncertain, the preferential localization to presynaptic nerve terminals and its interaction with vesicular phospholipids and proteins suggests a regulatory function associated with synaptic activity, dopamine (DA) production and metabolism, lipid vesicle trafficking, and chaperone-like activity (Maroteaux et al., 1988; Iwai et al., 1995; Souza et al., 2000b; Cabin et al., 2002; Chandra et al., 2005).
Data from postmortem evaluations of Parkinson's disease (PD) brains revealed that α-syn aberrantly forms highly organized, linear amyloid fibrils that constitute part of the characteristic inclusions found in neuronal perikarya (Lewy bodies) and dystrophic neurites (Lewy neurites). (Forno, 1996; Goedert, 2001; Norris et al., 2004; Mori et al., 2008). Despite the ubiquitous expression of α-syn throughout the CNS, these inclusions are found in certain susceptible neuronal subtypes of specific brain nuclei (Braak et al., 2003). Biochemically, α-syn within inclusions is resistant to extraction with non-ionic detergents. However, during extraction with formic acid or SDS/urea, α-syn collapses into monomers and SDS and heat-stable oligomers (Baba et al., 1998; Tu et al., 1998; Dickson et al., 1999).
In vitro evidence using purified recombinant α-syn has indicated that the conversion of monomers into amyloid fibrils progresses in a nucleation-dependent manner through an initial polymerization stage involving the formation of oligomeric intermediates (Conway et al., 2000b). The polymerization process is concentration dependent and can be accelerated by the PD-causing mutations A53T, A30P, and E46K (Conway et al., 1998, 2000b; Giasson et al., 1999; Narhi et al., 1999; Greenbaum et al., 2005). Although there is considerable confidence that accumulation and polymerization of α-syn plays an important role in neurodegeneration, the contribution of the different species that emerge through the aggregation process has not been fully delineated. Existing efforts identifying the potential pathogenic formations are based on studies in which oligomerization of the protein is forced in vitro (Goldberg and Lansbury, 2000; Volles et al., 2001; Danzer et al., 2007). Characterizing α-syn assemblies that are formed in the brain and in living cells is challenging because unstable conformations may be disrupted during the traditional biochemical extraction processes. Notwithstanding this important concern, there is considerable paucity in the biochemical and biological description of the oligomeric α-syn entities that are formed in mouse models and humans and are stable to standard isolation methodologies with mild non-ionic detergents.
In this study, we provide a brain-region-specific biochemical and biological characterization of the relatively stable α-syn oligomeric conformations that are formed in the transgenic mouse line expressing human A53T α-syn driven by the mouse prion protein (PrP) promoter (Giasson et al., 2002). The data indicate that, despite similarities in basic biochemical properties, α-syn oligomeric intermediates obtained from different neural regions demonstrated unexpected divergence in promoting α-syn amyloid fibril formation and toxicity.
Materials and Methods
Mouse breeding.
The mice used in this study express human A53T α-syn (line M83) or human wild-type (WT) α-syn (line M20) driven by the murine PrP promoter and have been described previously (Giasson et al., 2002). To generate A53T+/+ and nontransgenic (nTg) control mice used in experiments, A53T+/− females were mated with A53T+/− or A53T+/+ males, because A53T+/+ females were found to produce small litters and exhibit poor motherly behavior. Genotyping was performed by both end-point PCR using GeneAmp PCR system 9700 thermal cycler (Applied Biosystems) and quantitative PCR using Applied Biosystems 7500 real-time PCR system (Applied Biosystems) with the ABI MGB primer-probe set for human SNCA (assay ID Hs00240907_m1). SNCA values were normalized to mouse β-actin (4352341E). Mice identified as homozygous for the α-syn transgene were verified through backcrosses or validated by quantitative Western blot analysis of α-syn protein.
Activity measurements.
Analysis was performed on 5-month-old A53T+/+ males and age-matched nTg male littermates. Motor activity measurements were performed with an Opto-M3 activity meter (Columbus Instruments) by quantifying the number of infrared beam breaks over a 24 h period using the Multi Device Interface software (version 1.34; Columbus Instruments). Testing was done in the home holding room and home cage under controlled environmental conditions, and food/water was available ad libitum. The animals were acclimated to the apparatus for 2 d, followed by 3 consecutive days of beam-break data collection. The 3 d average was used as n = 1 for each mouse, with 5 mice used for each group.
Food intake and body weight measurements.
Testing was performed on 4- to 6-month-old A53T+/+ males and age-matched nTg male littermates in the home holding room and home cage under controlled environmental conditions. Singly housed mice were given 100 g of fresh standard animal chow pellets (laboratory rodent diet; catalog #5001; LabDiet) placed in a suspended wire-cage top feeder at the start of the testing period, and food consumption was determined by weighing pellets every 24 h for 5 consecutive days. Mouse body weight was also determined every day during the testing period and used to normalize food intake measurements. The average value of food consumed over the 5 d testing period was used as n = 1 for each mouse. The analysis was performed with six different mice from two separate litters.
Immunohistochemistry.
Brain sections from symptomatic A53T+/+ α-syn transgenic mice and age-matched nTg controls were stained with syn505 mouse monoclonal antibody specific for human α-syn (Duda et al., 2002). Perfusions, tissue processing, and staining were performed as described previously (Giasson et al., 2002). Adjacent sections were stained with rabbit polyclonal anti-glial fibrillary acidic protein (GFAP) (ab7260; Abcam). Incubation with secondary antibodies conjugated to biotin was followed by addition of avidin–biotin–peroxidase complex (ABC Elite; Vector Laboratories), visualized with 3,3′-diaminobenzidine as the chromogene, and counterstained with hematoxylin (SH30-500D; Thermo Fisher Scientific).
Mouse brain dissection for biochemical analysis.
Four- to six-month-old asymptomatic A53T+/+ α-syn transgenic mice, 11- to 12-month-old symptomatic A53T+/+ α-syn transgenic mice, and 11- to 12-month-old human WT α-syn transgenic mice were used for analysis. A53T+/+ symptomatic mice were killed 1–2 d after initial motor symptom onset. A53T+/+ mice were monitored on a daily basis, and symptomatic mice were identified by spine stiffness, weight loss, and hindlimb paralysis as described previously (Giasson et al., 2002). After removal of the cerebellum (Crb) (including the deep cerebellar nuclei; bregma, −5.46 to −7.70), olfactory bulbs (OB) (bregma, +4.74 to +3.14), and spinal cord (SC) (cervical region), the remainder of the brain was dissected into 1-mm-thick coronal sections using a vibratome (World Precision Instruments) submerged in ice-cold PBS, to dissect the substantia nigra (SN), hippocampus (Hipp), visual cortex (bregma, −2.44 to −3.88), and the striatum (+0.66 to −1.34). Dissected brain regions were weighed, rapidly frozen on dry ice, and stored at −80°C until analysis.
Sequential extraction.
Tissues derived from different regions of individual mice were homogenized with 10 vol of lysis buffer [20 mm HEPES, pH 7.4, 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm EGTA, 1.5 mm MgCl2, 1 μm lactacystin, 50 mm NaF, 10 mm Na-pyruvate, 2 mm Na3VO4, 1 mm PMSF, and protease inhibitor cocktail (P2714; Sigma)]. The tissues were grinded with the aid of mechanical homogenizer and then subjected to four consecutive freeze and thaw cycles of 2 min each (−80°C ethanol bath/37°C water bath). The Triton X-100-soluble fraction was obtained after ultracentrifugation at 100,000 × g for 30 min at 4°C. The resulting pellets were further extracted in 5 vol of 2% SDS, 50 mm Tris, pH 7.4, and a protease inhibitor cocktail by sonication and then boiling for 20 min. Samples were centrifuged for 10 min, 16,000 × g at 25°C to obtain the Triton X-100-insoluble (SDS-soluble) fractions. Protein concentration was determined by the microBCA assay (Pierce).
