

Abstract
Purpose
Alterations in transforming growth factor-β (TGF-β) signaling occur early during malignant transformation of renal epithelial cells and are associated with loss of type III TGF-β receptor (TβRIII) expression. We evaluated the role of TβRIII in mediation of apoptosis using in vitro cell culture and in vivo animal models of clear cell renal cell carcinoma.
Experimental Design
TβR3 expression was manipulated with adenoviral gene vector delivery system in vitro and in vivo. Induction of apoptosis and signaling through the Smad and mitogen-activated protein kinase (MAPK) pathways were examined at various time points after infection. To study viral oncolysis in vivo, human renal cell carcinoma cells were implanted s.c. in the flanks of nude mice and treated with intratumoral injections of adenovirus.
Results
Restoring TβRIII expression in clear cell renal cell carcinoma resulted in a marked induction of apoptosis using in vitro cell culture and in vivo animal models. The expression of the cytoplasmic domain, but not the extracellular domain, of TβRIII mimicked the induction of apoptosis by full-length TβRIII in cell culture and the growth inhibition of tumors in athymic nude mice. TβRIII-associated apoptosis was not dependent on signaling through the canonical TGF-β/Smad pathway but was mediated through p38 MAPK.
Conclusion
These findings indicate a novel mechanistic antitumor function forTβRIII and further support its role as an important tumor suppressor in clear cell renal cell carcinoma.
Approximately 210,000 people worldwide were diagnosed with renal cell carcinoma in 2007, and roughly one third of these patients will die from their disease (1). Although surgical resection cures 70% to 90% of patients with clinically localized disease, patients with metastatic renal cell carcinoma face a poor prognosis, with a median survival duration of 1 year and a 2-year survival rate of only 10% to 20% (2). The lack of effective systemic therapy for metastatic renal cell carcinoma is, in part, due to a fundamental lack of understanding of the molecular events that result in cellular transformation, carcinogenesis, and progression in the human kidney.
Through the use of genomic profiling in human clear cell renal cell carcinoma, we have previously shown that alterations in transforming growth factor-β (TGF-β) signaling occur as an early event in malignant transformation of renal epithelial cells and that dysregulation of TGF-β signaling is associated with loss of type III TGF-β receptor (TβRIII) expression (3). Furthermore, we and others have shown that reexpression of TβRIII in in vitro models of human cancer attenuated the malignant phenotype, which suggests an important role for TβRIII in renal epithelial homeostasis that must be abrogated for renal cell carcinogenesis and progression to occur (3-6). Similarly, TβRIII has been shown to be an important tumor suppressor, the expression of which is attenuated in prostate, breast, ovarian, pancreas, lung, and endometrial cancers (4-9).
TβRIII, an 853-amino-acid transmembrane proteoglycan, is the most abundantly expressed TGF-β receptor, which binds all three TGF-β isoforms with high affinity, and due to its structural characteristics and lack of obvious signaling motifs, is thought to function primarily as a TGF-β coreceptor (10-13). Nonetheless, recent evidence suggests that the function of TβRIII may be more complex (14-17). In this study, we show that reexpression of TβRIII in in vitro and in vivo models of localized and metastatic clear cell renal cell carcinoma results in marked induction of apoptotic-mediated cell death. We further show that these effects occur independently of TGF-β or the presence of signal transduction through the canonical type II TGF-β receptor (TβRII)/type I TGF-β receptor (TβRI)/Smad signaling complex. Finally, we show that intact signaling through the p38 mitogen-activated protein kinase (MAPK) pathway is essential for TβRIII-induced apoptosis. These findings suggest a novel and important function for TβRIII and further strengthen our observations that TβRIII is an essential tumor suppressor in clear cell renal cell carcinoma.
Translational Relevance.
In this investigation, we have shown that reexpression of TβRIII in in vitro and in vivo renal cell carcinoma models induces striking apoptosis in a manner independent of the TGF-β ligand or the presence of a functional type II TGF-β receptor/Smad signal transduction pathway. These findings suggest that TβRIII is an important tumor suppressor in renal cell carcinoma and provide a strong rationale for therapeutic modulation of theTβRIII/p38 MAPK axis as a treatment strategy for renal cancer.
Materials and Methods
Patient sample collection
Tissues were collected from The University of Texas M.D. Anderson Cancer Center according to Institutional Review Board protocols. On ice and under sterile conditions, renal tissue (normal and tumor) was transported to a sterile hood within the pathology department. Tissue was dissected under the direction of a pathologist, and a portion of tissue was processed to establish primary cell cultures.