Gel filtration HPLC.
Triton X-100-soluble protein (400 μg) from each tissue in a total volume of 250 μl were loaded onto a Superdex 200 HR10/30 column (GE Healthcare) connected to an Agilent 1100 series HPLC system for size exclusion chromatographic (SEC) analysis. The mobile phase used to resolve the protein lysates consisted of 25 mm HEPES and 150 mm NaCl, pH 7.25. The fractions were collected and concentrated with 3000 nominal molecular weight limit (NMWL) Ultracel Microcon filters (Millipore). Fractions 2–5 (99–63 Å), 6–9 (63–48 Å), 18–20 (25–20 Å), and 21–24 (20–11 Å) were combined before being loaded on the SDS-PAGE gels to enhance the signals. To isolate the 53-Å-sized soluble α-syn oligomers, the corresponding fractions were separated and concentrated using 3000 NMWL Ultracel Microcon filters. The gel filtration column was calibrated using a mixture of globular proteins (HMW standards kit; GE Healthcare).
Proteinase K and urea treatments of α-syn oligomers.
The 53-Å-sized Triton X-100-soluble α-syn oligomers isolated by SEC from various neuronal regions were treated with 100 μg/ml proteinase K (PK) for 30 and 60 min at 37°C, and another set of samples was treated with 8 m urea for 30 and 60 min at room temperature. The protein concentration for each sample was adjusted to 0.5 mg/ml. The reactions were stopped by heating to 100°C for 10 min after adding SDS sample buffer. The samples were resolved on SDS polyacrylamide gels.
Seeding assay.
Human recombinant α-syn protein used for the assay was expressed and purified as described previously (Giasson et al., 1999). In vitro α-syn aggregation was performed as described (Mazzulli et al., 2007). Briefly, 2.5 mg/ml purified protein was incubated for 4 d in conditions promoting aggregation (shaking at 1000 rpm, 37°C) with 0.1 μg/μl fractionated lysate corresponding to the 53-Å-sized Triton X-100-soluble α-syn oligomers from the OB and SC regions. Control experiments were conducted in parallel by using samples immunodepleted from α-syn (immunodepletion described below), the fractions corresponding to the monomer of α-syn, and also using the purified protein with no addition of fractionated lysates. The corresponding fractions from lysates of WT α-syn transgenic mice were also assessed as additional controls. The progress of fibril formation was monitored every 12 h by addition of 10 μm Thioflavin T (Sigma) solution in the reaction and measuring fluorescence emission at 490 nm during excitation at 450 nm.
Western blotting.
For Western blot analysis of samples sequentially extracted, the following primary antibodies were used: monoclonal anti-α-syn LB509 (1:1000) (Baba et al., 1998; Jakes et al., 1999), monoclonal anti-tyrosine hydroxylase (TH) (EC 1.14.16.2) (1:4000; catalog #657010; EMD Biosciences), polyclonal anti-neuronal specific enolase (NSE) (EC 4.2.1.11) (1:2000; catalog #16625; Polysciences), and monoclonal anti-vimentin (1:500; catalog #550513; BD Pharmingen). For Western blot analysis of fractions from gel filtration profiles, the following antibodies were used: monoclonal anti-α-syn syn211 (1:1000) (Giasson et al., 2000) and polyclonal anti-NSE (1:2000; Polysciences). Fractionated lysates were also blotted with monoclonal anti-α-syn syn208 and syn303 (1:1000) (Giasson et al., 2000; Duda et al., 2002). Dopaminergic markers l-3,4-dihydroxyphenylalanine decarboxylase (DDC) (EC 4.1.1.28) and dopamine transporter (DAT) were detected with mouse monoclonal antibodies DDC-11-M (1:500; Alpha Diagnostic) and MAB369 (1:800; Millipore Bioscience Research Reagents), respectively. Primary antibodies were detected with either goat anti-mouse IgG Alexa Fluor 680 (1:5000; Invitrogen) or goat anti-rabbit IgG IRDye 800 (1:5000; Rockland) conjugated secondary antibodies and scanned at intensity level 2 with an Odyssey Infrared Imaging System (Li-Cor Biosciences). The bands from the Western blots were quantified by densitometry using Odyssey infrared imaging system software (Odyssey version 2.1; Li-Cor Biosciences).
Quantification of α-syn oligomers.
All individual α-syn-immunoreactive bands from SEC/SDS-PAGE/syn211 Western blot analysis were quantified by densitometry using the Odyssey version 2.1 software (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (Li-Cor Biosciences). Our previous SEC analysis of WT and A53T α-syn extracted from transfected neuroblastoma cells has shown that in vitro oligomerization does not occur as a result of the extraction and analysis procedure used here (Mazzulli et al., 2006), thus ruling out the possibility that oligomers detected in A53T α-syn mice form in vitro. α-Syn-immunoreactive bands that comprised the 53-Å-sized α-syn oligomer peaked at a volume corresponding to a 225 kDa globular protein in the gel filtration column and were found to be made of a 59, 52, and 31 kDa species by SDS-PAGE. A minor oligomer eluted at a volume corresponding to a 36-Å-sized particle and comprised 64 and 38 kDa bands as determined by SDS-PAGE. The bands eluting at a volume corresponding to a 34-Å-sized particle and migrating at 19 kDa in the SDS-PAGE gels were quantified as the α-syn monomer. Each species was quantified and divided by the total α-syn signal (α-syn 53 Å plus minor oligomer plus α-syn monomer) to determine the percentage of each.
Catechol quantification by HPLC with electrochemical detection.
Catechols were resolved on a reverse-phase C18 Luna column (150 × 4.6 mm, 5 μm; Phenomenex) connected to an 1100 series Agilent HPLC system and detected by a Coularray detector (ESA Inc.) as described previously (Mazzulli et al., 2006).
Preparation of mouse primary neuronal cortical cultures.
Cultures were prepared from C57BL/6 mouse embryos at day 17 (stock #3704732; Charles River). After dissection from embryos in HEPES-buffered saline [supplemented with 1 mm sodium pyruvate (Invitrogen), 6 mg/ml d-glucose (Sigma), 100 U/ml penicillin, and 100 μg/ml streptomycin], the cortex was mechanically dissociated, trypsinized, and seeded at 50,000 cells per well into 96-well plates (catalog #35-4640; BD Biosciences) freshly coated with 50 μg/ml poly-d-lysine (Sigma). After seeding, neurons were cultured for 2 h in serum media [10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mm glutamine, in Neurobasal media (all from Invitrogen)]. After 2 h, the media was changed to B-27 media (Neurobasal media containing 1× B-27 supplement, 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin). At 4 d in vitro (DIV), half of the media was replaced with fresh B-27 media.
Treatment of primary cortical cultures with α-syn oligomers derived from A53T α-syn mice.