Cell culture
Primary renal cell carcinoma cultures from index tumors, lymph node metastases, and systemic metastases were established using standard collagenase/DNase techniques to digest tissue and isolate single cells, and were further analyzed for homogeneity with regard to epithelial population using appropriate immunohistochemical markers (vimentin, cytokeratin, and megalin; data not shown). UMRC3 and UMRC6 cells were a kind gift from Dr. Bart Grossman (The University of Texas M.D. Anderson Cancer Center, Houston, TX; ref. 18). UMRC6 cells are representative of localized clear cell renal cell carcinoma and express TβRII and TβRI, but not TβRIII, whereas UMRC3 cells are representative of metastatic clear cell renal cell carcinoma having lost expression of TβRII and TβRIII protein expression (3). Renal cell carcinoma cell lines were maintained in αMEM containing 5% heat-inactivated fetal bovine serum, 1% penicillin-streptomycin-amphotericin B, HEPES, sodium pyruvate, insulin/transferrin/selenium, and epidermal growth factor. Stable transfection of UMRC6 cells using pUHD10-3/ΔTβRII, a gift of Dr. Joan Massague (Howard Hughes Medical Institute, Memorial Sloan-Kettering Cancer Center, New York, NY), was done using Fugene 6 (Roche) according to the manufacturer’s instructions. Stable clones were selected in the above media containing 0.05 mg/mL G418.
Generation of recombinant adenoviral vectors
Human cDNAs encoding TβRII, TβRIII, TβRIII extracellular domain deleted (TβRIIICD), and TβRIII cytoplasmic domain deleted (TβRIIIECD; generous gifts of Dr. Gerard Blobe, Duke University Medical Center, Durham, NC; ref. 16) were cloned into the EcoR1/Sma1 site of shuttle plasmid pDC316 (Microbix). The shuttle vectors and the pBHGlox (delta)E1,3Cre vector containing the adenoviral genome (Microbix) were cotransfected into the packaging (HEK293) cells by using Fugene 6 reagent (Roche). After 48 h of transfection, medium was removed and cells were layered with a 1:1 mixture of 2× media with 1.6% agarose and incubated for 2 wk. Enrichment and purification of selected adenoviral plaques were carried out according to standard methods. A similarly constructed adenovirus vector expressing β-galactosidase construct HCMVSP1lacZ was used as infection control.
Apoptosis assay
For the Annexin V/propidium iodide assay, cells were plated, allowed to adhere overnight, infected with controls (PBS or Ad-βGal) or appropriate adenoviral vectors at 1:100 multiplicity of infection, and incubated for 72 h. Under these conditions, the following reagents were added, depending on the experiments: 2 ng/mL TGF-β1 (R&D Systems), 50 μg/mL TGF-β antibodies (R&D Systems); SB202190, the specific inhibitor of p38 MAPK (Calbiochem); or TβRI kinase inhibitor, LY2109761 (a kind gift from Dr. Jonathan Yingling, Eli Lilly and Company). After 72 h of incubation, floating cells were combined with adhered cells, washed with PBS, stained with Annexin V and propidium iodide (Molecular Probes, Inc.) as indicated by the manufacturer, and evaluated by flow cytometry.
Transient transfections and luciferase assays
UMRC3 cells, UMRC6 cells, and stable clones were plated at 80% confluence on six-well plates and grown overnight. Infection with appropriate adenoviral vectors was carried out as described above, and medium changed 24 h after incubation with adenovirus. Transient transfections were done using Fugene 6 (Roche) with a control plasmid (pRL-CMV) and TGF-β–responsive construct p(CAGA)12MPL-Luc, provided by (Dr. J-M. Gauthier, GlaxoWellcome). Briefly, cells were transfected with 2 μg of (CAGA)12MPL-Luc per well or 2 μg of control plasmid for 15 min. Approximately 12 h after transfection, TGF-β1, SB202190, or LY2109761 was added to the culture medium, depending on the experiments. Cells were harvested 48 h later and assayed for dual luciferase activity (Promega). Total light emission was measured with a luminometer. The data presented are representative experiments of mean values carried out in triplicates (± SD) of the relative light units normalized to the luciferase activity of the internal control vector.