Triton X-100-soluble lysates from the hippocampus, spinal cord, or olfactory bulbs of symptomatic A53T α-syn were subjected to SEC, and fractions corresponding to 53-Å-sized α-syn oligomers were used for experiments. The fractions were concentrated, and protein concentration was determined by the BCA protein assay (Pierce). To immunodeplete α-syn oligomers for control samples, 2 μg of syn211 antibody (Sigma) was incubated with 100 μg of fractionated lysate in a total volume of 200 μl for 6 h at 4°C. Another set of samples was also incubated with mouse IgG (Santa Cruz Biotechnology) in parallel. Protein G-conjugated beads (4% final, Fast Flow; Sigma) were equilibrated in 25 mm HEPES and 150 mm NaCl, pH 7.4, and then incubated with the immunocomplexes for 1 h at 4°C. α-Syn oligomers were then pulled down by centrifugation at 2000 × g for 2 min, and the protein concentration of the supernatant was determined. One microgram of total protein per well of this supernatant was directly added to primary cultures grown in a 96-well plate at 9 DIV, in a total media volume of 200 μl/well. For samples incubated with IgG, the concentration of α-syn oligomers in the supernatant was estimated by quantitative Western blot, using purified recombinant α-syn as a standard, and found to be ∼10 ng oligomers/μg total protein. In addition to fractions immunodepleted of α-syn, total fractionated hippocampal lysate was used as a negative control (1 μg lysate/well), because this tissue contains a low level of α-syn oligomers (see Figs. 1, 2). Cells were harvested 1 and 3 d after addition of the oligomers or immunodepleted supernatants and analyzed for neurodegeneration.
Figure 1.

Region-specific accumulation of Triton X-100-insoluble α-syn in symptomatic A53T α-syn transgenic mice. a, Tissues from asymptomatic (4–6 months old) and symptomatic (11–12 months old) A53T α-syn transgenic mice, as well as 11-month-old transgenic mice expressing human wild-type α-syn, were subjected to extraction with 1% Triton X-100 buffer (T-soluble), followed by Western blot analysis using the human-specific anti-α-syn monoclonal antibody LB509. NSE was used as a loading control. TH served as a marker of catecholaminergic regions. Regions containing α-syn lesions [SC and cerebellum (Cr)] as well histologically unaffected regions [SN, OB, and hippocampus (H)] were analyzed. b, The insoluble pellets obtained from Triton X-100 fractions were extracted with buffer containing 2% SDS (T-insoluble). Vimentin (Vim) was used as a loading control. The markers on the left indicate the mobility of standards with known molecular masses on SDS-PAGE in kilodaltons. c, Immunohistochemical analysis demonstrating gliosis in areas of the nervous system associated with α-syn pathological inclusions. Tissue sections from olfactory bulb (A, B, E, F ) and spinal cord (C, D, G, H) from 12-month-old nTg (A, C, E, G ) and symptomatic A53T α-syn transgenic (B, D, F, H ) mice were stained with an antibody to GFAP (A–D ) or α-syn (syn505) (E–H ). Staining with syn505 reveals abundant α-syn pathological inclusions in spinal cord of the A53T α-syn transgenic mice (H) and the lack thereof in the olfactory bulb (F) and in the tissues from the nTg mice (E, G). Staining with an anti-GFAP antibody demonstrates significant gliosis in the spinal cord of the symptomatic A53T α-syn transgenic mice (D), whereas the olfactory bulbs of A53T α-syn transgenic mice (B) as well as tissues from nTg mice (A, C) demonstrate less gliosis. Scale bar, 100 μm.
Figure 2.

Identification and regional distribution of α-syn oligomers in A53T α-syn transgenic mice. Triton X-100-soluble extracts from the indicated regions of the nervous system of symptomatic mice were subjected to native SEC. The SEC fractions were then analyzed by SDS-PAGE/Western blot for α-syn using the human-specific anti-α-syn monoclonal antibody syn211. NSE was used as loading control. α-Syn monomer eluted at a volume corresponding to a 34-Å-sized particle, migrating as a 19 kDa species by SDS-PAGE. High-molecular-weight oligomers eluted between 36 and 113 Å. The horizontal marker indicates the apparent molecular radius in angstroms (Å) that corresponds to the elution volume of globular protein standards analyzed by SEC, whereas the vertical marker indicates the mobility of protein standards with known molecular mass on SDS-PAGE in kilodaltons. Representative blots of each region analyzed are shown. The graph on the bottom right shows a quantitative densitometry analysis of the SEC/SDS-PAGE blots from symptomatic and asymptomatic mice, revealing equal relative levels of soluble oligomeric α-syn from tissues of asymptomatic and symptomatic mice. A quantitative analysis of each oligomeric form is shown in supplemental Figure 2 (available at www.jneurosci.org as supplemental material). Total α-syn oligomers levels are expressed as percentage of total soluble α-syn. Mean ± SEM values; n = 3–5 for all tissues were plotted. *p = 0.02, SC symptomatic versus Hipp symptomatic, using a one-way ANOVA with Tukey's post hoc test. Combination of oligomer levels from all inclusion-bearing regions were also compared with those found in all non-inclusion-bearing regions and also did not reveal a statistically significant difference (percentage of total oligomers: regions with inclusions, 44.1 ± 9.4%, n = 9; regions without inclusions, 41.5 ± 4.6%, n = 28; mean ± SEM values; p = 0.63, Student's t test analysis). MW, Molecular weight.
Assessment of neurodegeneration.
Primary cortical neurons grown on a 96-well plate were washed briefly in warm PBS, followed by immediate fixation in 4% formaldehyde for 15 min. Cells were incubated with PBS containing 0.3% Triton X-100 for 20 min with gentle shaking and then with Odyssey blocking buffer (Li-Cor Biosciences) for 1 h. Anti-neurofilament antibody (1:1000; mouse IgG 2H3; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) was then incubated overnight in blocking buffer at 4°C with gentle shaking, followed by washing in PBS with 0.1% Tween 20 for 20 min. IRdye800-conjugated anti-mouse IgG antibodies (1:1000 dilution; Li-Cor Biosciences) were incubated in blocking buffer for 1 h. Draq5 (nuclear stain; 1:10,000 dilution; Li-Cor Biosciences) and Sapphire700 (nonspecific cytosolic stain; 1:1000 dilution; Li-Cor Biosciences) were added with the secondary antibody for determination of cell volume and number. Cells were washed four times in PBS and 0.1% Tween 20 and scanned on an Odyssey infrared imaging system (Li-Cor Biosciences). Neurofilament intensity was determined using Odyssey software (version 2.1; Li-Cor Biosciences). Toxicity assays were repeated with two separate cell culture preparations with n = 6 for each preparation.
Statistical analysis.
One-way ANOVA with Tukey's post hoc test was used to assess neurodegeneration in primary cortical cultures as well as SEC analysis of α-syn oligomers, and p < 0.05 was considered significant. For determination of values that varied significantly from the mean, Grubbs test was used to determine Z values. α-Syn oligomer levels that had Z values over 1.15 (n = 3), 1.48 (n = 4), and 1.71 (n = 6) were excluded from the analysis. For direct tissue-to-tissue comparisons as well as asymptomatic/symptomatic comparisons of oligomers from the SC (shown in supplemental Fig. 2, available at www.jneurosci.org as supplemental material), a paired Student's t test was used, and p values <0.05 were considered significant. Two-way ANOVA with a Bonferroni's post hoc test was used for the seeding assays shown in Figure 4 (n = 4), and p values <0.05 were considered significant.
Figure 4.
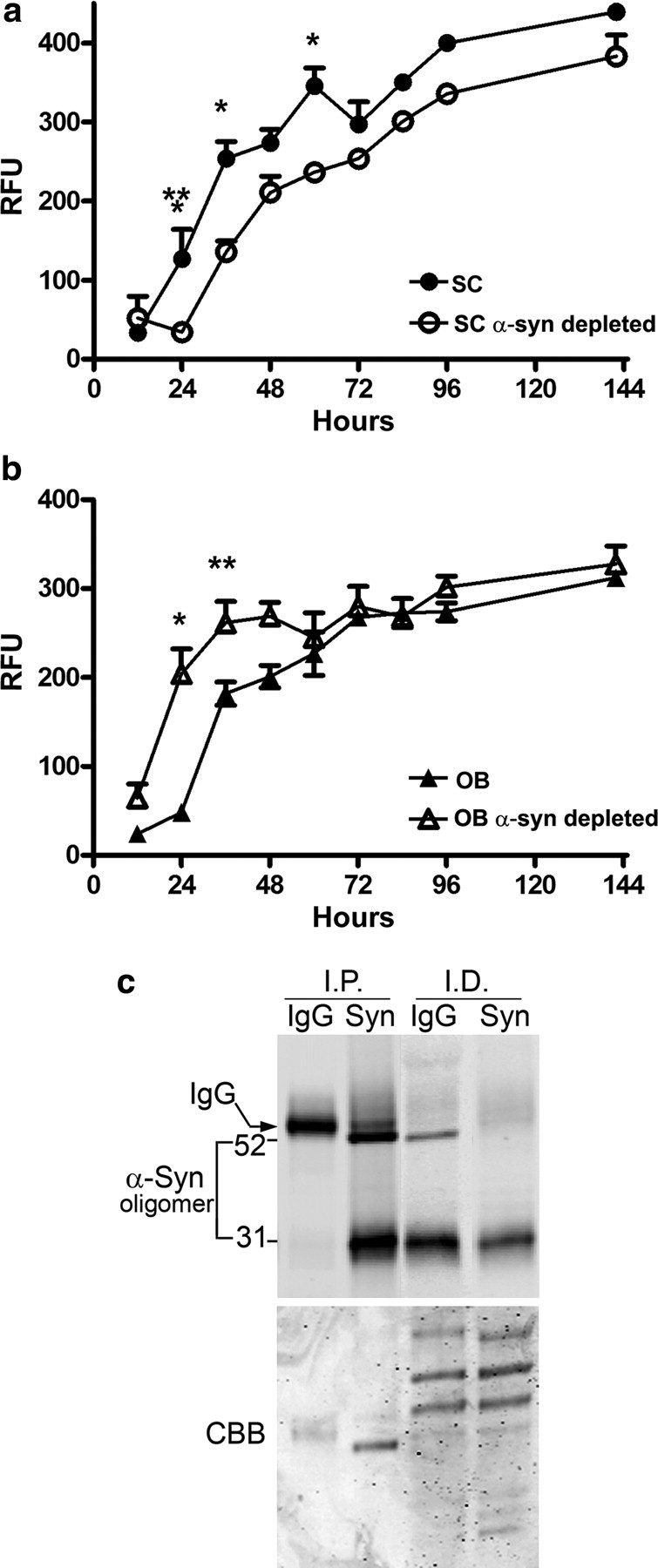
Effects of α-syn oligomers derived from symptomatic A53T α-syn transgenic mice on the kinetics of α-syn fibril formation. The kinetics of forming fibrils from recombinant purified α-syn in vitro was assessed using the amyloid binding fluorescent dye Thioflavin T and expressed as relative fluorescence units (RFU). a, Oligomers isolated from SC significantly decreased the lag phase of fibril formation of recombinant α-syn; b, oligomers isolated from the OB prolong the lag phase of fibril formation. α-Syn was depleted from oligomeric fractions by immunoprecipitation with the monoclonal antibody syn211. Values represent the mean ± SEM (n = 4–5; two-way ANOVA, *p = 0.001, **p = 0.05, ***p = 0.01). c, Western blot analysis using syn211 of immunoprecipitated (I.P.) α-syn and the corresponding immunodepleted (I.D.) fractions from the OB used in the seeding assays (similar results were obtained with the SC fractions). Total protein was visualized by Coomassie brilliant blue (CBB) staining showing equal loading of samples.
Results
Regional distribution and biochemical properties of α-synuclein oligomers extracted from A53T α-synuclein mice
To characterize the regional distribution of aggregated forms of α-syn in A53T α-syn mice, we first sought to biochemically determine the distribution of insoluble α-syn in different neural regions. Tissues from various regions of the nervous system of asymptomatic (4–6 months old) and symptomatic (11–12 months old) mice were sequentially extracted in detergents of increasing strength. Western blot analysis using an antibody specific for human α-syn, LB509, revealed that the SC and Crb from symptomatic mice contained more Triton X-100-insoluble α-syn compared with asymptomatic mice (Fig. 1 b). Triton X-100-insoluble α-syn from these tissues was present primarily as a 19 kDa monomer but also contained higher molecular weight bands stable to SDS/heat that likely correspond to dimers, trimers, and multimers. Conversely, in other brain regions, such as the SN, OB, and Hipp, α-syn was mainly extracted in the Triton X-100-soluble fraction with only a modest change in solubility as the mice presented with motor symptoms (Fig. 1 a). This is consistent with histological analysis, which demonstrated a paucity of inclusions in these three regions (Giasson et al., 2002). Analysis of age-matched transgenic mice expressing human WT α-syn, which do not exhibit α-syn pathology or motor symptoms at any age, revealed undetectable levels of Triton X-100-insoluble α-syn in all brain regions examined (Fig. 1 b) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These biochemical findings were corroborated by histological examination. In accordance with previous reports (Giasson et al., 2002), immunohistological analysis revealed abundant α-syn inclusions (both perikaryal and neuritic) in the spinal cord of symptomatic A53T α-syn mice (Fig. 1 c, H). To assess whether indications of neuronal injury are present in areas that contain α-syn inclusions, tissue sections from symptomatic A53T α-syn transgenic mice and nTg mice were histologically examined for gliosis with an anti-GFAP antibody. Robust astrocytic gliosis is observed in the spinal cord of symptomatic A53T α-syn mice (Fig. 1 c, D) but not in the olfactory bulb (Fig. 1 c, B), a brain region in which inclusions of α-syn are scarcely detected. These data suggest that formation of insoluble α-syn temporally correlates with motor symptom onset and occurs in regions that contain histological α-syn inclusions.
To further investigate α-syn species present in the nervous system of A53T α-syn mice, we initially focused on the SC because of the neurodegenerative changes that occur in this region once the mice develop symptoms (Fig. 1 c) (Giasson et al., 2002). Triton X-100-soluble extracts from the SC of symptomatic A53T α-syn transgenic mice were analyzed by SEC, followed by SDS-PAGE/Western blot analysis. Previous SEC analysis of recombinant purified α-syn had shown that although the monomeric form elutes from a gel filtration column at a volume corresponding to a 34-Å-sized particle (or 58 kDa globular protein), in SDS-PAGE, it migrates to an apparent molecular weight of nearly 19 kDa (Weinreb et al., 1996). This highly reproducible observation reflects the elongated, unstructured conformation of α-syn in solution. SEC analysis of Triton X-100-soluble SC extracts from A53T α-syn transgenic mice revealed the presence of soluble 53-Å-sized oligomers, with apparent molecular weights of 59, 52, and 31 kDa on SDS-PAGE (Fig. 2) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These high-molecular-weight α-syn species were the most abundant oligomeric intermediates detected in the SC of symptomatic mice (Fig. 2). In addition to the 53-Å-sized α-syn oligomers, a minor amount of α-syn oligomers eluted at nearly the same volume as the monomeric form, corresponding to a 36-Å-sized particle, and migrated at 64 and 38 kDa on SDS-PAGE (Fig. 2) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Next, SEC profiles of the SC of symptomatic mice were compared with those of asymptomatic mice (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material) and revealed a similar SEC profile, indicating that soluble α-syn oligomers accumulate before the formation of insoluble α-syn inclusions and presentation of motor deficits. Additionally, the amounts of oligomers in symptomatic and asymptomatic mice were not significantly different by quantitative densitometry analysis of the SEC/SDS-PAGE blots (Fig. 2). Despite the detection of various oligomeric α-syn species, the SEC analysis did not identify a soluble oligomeric form of α-syn that is specifically correlated with symptom onset (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
To determine whether α-syn oligomers exist in other histologically unaffected regions of symptomatic A53T α-syn transgenic mice, Triton X-100-soluble extracts from the SN, OB, and Hipp were analyzed by SEC. Unexpectedly, this analysis revealed a similar elution profile compared with the inclusion-bearing regions (SC and Crb), with the 53-Å-sized α-syn oligomers as the most prominent species (Fig. 2). Quantification indicated that the relative levels of oligomeric α-syn in the SN and OB were similar to those in the SC (Fig. 2). Additionally, SEC analysis of hippocampal lysates of A53T α-syn transgenic mice as well as most tissues from control mice expressing WT α-syn showed very low levels of soluble oligomeric α-syn (Fig. 2) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), indicating that soluble oligomers detected by SEC form as a result of in vivo processes.