Antibodies and Western blot analyses
Goat anti–phospho-Smad2/3 (Ser433/435), goat anti-TBRII, rabbit anti-TBRIII, and goat anti-Smad2/3 (N-19) antibodies were purchased from Santa Cruz Biotechnology, Inc. Rabbit anti–phospho-p38 MAPK (Thr180/Tyr182), rabbit anti–p38 MAPK, rabbit anti–phospho-p44/42 MAPK (Thr202/Tyr204), rabbit anti – p44/42 MAPK, rabbit anti – phospho-stress-activated protein kinase (SAPK)/c-jun NH2-terminal kinase (JNK) (Thr183/Tyr185), rabbit anti–SAPK/JNK, rabbit anti–phospho-MAPKAPK-2 (Thr222), rabbit anti–MAPKAPK-2, and rabbit anti–β-actin antibodies were purchased from Cell Signaling Technology, Inc. Proteins were harvested in radioimmunoprecipitation assay buffer [50 mmol/L HEPES (pH 7.0), 150 mmol/L NaC1, 0.1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS] containing proteinase and phosphatase inhibitor cocktails (Pierce). Cell extracts were quantitated using the Bradford assay reagent (Bio-Rad) according to the manufacturer’s instructions. Proteins were loaded on SDS-polyacrylamide gels and transferred onto Immobilon-P membrane (Millipore). The membranes were blocked with 5% nonfat milk and hybridized with primary antibodies overnight at 4°C. The secondary antibodies used were horseradish peroxidase–conjugated antigoat or antirabbit secondary antibodies (Cell Signaling Technology). Detection was done using chemiluminescent detection substrate (ECL, Amersham Pharmacia Biotech.).
Ectopic mouse model
Female athymic nude mice (4-5 wk old; BALBc nu /nu) were used in all experiments (National Cancer Institute). UMRC3 cells were briefly treated with trypsin-EDTA, washed twice with PBS, and resuspended in serum-free medium. Cell suspensions of 1 × 106/0.1 mL were injected s.c. in each flank, and tumor growth was monitored twice a week by measuring two perpendicular tumor diameters using a precision caliper. The tumor volumes were calculated using the following equation: volume (mm3) = A × B2 × 0.5236, where A is the largest dimension and B is the perpendicular diameter. When tumors reached 100 mm3, intratumoral injections with 50 μL of PBS, Ad-βGal, Ad-TβRII, Ad-TβRIII, Ad-TβRIIICD, and Ad-TβRIII + Ad-TβRII were done, with five animals randomly assigned to each treatment group. At total of three injections were done on days 1, 7, and 14 for a total dose of 1.5 × 1010 plaque-forming units/tumor.
Immunohistochemistry and apoptosis analysis
Tumors were removed 7 d after the last adenoviral injection, and representative portions embedded in paraffin blocks. For cell lines, cells were plated on glass coverslips in wells. Before the detection of TβR expression, cells were fixed onto the coverslips with 3% formalin. For terminal deoxynucleotidyl transferase–mediated dUTP end labeling (TUNEL) assays, the tissue sections were evaluated with the TumorTACS in situ apoptosis detection kit (R&D Systems). For immunohistochemical analyses, antigen retrieval was first done by incubating the slides in Target Retrieval Solution (DAKO) at 100°C in a steam bath for 20 min. Immunoperoxidase staining was done with the Vectastatin Elite ABC kit (Vector Laboratories, Inc.) with TβRII, TβRIII, phospho-Smad2/3 (Santa Cruz Biotechnology), and phospho-p38 MAPK (Cell Signaling Technology). Microscopy was done using a Zeiss universal microscope (Carl Zeiss) equipped with Sony three-chip camera (Sony Corporation of America). For the quantification of TUNEL, the number of positive tumor cells was counted in 10 random fields at ×100 and divided by the total number of cells per field.
Statistical analyses
Differences between the groups were statistically evaluated using the unpaired Student t test. The results are presented as mean ± SD. All P values were two tailed, and P < 0.05 was considered to be statistically significant.