A lack of correlation between the relative abundance of soluble oligomers and the formation of insoluble inclusions is unexpected, given the assumption that these oligomeric species are intermediates of the fibril generating process. Therefore, the biochemical nature of the 53-Å-sized α-syn Triton X-100-soluble oligomers was further characterized. For this analysis, we focused on the OB and SC because of the clear histological differences between these regions, as well as tissue availability. After SEC, the fractions corresponding to Stokes radii of 53 Å were separated from the rest of the fractions and concentrated to obtain an enriched solution of oligomeric species. Treatment of 53-Å-sized oligomers derived from the SC or OB with urea did not affect the 59, 52, and 31 kDa bands, suggesting that these species represent self-associated α-syn molecules that are held together by urea-resistant bonds (Fig. 3 a). Incubation with PK was then performed to reveal potential regions of α-syn that might be protected from proteolysis. Oligomers of the SC and OB were equally sensitive to PK treatment, indicating a lack of buried peptide regions protected from digestion (Fig. 3 b). Together, these data indicate that α-syn oligomers in the brain of A53T α-syn transgenic mice are Triton X-100 soluble, SDS and heat stable, resistant to denaturation by urea, and sensitive to PK. Despite the similarity in these biochemical properties, an interesting distinction between oligomers of the OB and SC was evident during Western blot analysis using syn303, a monoclonal antibody generated against oxidized/nitrated recombinant α-syn (Duda et al., 2002). Immunoblotting with syn303 revealed more prominent immunoreactivity with SC oligomers as opposed to the OB in the bands corresponding to dimers and trimers having an apparent molecular weight of 59, 52, and 31 kDa (Fig. 3 c). The same samples were analyzed by immunoblot using LB509, a human-specific antibody that does not preferentially detect oxidized forms of α-syn, showing approximately equal amounts of oligomers in each region (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Because syn303 preferentially recognizes oxidized and/or nitrated α-syn in human inclusions (Duda et al., 2002), these data suggest that oxidative modifications to soluble oligomers occur to a greater extent in the SC compared with the OB.
Figure 3.

Biochemical analysis of the 53 Å α-syn oligomers isolated by SEC from A53T α-syn transgenic mice. The fraction corresponding to the 53-Å-sized oligomers is shown in lanes 2 and 4, and the monomer is shown in lanes 1 and 3 of each blot. a, Fractionated lysates at 0.5 mg/ml were incubated with 8 m urea for 1 h at 25°C; b, proteinase K at 100 μg/ml for 30 min at 37°C. The results of the treatments were the same for oligomers isolated either from the SC (shown here) or OB from both symptomatic and asymptomatic mice. The human-specific α-syn monoclonal antibody LB509 (shown here) recognizes an epitope at the C terminus of the protein (residues 115-122). Analysis with the monoclonal antibody syn208 recognizing an epitope at the amyloidogenic region of the protein (residues 87-110) yielded the same results. c, Indication of biochemical diversity between oligomers isolated from regions bearing inclusions (SC) and pathology free (OB) is revealed by the monoclonal antibody syn303, which was generated against oxidized α-syn and preferentially recognizes oligomeric forms of α-syn in human brain tissue with α-syn aggregates (Duda et al., 2002).
Functional and biological properties of α-synuclein oligomers
To determine whether additional differences exist between oligomers of the SC compared with the OB, we compared the ability of these oligomers to influence the kinetics of the in vitro polymerization of α-syn. Based on previous structural analysis of in vitro formed oligomers (Conway et al., 2000a; Apetri et al., 2006), the size of the oligomers detected here (53 Å) is consistent with an intermediate species in the pathway of forming α-syn amyloid fibrils. Therefore “on-pathway” oligomers should act as “seeds” in the in vitro polymerization of α-syn and thus accelerate the initial phase of this process (Jarrett and Lansbury, 1993; Wood et al., 1999). Indeed, enriched oligomeric fractions from SC shortened the lag phase and increased the rate of recombinant purified α-syn forming amyloid fibrils as indicated by the changes in Thioflavin T fluorescence (Fig. 4 a) when compared with identical SC oligomer-enriched fractions that were immunodepleted for α-syn. In contrast, oligomeric α-syn extracted from the OB prolonged the lag phase of purified α-syn amyloid fibril formation (Fig. 4 b). These effects on the in vitro kinetics of fibril formation were dependent on α-syn because immunodepletion of the oligomers restored the kinetic profile of the purified α-syn aggregation. Moreover, addition of α-syn monomers extracted from SC or OB of A53T α-syn transgenic mice as well as of SEC fractions from SC or OB of mice expressing WT α-syn corresponding to ≥53 Å did not alter the kinetics of α-syn aggregation (Table 1) (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Collectively, the data indicate that oligomers derived from the OB either represent “off-pathway” oligomers or contain an oligomer-associated factor that kinetically delays the formation of α-syn fibrils. The absence of the detectable α-syn inclusions in the OB of the A53T α-syn transgenic mice supports these findings.
Table 1.
Effects of α-syn monomer and oligomers extracted from different brain regions of A53T and WT α-syn transgenic mice on the kinetics of recombinant α-syn fibril formation in vitro
| Brain region/fraction | RFU (ThioT) |
|---|---|
| α-syn alone | 181.1 ± 9.8 |
| α-syn + A53T SC 53Å oligomer | 223.5 ± 8.7* |
| α-syn + A53T SC monomer | 159.0 ± 10.7 |
| α-syn + A53T OB 53Å oligomer | 136.8 ± 11.3* |
| α-syn + A53T OB monomer | 156.8 ± 18.1 |
| α-syn + WT SC 53Å fraction | 187.4 ± 11.3 |
| α-syn + WT OB 53Å fraction | 182.8 ± 5.3 |
The values represent the mean ± SEM (n = 4–6). Fluorescence intensity was recorded after 24 h incubation.
*p < 0.05, Student's t test analysis compared with purified recombinant α-syn protein alone. RFU, Relative fluorescence units.