Results
Expression of TβRIII in renal cell carcinoma cell lines induces apoptosis in a TGF-β– and TβRII/Smad–independent manner
Our previous observations in clear cell renal cell carcinoma models representing localized tumor (UMRC6; expressing TβRI and TβRII) and metastatic tumor (UMRC3; expressing TβRI) have suggested a growth inhibitory phenotype of TβR3, which is independent of TGF-β and TβR2 (3). Based on these preliminary observations, we evaluated the potential role of TGF-β and presence of functional TβRII and TβRIII as potential inducers of apoptosis by overexpressing wild-type TβRIII in various clear cell renal cell carcinoma cell lines using the adenoviral construct Ad-TβRIII. A significant induction of apoptosis was observed in UMRC3, UMRC6, and primary clear cell renal cell carcinoma cell lines isolated from index tumors and nodal and systemic metastases where TβRIII was overexpressed, in cultures supplemented with TGF-β1 or with TGF-β blocking antibodies (Fig. 1A). The observed induction of apoptosis was not a consequence of the adenoviral infection itself as revealed by the infection with an adenovirus control (Ad-βGal). Moreover, TβRIII-induced apoptosis was not dependent on the presence of functional extracellular TβRIII domain. All tested clear cell renal cell carcinoma cell lines showed significant induction of apoptosis after infection with the adenoviral vector carrying the deleted extracellular domain TβRIII mutant (Ad-TβRIIICD; Fig. 1B). Conversely, no increased apoptosis was evident in cell models lacking functional TβRII receptor [UMRC3 and UMRC6 stably transfected with a dominant negative TβRII receptor (UMRC6/ΔTBRII)] when infected with a mutant form of TβRIII, which lacks the cytoplasmic domain portion of this receptor (Ad-TβRIIIECD; Fig. 1C).
Fig. 1.
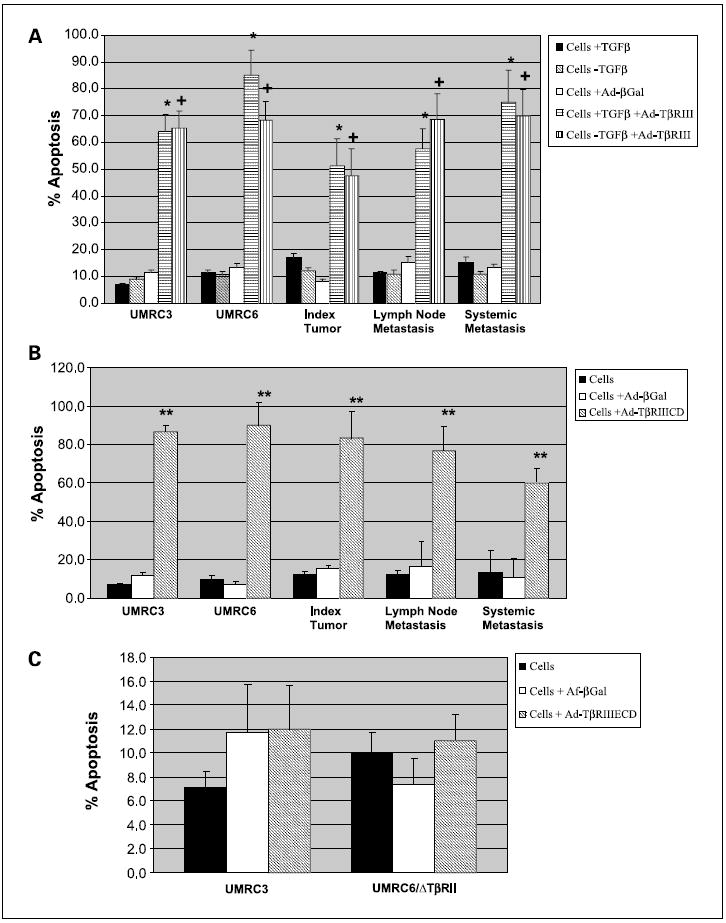
TβRIII induces apoptosis in renal cell carcinoma cell lines in a ligand-independent manner. A, UMRC3, UMRC6, and primary renal cell carcinoma cell lines from index tumors, lymph node metastases, and systemic metastases were plated; allowed to adhere overnight; infected with controls (PBS orAd-βGal) or appropriate adenoviral vectors at 1:100 multiplicity of infection; and incubated for 72 h. Medium was supplemented with 2 ng/mL TGF-β (+TGF-β) or 50 μg/mL TGF-β neutralizing antibody (-TGF-β). After incubation, floating cells were combined with adhered cells, washed with PBS, stained with AnnexinV and propidium iodide, and measured in a flow cytometer. *, P < 0.001, Ad-TβRIII + TGF-β versus Ad-βGal; +, P < 0.001, Ad-TβRIII-TGF-β versus Ad-βGal. Columns, mean; bars, SD. B, renal cell carcinoma cell lines were infected with adenovirus containing recombinant TβRIII lacking extracellular domain (Ad-TβRIIICD). **, P < 0.001, Ad-TβRIIICD versus Ad-βGal. Columns, mean; bars, SD. C, renal cell carcinoma cell lines lacking functional TβRII receptor, UMRC3 and UMRC6 stably transfected with a dominant negativeTβRII receptor (UMRC6/ΔTBRII), were infected with a mutant form ofTβRIII lacking the cytoplasmic domain portion of this receptor (Ad-TβRIIIECD). Columns, mean; bars, SD.