To determine the toxicity of SC and OB oligomers, SEC fractions containing oligomers were added to primary cortical cultures, and neuronal toxicity was assessed at 1 and 3 d after treatment by measuring total cell number using a fluorescent nonspecific cell stain in combination with a nuclear stain. Neurodegeneration was also assessed by immunostaining for neurofilament protein, which is a more sensitive assay that can measure degeneration of neurites before occurrence of more severe nuclear toxicity (Zala et al., 2005). Although toxicity of α-syn is presumed to occur through intracellular events in PD, recent studies have indicated that α-syn aggregates are taken up by neurons in culture through endocytosis and can be disseminated from cell to cell, causing widespread neurotoxicity (Desplats et al., 2009). Furthermore, other studies have shown that in vitro formed α-syn oligomers exhibit toxicity when applied externally to cell cultures, likely through disruptions in lipid bilayers (Kayed et al., 2003, 2004). Therefore, we considered this experimental paradigm as a reliable proxy for the potential toxicity that may occur in vivo. SEC fractions containing the 53-Å-sized oligomers were added to cultures, and neurodegeneration was assessed. We found that addition of this SEC fraction containing α-syn oligomers derived from the SC resulted in a significant decline in cell viability as determined by analysis of cell volume and total cell number, whereas the same SEC fraction obtained from the OB had no effect (Fig. 5 a). We also observed a decrease in neurofilament immunostain intensity, attributed to neurite degeneration, in cultures treated with SC oligomers but not oligomers obtained from the OB (Fig. 5 b). The toxicity of SC oligomers was observed 1 d after treatment and persisted until 3 d after addition of the oligomers. This effect is presumed to be attributable to the presence of oligomeric α-syn because depletion with an anti-α-syn antibody reversed the neurodegeneration observed in the SC-treated cultures (Fig. 5 a,b). Additionally, the observed toxicity differences between the OB and SC did not result from addition of different quantities of oligomers, because we quantified the amount of α-syn oligomers that were added to the primary cultures by immunoprecipitation and Western blot (Fig. 5 c). These data suggest, that despite similar SEC profiles and biochemical properties, α-syn oligomers display different cellular toxicities. The histological analysis shown in Figure 1 also supports the neuron toxicity findings, because astrogliosis is more prominent in the SC.
Figure 5.
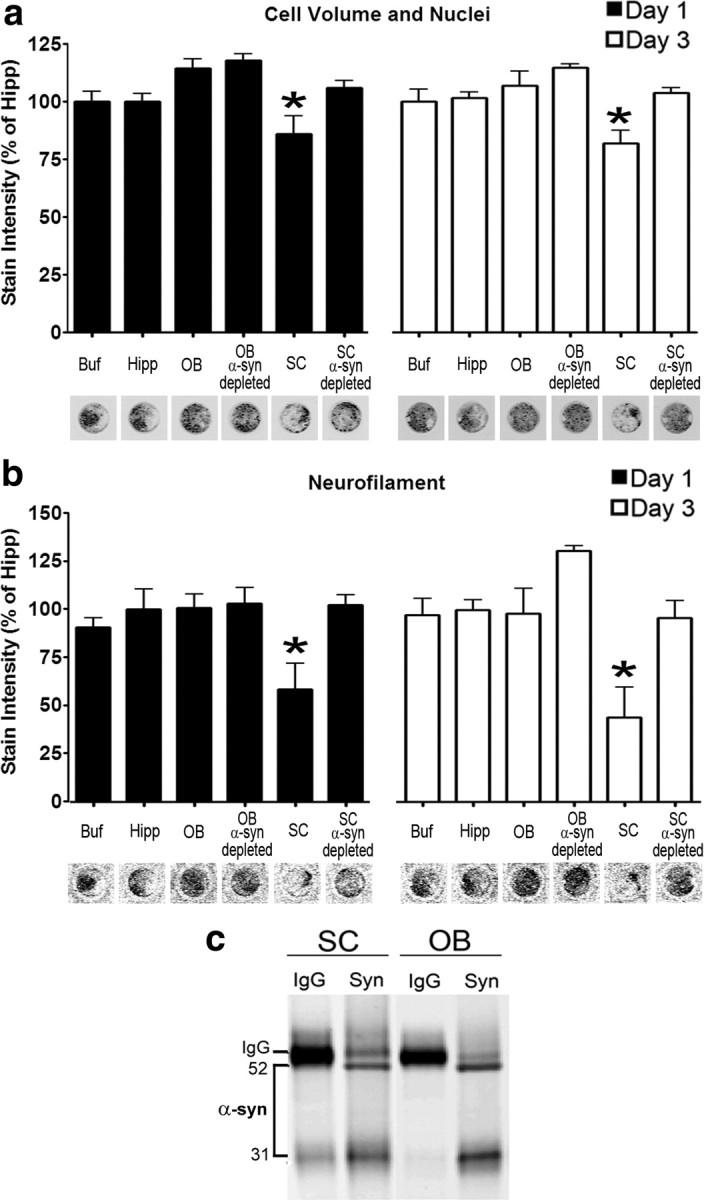
In vitro toxicity of α-syn oligomers derived from the spinal cord of symptomatic A53T α-syn transgenic mice. a, Total cell number and volume of primary mouse cortical cultures was determined by a combination of Sapphire700 (nonspecific cytosolic stain) and Draq5 (nuclear stain) after the addition of SEC fractions containing α-syn oligomers. b, Neurite degeneration was assessed by quantifying neurofilament levels by immunostaining. Twenty-five millimolar HEPES/150 mm NaCl, pH 7.4 (Buf), and equivalent SEC fractions from the Hipp were used as controls. SEC fractions from the SC and OB were incubated with either IgG or anti-α-syn antibodies (syn211) to deplete α-syn oligomers and determine whether neurodegeneration is mediated by the presence of α-syn (n = 5–10 for each time point; *p < 0.05). Representative immunostaining of Sapphire700/Draq5 and neurofilament from scans of the 96-well culture plate are shown under the graph. c, The amount of α-syn oligomers obtained from SC and OB SEC fractions added to the primary cultures were determined by immunoprecipitation/Western blot analysis. Samples were immunoprecipitated and blotted using the monoclonal antibody syn211 or mouse IgG as a control. The bands were analyzed by densitometry and found to be equal (integrated intensity: OB, 15.2; SC, 15.3).
Although previous histological analysis of A53T α-syn transgenic mice at 4–5 months indicated normal cell structure without neurodegenerative changes (Giasson et al., 2002), we sought to determine whether the presence of detergent-soluble oligomers detected by SEC/SDS-PAGE is reflected on behavioral changes in the mice. Ambulatory activity measurements revealed that A53T α-syn transgenic mice had similar motor activity compared with nTg mice (Table 2 A). Food intake and body weight of A53T α-syn transgenic mice were also assessed, because it has been well established that feeding behavior critically depends on the function of the substantia nigra pars compacta (SNpc) → caudate–putamen (CPu) circuit (Zigmond and Stricker, 1972; Szczypka et al., 2001). We found that A53T α-syn transgenic mice had similar body weights and consumed slightly more food compared with nTg age-matched controls, suggesting that the SNpc → CPu circuit is functional (Table 2 A). Growth curves, by measuring body weight starting at 2 weeks, did not reveal any differences in the development of A53T α-syn transgenic mice for up to 11 months of age (data not shown). After 11 months, A53T α-syn transgenic mice experience motor deficits and a rapid decline in body weight at variable time points. These data show that the presence of detergent-soluble α-syn oligomers in the SN is associated with only mild behavioral changes.
Table 2.
Dopamine-dependent activities of 4- to 6-month-old A53T α-syn and age-matched nontransgenic mice (A) and catechol levels in symptomatic A53T α-syn transgenic mice and age-matched controls (B)
| A53T | Nontransgenic | |
|---|---|---|
| A | ||
| Body weight (g) (n = 6) | 33.8 ± 1.9 | 33.7 ± 0.7 |
| Food intake (g/g body weight) (n = 6) | 0.16 ± 0.03* | 0.12 ± 0.03 |
| Activity (beam breaks/24 h) (n = 4–5) | 78,941 ± 12,280 | 76,610 ± 11,859 |
| B | ||
| Dopamine (fmol/μg protein) (n = 9) | 1000.2 ± 78.3* | 612.3 ± 71.5 |
| DOPAC (fmol/μg protein) (n = 9) | 157.6 ± 28.0 | 105.4 ± 10.0 |
| DOPAC/dopamine (n = 9) | 0.17 ± 0.04 | 0.19 ± 0.04 |
| Serotonin (fmol/μg protein) (n = 7) | 56.1 ± 4.0 | 44.5 ± 2.1 |
| Norepinephrine (fmol/μg protein) (n = 7) | 56.2 ± 6.2 | 42.2 ± 7.0 |
The values represent the mean ± SEM.