We have assessed the expression of TGF-β receptors in all renal cell carcinoma in vitro models and found no significant differences in expression of TβRIII between normal renal epithelial cells and renal cell carcinoma cell lines infected with Ad-TβRIII (Fig. 2). Moreover, we have evaluated the protein expression levels of TβRII and TβRIII in primary clear cell renal cell carcinoma cultures and renal cell carcinoma cell lines infected with Ad-TβRII, Ad-TβRIII, Ad-TβRIIICD, and Ad-TβRIIIECD (Fig. 2). Results of immunohistochemical assessment of TβRIIIECD were identical to those of full-length TβRIII (data not shown). To the best of our knowledge, there is no specific antibody to the short cytoplasmic domain of TβRIIICD; consequently, Western blots and immunohistochemistry for TβRIIICD using commercially available antibodies to TβRIII were inconclusive. We have routinely done construct-specific PCR after viral infection and have confirmed transcription of full-length TβRIII and TβRIII mutants in our in vitro models.
Fig. 2.
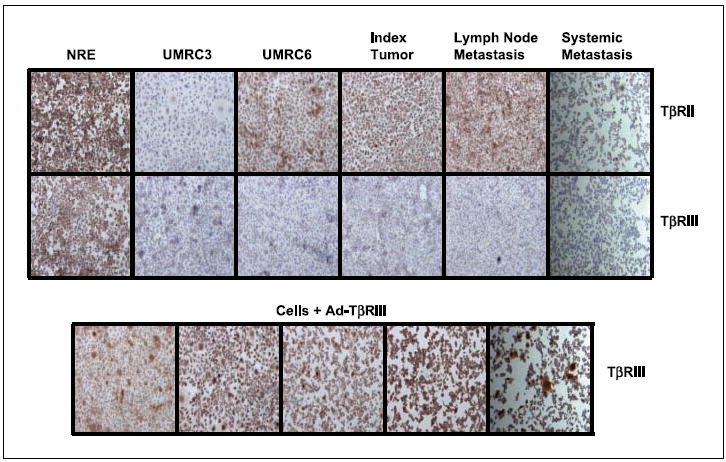
Immunohistochemical assessment ofTβRII andTβRIII expression in renal cell carcinoma cell lines before and after infection with Ad-TβRIII. Staining was done after antigen retrieval, as described in Materials and Methods. Magnification, × 400.
To determine if the presence of functional TβRII receptor was necessary for TβRIII-induced apoptosis, UMRC3 cells, which lack detectable TβRII mRNA and protein, UMRC6/ΔTBRII, and UMRC6 cells pretreated with a small-molecule inhibitor of TβRI/II were infected with Ad-TβRIII and Ad-TβRIIICD. Significant induction of apoptosis was shown in the absence or with the inhibition of TβRII after overexpressing the cytoplasmic portion of the TβRIII receptor (Fig. 3A).
Fig. 3.
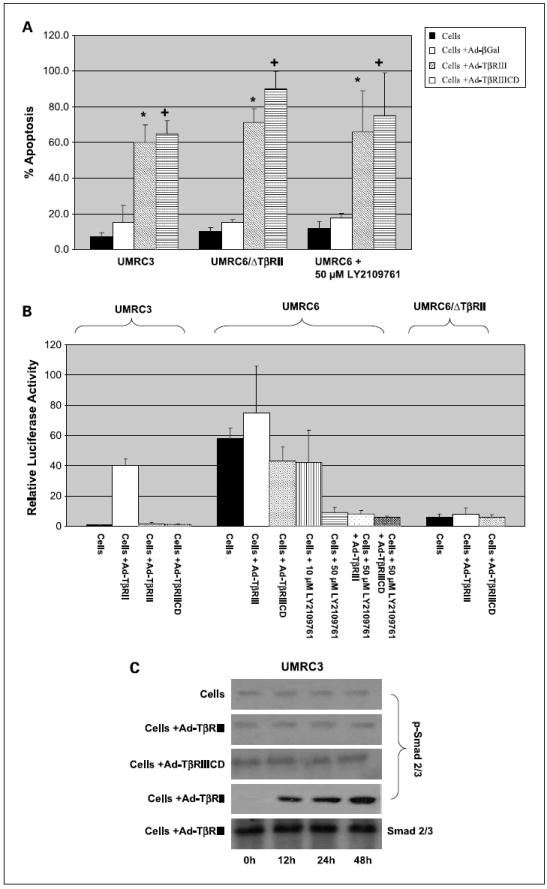
TβRIII-mediated apoptosis is not dependent on signal transduction through the TβR/Smad pathway. A, UMRC3 cells, which lack detectableTβRIII, UMRC6 cells stably transfected with a dominant negative TβRII receptor (UMRC6/ΔTBRII), and UMRC6 cells pretreated with a small-molecule inhibitor ofTβRI/II (50 μmol/L LY2109761) were infected with Ad-TβRIII and Ad-TβRIIICD as previously described. After 72 h of incubation, floating cells were combined with adhered cells, washed with PBS, stained with AnnexinV and propidium iodide, and measured in a flow cytometer.*, P < 0.001, Ad-TβRIII versus Ad-βGal; +, P < 0.001, Ad-TβRIIICD versus Ad-βGal. Columns, mean; bars, SD. B, signaling through the TβR/Smad pathway was quantitated after transient transfection of (CAGA)12MPL-Luc reporter plasmid, done 24 h after adenoviral infection. Culture medium was supplemented with 2 ng/mL TGF-β1for 24 h. Cells were harvested 48 h later after transient transfection and assayed for dual luciferase activity, as described in Materials and Methods. Columns, mean of triplicates of the relative light units normalized to the luciferase activity of the internal control vector (cells not treated with 2 ng/mL TGF-β1); bars, SD. C, Western blot analysis was used to evaluate the phosphorylation state of Smad2/3. Cells were grown under condition of supplementation with 2 ng/mL TGF-β1. Proteins were harvested in radio immuno precipitation assay buffer, containing proteinase and phosphatase inhibitor cocktails, at 0,12, 24, and 48 h after infection of renal cell carcinoma lines with appropriate adenovirus. Amounts of phospho-Smad2/3 and total Smad2/3 (used as loading control) were determined as described in Materials and Methods.
To confirm lack of TβR/Smad signaling in the TβRII knockdowns following infection with Ad-TβRIII/TβRIIICD, we transiently transfected the above-mentioned models with (CAGA)12MPL-Luc reporter plasmid, 24 hours after infection with adenovirus. This plasmid contains 12 repeats of the Smad3 response element (CAGA) in front of the luciferase cDNA, thus allowing for monitoring of TβRII/ TβRI–dependent Smad3 activation (19). It should be noted that during transient transfection experiments, the culture medium was supplemented with 2 ng/mL TGF-β1. No increased signaling through the TβR/Smad pathway was seen in UMRC3 cells, UMRC6/ΔTβRII stable clones, and UMRC6 cells pretreated with 50 μmol/L of TβRI/II inhibitor LY2109761 after overexpression of Ad-TβRII/Ad-TβRIIICD (Fig. 3B). On the contrary, significant increase in Smad3 reporter activity was seen in positive controls, UMRC6 cells expressing wild-type TβRII, and UMRC3 cells infected with adenovirus carrying wild-type TβRII (Ad-TβRII; Fig. 3B).
Phosphorylation of Smad2/3, a well-known substrate of activated TβRII/ TβRI receptors, and its association with TβRIII mediated apoptosis were further evaluated by Western blot. Figure 3C shows the absence of increased phosphorylation of Smad2/3 under conditions of 2 ng/mL TGF-β1 supplementation in UMRC3 cells infected with Ad-TβRIII or Ad-TβRIIICD. Increased Smad2/3 phosphorylation was, however, observed in UMRC3 cells infected with Ad-TβRII supplemented with TGF-β1, used as positive controls. Smad2/3 phosphorylation levels were also assessed minutes and hours after adenoviral infection; however, the first observed increase in Smad phosphorylation occurred at 12 hours after viral infection, corresponding to the expected timeline of TβR reexpression. Taken together, these data suggest that in clear cell renal cell carcinoma, TβRIII mediates apoptosis through its cytoplasmic domain in a ligand-independent and TβRII-independent manner. Furthermore, induction of apoptosis associated with expression of TβRIII is not dependent on signaling through the canonical TβR/Smad pathway.