*p < 0.05, Student's t test analysis.
Additional biochemical evaluation was also performed to determine the integrity of the dopaminergic system in asymptomatic and symptomatic mice. Enzyme levels related to dopamine synthesis and metabolism were quantified, such as TH, DAT, and DDC, in symptomatic mice and showed no statistically significant difference compared with nTg mice (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Additionally, striatal catechol levels were measured in both asymptomatic and symptomatic A53T α-syn transgenic mice, as well as in nontransgenic control mice, by HPLC with electrochemical detection. Analysis of 4-month-old mice showed that striatal catechol levels of A53T α-syn transgenic mice were similar to nontransgenic age-matched controls [in fmol/μg protein: nontransgenic mice, DA, 598 ± 82.5; 3,4-dihydroxyphenylacetic acid (DOPAC), 100.4 ± 27.9; A53T α-syn transgenic mice, DA, 703 ± 30; DOPAC, 89.9 ± 11.5; n = 3–4; mean values ± SEM]. Analysis of symptomatic A53T α-syn transgenic mice revealed slightly elevated levels of striatal dopamine, whereas the levels of other analytes tested, including DOPAC, norepinephrine, and serotonin, did not significantly change (Table 2 B). Because the ratio of DOPAC to dopamine reflects the relative rate of DA turnover, these data suggest that the increased concentration of DA may be attributable to increased synthesis or storage. Catechol levels in the OB of symptomatic A53T α-syn transgenic mice were also similar to age-matched nontransgenic controls (data not shown). Collectively, these data suggest that the presence of Triton X-100-soluble oligomeric α-syn in dopaminergic regions is not associated with alterations in the levels of enzymes involved in dopamine metabolism and coincides with subtle differences in the production of catechols in vivo as the levels of dopamine increase with aging in A53T α-syn transgenic mice.
Discussion
The documentation that Lewy bodies and other neuronal inclusions are composed of aggregated α-syn has positioned the process of α-syn aggregation as a critical biochemical event in the pathogenesis of neurodegeneration (Spillantini et al., 1997; Baba et al., 1998; Goedert, 2001). Considerable advances in cell-free systems and cell-model systems have indicated that the process of aggregation includes a committed step of α-syn dimer formation that is held together by hydrophobic interactions induced by a conformational transition to β-sheet structure (Conway et al., 2000b; Serpell et al., 2000; Uversky et al., 2001). The dimer represents a thermodynamically stable entity allowing the formation of oligomers that will continue to grow in linear β-sheets, forming polarized protofibrils and eventually the typical amyloid fibrils. Alternatively, the transition of oligomers to protofibrils can be kinetically arrested by interactions with small molecules, other proteins, or unidentified cellular constituents, allowing these oligomers to assume secondary structures such as annular pores or spheres (Conway et al., 2001; Li et al., 2004; Norris et al., 2005).
The emerging appreciation of conformationally distinct oligomers of not only α-syn but also other proteins that form amyloid structures in human neurodegenerative disorders has sparked a considerable interest in the pathobiological role of oligomers and the mechanism by which they induce neuron dysfunction and neurodegeneration. However, in part because of methodological challenges, there is limited insight regarding the biochemical and biological properties of oligomeric species formed in vivo. Therefore, the in vivo regional distribution and temporal formation of α-syn oligomeric intermediates was investigated in the previously characterized transgenic mouse model expressing human A53T α-syn (Giasson et al., 2002) that recapitulates the aggregation of α-syn and inclusion formation. Detailed biochemical and biological analysis of the in vivo formed α-syn oligomers revealed unexpected new insights regarding regional differences in conformation and composition that may govern their ability to promote inclusion formation and induce neuron dysfunction.
Using the previously validated two-dimensional analysis, the presence of detergent-soluble oligomeric forms of α-syn was confirmed in brain regions that contain inclusions and unexpectedly in brain regions devoid of α-syn inclusions, such as the SN and OB. The presence of oligomeric intermediates in regions containing insoluble α-syn inclusions is expected assuming that these α-syn oligomers represent transient intermediates of the polymerization process. The presence of these oligomers in regions that do not form inclusions, at least up to the lifespan of the symptomatic mouse, indicates that the process of aggregation has been initiated in these regions but has not progressed beyond the oligomeric stages. Moreover, the data indicated that oligomers are formed before the formation of inclusions and onset of symptoms (at 4–6 months) and continue to be present at similar levels during aging and symptom onset.
The regional variation is not a result of differences in the levels of human A53T α-syn protein because previous studies using the mouse PrP promoter have shown similar expression levels in various brain regions (Lee et al., 2002), which was confirmed by quantitative Western blot analysis for all regions analyzed (Figs. 1, 2). Furthermore, regional variation was not attributable to generation of oligomers with divergent basic biochemical properties. Detergent-soluble α-syn oligomers isolated from either inclusion-bearing or inclusion-free regions were composed primarily of 53-Å-sized species that are SDS, heat, and urea stable and are sensitive to digestion by proteinase K. However, subtle differences in conformation are evident by the differential reactivity toward oligomer-specific antibodies (Fig. 3 c). These antibodies were generated against oxidized and tyrosine nitrated α-syn oligomers and were shown to preferentially recognize detergent-soluble α-syn oligomers as well as pathological α-syn inclusions in human brain tissue (Duda et al., 2002). These antibodies also recognize oxidative posttranslationally modified α-syn, such as dityrosine crosslinked dimers, which has been considered a critical nucleation event for the assembly of α-syn monomers into fibrils (Souza et al., 2000a; Krishnan et al., 2003). It is possible that the oligomers extracted from SC contain a higher proportion of dityrosine crosslinked oligomers that facilitate amyloid fibril formation. This possibility is in part supported by the data documenting that SC-derived oligomers accelerated the aggregation of purified α-syn in a typical fibril formation assay (Fig. 4 a, Table 1). In contrast, oligomers isolated from the OB prolonged the lag phase of α-syn aggregation in the same assay (Fig. 4 b, Table 1). The contrasting biological function of the different oligomeric populations was also evident in primary neuron cultures in which oligomers derived from SC but not OB induced cell death and dramatic neurite degeneration as determined by neurofilament stain intensity (Fig. 5).
One candidate pathway that may explain the kinetic deceleration of fiber formation of the OB-extracted oligomers may relate to the previously recognized ability of oxidation products of catechols, such as the ortho-quinones of dopamine and DOPAC, to kinetically arrest the formation of α-syn fibrils (Conway et al., 2001; Li et al., 2005; Norris et al., 2005). This non-covalent interaction has been documented in cell-free systems and recently in cellular model systems (Mazzulli et al., 2006, 2007; Mosharov et al., 2009; Outeiro et al., 2009). Interestingly, we found that striatal catechol levels of A53T α-syn transgenic mice increase with age, although they remain stable in nontransgenic mice. This occurred without an apparent increase in the levels of tyrosine hydroxylase, the DA synthesizing rate-limiting enzyme. Tyrosine hydroxylase uses tyrosine to catalyze the formation of l-3,4-dihydroxyphenylalanine, a process that is dependent on molecular oxygen, Fe+2, and the cofactor BH4 [6(R)-l-erythro-5,6,7,8-tetrahydrobiopterin] (Fitzpatrick, 1989). It has been established that tyrosine hydroxylase activity is intricately regulated through feedback inhibition by catechols, allosteric regulation by various polyanions, and posttranslational modifications such as phosphorylation in the N-terminal region of the protein (Kumer and Vrana, 1996). Therefore, it is possible that the observed increase in striatal DA may result from altered levels of cofactors or posttranslational modifications that increase the enzymatic activity without affecting protein levels. Although an increase in striatal DA may provide a biochemical mechanism for preventing the formation of fibrils in the SNpc of A53T α-syn transgenic mice, genetic and pharmacological manipulations of dopamine levels are required to verify the biological importance of a potential α-syn–catechol interaction in vivo. In relation to PD, it is possible that either a decline in steady-state dopamine levels or cleavage of the DA-interacting C terminus of α-syn contribute to the formation of Lewy bodies in the SNpc, resulting in degeneration of the nigrostriatal pathway.
Although the dopamine interaction may be prominent for dopaminergic neurons, other intrinsic factors present in non-dopaminergic neurons must be also considered. Previous studies using cell models have shown that molecular chaperones have the ability to alter the levels of soluble oligomeric species (Tetzlaff et al., 2008). It is possible that some regions of A53T α-syn transgenic mice possess elevated levels of molecular chaperones or other intrinsic factors that may engage the soluble oligomers and possibly accelerate clearance or alter their conformation, thus preventing their conversion into insoluble fibrils within the lifespan of the A53T α-syn transgenic mice. This ability may be sustained with age in some brain regions (such as the Hipp) but decline or become overwhelmed in other susceptible neuronal regions (such as the Crb and SC) that also contain oxidatively modified α-syn species that have the potential to promote the polymerization process, resulting in the formation of insoluble inclusions (Souza et al., 2000a).
Overall, the data provide a systematic identification and biochemical characterization of in vivo formed oligomeric intermediates of α-syn in the A53T α-syn transgenic mice that exhibit neurodegeneration and α-syn inclusion formation. Although the analysis does not include transient unstable oligomeric conformations that remain unaffected through the extraction process, these more stable and well represented α-syn assemblies were shown to vary in conformation and possibly composition based on the region of origin. These differences in addition to other unrecognized regional specific factors may be responsible for the ex vivo and in vivo toxicity of the oligomers. The insights gained by the present analysis highlight the importance of therapeutic interventions with agents that selectively enhance the clearance of α-syn. Those compounds are likely to prevent the formation of both soluble and insoluble toxic α-syn assemblies, providing therapeutic benefit in PD and related disorders.
Footnotes
This work was supported by the National Institutes of Health Grants AG09215 (B.I.G), NS053488 (B.I.G.), and R01NS051303 (D.K.), United States Public Health Service Grant AG13966 (H.I.), National Institute of Environmental Health Sciences Center of Excellence in Environmental Toxicology Grant ES013508 (H.I), and National Institute of Neurological Disorders and Stroke Award F32NS066730 (J.R.M.). H.I. is the Gisela and Dennis Alter Research Professor of Pediatrics.
References
- Apetri MM, Maiti NC, Zagorski MG, Carey PR, Anderson VE. Secondary structure of α-Synuclein oligomers: characterization by Raman and atomic force microscopy. J Mol Biol. 2006;355:63–71. doi: 10.1016/j.jmb.2005.10.071. [DOI] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000a;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000b;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of α-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Liu W, Hardy J, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Sima A, D'Amato C, Albin R, Gilman S, Yen SH. Widespread alterations of α-synuclein in multiple system atrophy. Am J Pathol. 1999;155:1241–1251. doi: 10.1016/s0002-9440(10)65226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol. 2002;52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick PF. The metal requirement of rat tyrosine hydroxylase. Biochem Biophys Res Commun. 1989;161:211–215. doi: 10.1016/0006-291x(89)91582-9. [DOI] [PubMed] [Google Scholar]
- Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VM. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson's disease. J Neurosci Res. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Lansbury PT., Jr Is there a cause-and-effect relationship between α-synuclein fibrillization and Parkinson/'s disease? Nat Cell Biol. 2000;2:E115–E119. doi: 10.1038/35017124. [DOI] [PubMed] [Google Scholar]
- Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in α-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Jakes R, Crowther RA, Lee VM, Trojanowski JQ, Iwatsubo T, Goedert M. Epitope mapping of LB509, a monoclonal antibody directed against human alpha-synuclein. Neurosci Lett. 1999;269:13–16. doi: 10.1016/s0304-3940(99)00411-5. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, Citron M, Carpenter JF. Oxidative dimer formation is the critical rate-limiting step for Parkinson's disease alpha-synuclein fibrillogenesis. Biochemistry. 2003;42:829–837. doi: 10.1021/bi026528t. [DOI] [PubMed] [Google Scholar]
- Kumer SC, Vrana KE. Intricate regulation of tyrosine hydroxylase activity and gene expression. J Neurochem. 1996;67:443–462. doi: 10.1046/j.1471-4159.1996.67020443.x. [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HT, Lin DH, Luo XY, Zhang F, Ji LN, Du HN, Song GQ, Hu J, Zhou JW, Hu HY. Inhibition of alpha-synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha-synuclein. Implication for dopaminergic neurodegeneration. FEBS J. 2005;272:3661–3672. doi: 10.1111/j.1742-4658.2005.04792.x. [DOI] [PubMed] [Google Scholar]
- Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL. Dopamine and l-dopa disaggregate amyloid fibrils: implications for Parkinson's and Alzheimer's disease. FASEB J. 2004;18:962–964. doi: 10.1096/fj.03-0770fje. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Mishizen AJ, Giasson BI, Lynch DR, Thomas SA, Nakashima A, Nagatsu T, Ota A, Ischiropoulos H. Cytosolic catechols inhibit α-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J Neurosci. 2006;26:10068–10078. doi: 10.1523/JNEUROSCI.0896-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Armakola M, Dumoulin M, Parastatidis I, Ischiropoulos H. Cellular oligomerization of α-synuclein is determined by the interaction of oxidized catechols with a C-terminal sequence. J Biol Chem. 2007;282:31621–31630. doi: 10.1074/jbc.M704737200. [DOI] [PubMed] [Google Scholar]
- Mori F, Tanji K, Zhang H, Kakita A, Takahashi H, Wakabayashi K. α-Synuclein pathology in the neostriatum in Parkinson's disease. Acta Neuropathol. 2008;115:453–459. doi: 10.1007/s00401-007-0316-4. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. Both familial Parkinson's disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Lee VM. Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 2004;60:17–54. doi: 10.1016/S0070-2153(04)60002-0. [DOI] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VM. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem. 2005;280:21212–21219. doi: 10.1074/jbc.M412621200. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Klucken J, Bercury K, Tetzlaff J, Putcha P, Oliveira LM, Quintas A, McLean PJ, Hyman BT. Dopamine-induced conformational changes in alpha-synuclein. PLoS ONE. 2009;4:e6906. doi: 10.1371/journal.pone.0006906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000a;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Lee VM, Ischiropoulos H. Chaperone-like activity of synucleins. FEBS Lett. 2000b;474:116–119. doi: 10.1016/s0014-5793(00)01563-5. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, Palmiter RD. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron. 2001;30:819–828. doi: 10.1016/s0896-6273(01)00319-1. [DOI] [PubMed] [Google Scholar]
- Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT, McLean PJ. CHIP targets toxic α-synuclein oligomers for degradation. J Biol Chem. 2008;283:17962–17968. doi: 10.1074/jbc.M802283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanoswki JQ, Lee VM. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 1998;44:415–422. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- Zala D, Benchoua A, Brouillet E, Perrin V, Gaillard MC, Zurn AD, Aebischer P, Déglon N. Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol Dis. 2005;20:785–798. doi: 10.1016/j.nbd.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Stricker EM. Deficits in feeding behavior after intraventricular injection of 6-hydroxydopamine in rats. Science. 1972;177:1211–1214. doi: 10.1126/science.177.4055.1211. [DOI] [PubMed] [Google Scholar]