TβRIII-mediated apoptosis involves signaling through p38 MAPK
Because TβRIII-mediated apoptosis did not require the presence of a functional TβR/Smad signaling pathway, we evaluated the effect of TβRIII expression on the activation of the extracellular signal – regulated kinase (ERK), SAPK/ JNK, and p38 MAPK pathways. Figure 4A shows increased phosphorylation of p38 MAPK after infection with Ad-TβRIII or Ad-TβRIIICD in cells lacking functional TβRII. Similar results were obtained when growth medium was supplemented with TGF-β or with TGF-β blocking antibodies (data not shown). Given these findings, the specific inhibitor of p38 MAPK, SB202190, was used to test its effect on TβRIII-induced apoptosis. Complete inhibition of TβRIII-mediated apoptosis was observed in cells pretreated for 24 hours with SB202190 before infection with Ad-TβRIII (Fig. 4B). To show functional inactivation of p38 MAPK signaling, we evaluated the phosphorylation status of a known p38 MAPK effector, MAPKAPK-2, after pretreatment with SB202190. Figure 4C shows that increased phosphorylation of MAPKAPK-2 observed after overexpression of TβRIII was completely abolished in cells pretreated with SB202190.
Fig. 4.
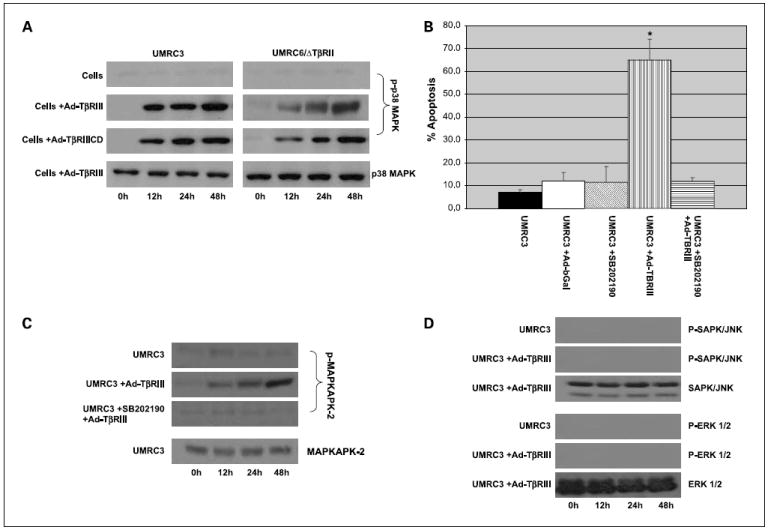
Signal transduction through the p38 MAPK pathway is essential for TβR/Smad – independentTβRIII-induced apoptosis. A, Western blot analysis of p38 MAPK phosphorylation state. Proteins were harvested in radioimmun oprecipitation assay buffer, containing proteinase and phosphatase inhibitor cocktails, at 0,12, 24, and 48 h after infection of renal cell carcinomalines with adenovirus. Amounts of phospho-p38MAPKandtotalp38MAPK (used as loading control) were determined as described in Materials and Methods. B, UMRC3 cells were pretreated for 24 h withp38 MAPK inhibitor (SB202190) before infection with Ad-TβRIII. After 72 h of incubation with adenovirus, floating cells were combined with adhered cells, washed with PBS, stained with Annexin V and propidium iodide, and measured by flow cytometry.*, P <0.001, Ad-TβRIII versus Ad-TβRIII + SB202190. Columns, mean; bars, SD. C, Western blot analysis of phosphorylation status of p38 MAPK effector, MAPKAPK-2. UMRC3 cells were pretreated for 24 hwithp38 MAPK inhibitor (SB202190) before infection with Ad-TβRIII. Protein was isolatedas described above. Amounts of phospho-MAPKAPK-2 and total MAPKAPK-2 (used as loading control) were determined as described in Materials and Methods. D, in controls and in cells infected with Ad-TβRIII, the amounts of phospho-ERK1/2 and phospho-SAPK/JNK were determined by Western blot analyses as described above. Total ERK1/2 and total SAPK/JNK were used as loading controls.
In contrast, Fig. 4D shows that no effect was noted on the phosphorylation of ERK and SAPK/JNK under conditions of TβRIII overexpression. Combined, these results indicate that signal transduction through the p38 MAPK pathway is essential for TβRII/Smad–independent, TβRIII-induced apoptosis in clear cell renal cell carcinoma, and that this activity is independent of the ERK and SAPK/JNK pathways.
TβRIII blocks tumor growth and mediates apoptosis in human renal cell carcinoma xenografts lacking functional TβR/Smad pathway
To evaluate whether reintroduction of TβRIII expression resulted in TβR/Smad–independent apoptosis in vivo, UMRC3 xenografts were successfully established in the flanks of athymic nude mice. As shown in Fig. 5A, administration of Ad-TβRIII and Ad-TβRIIICD resulted in significant suppression of tumor growth compared with the control groups, suggesting that the cytoplasmic domain of TβRIII was essential for the in vivo growth inhibitory effect of TβRIII. Coinfection with Ad-TβRII and Ad-TβRIII did not result in enhanced growth inhibition compared with infection with Ad-TβRIII alone, whereas reexpression of TβRII resulted in less efficient growth inhibition compared with TβRIII/TβRIIICD. There was no significant effect on growth kinetics in tumors treated with Ad-βGal compared with untreated controls group. In addition, there was no difference in body weight of animals between groups (data not shown), indicative of low general toxicity.
Fig. 5.
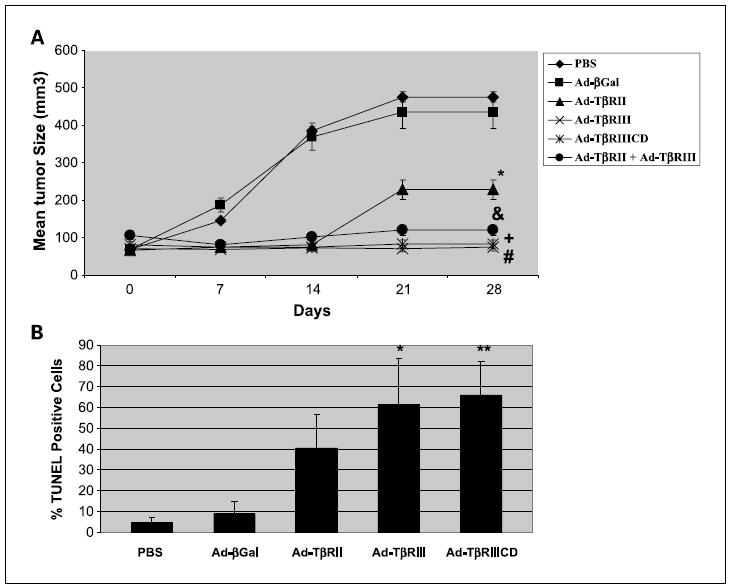
TβRIII mediates apoptosis in human renal cell carcinoma xenografts. A, UMRC3 cell suspensions of 1 ×106/0.1mL were injected s.c. into flanks of female nude mice and tumor growth was monitored twice a week. Intratumoral injections of PBS and appropriate adenovirus were done, with five animals randomly assigned to each treatment group once tumors reached 100 mm3. A total of three injections were done on days 1, 7, and 14 for a total dose of 1.5 3 1010 plaque-forming units/tumor., P < 0.05, Ad-TβRII versus Ad-βGal; &, P < 0.05, Ad-TβRII + Ad-TβRIII versus Ad-βGal; +, P < 0.05, Ad-TβRIII versus Ad-βGal; #, P < 0.05, Ad-TβRIIICD versus Ad-βGal. Points, mean; bars, SD. B, tumors were removed 7 d after the last adenoviral injection and representative portions embedded in paraffin blocks. For TUNEL assays, the tissue sections were evaluated with theTumorTACS in situ apoptosis detection kit. TUNEL quantification was done by counting the number of positive tumor cells in 10 random fields at ×100 and dividing by the total number of cells per field. Columns, mean; bars, SD.*, P < 0.05, Ad-TβRIII versus Ad-βGal; **, P < 0.05, Ad-TβRIIICD versus Ad-βGal.
To further evaluate the effects of the TβRIII, TβRIIICD, and TβRII adenovirus vectors in UMRC3 tumors, we conducted histochemical analysis of tumors harvested 7 days after injection with each vector. In situ TUNEL staining confirmed that injection of tumors with either the Ad-TβRIII or Ad-TβRIIICD vector, but not the control Ad-βGal, induced striking apoptosis of tumor cells (Figs. 5B and 6). The observed apoptotic effects of TβRIII/TβRIIICD were associated with increased phospho-p38 MAPK staining of treated tumor tissues, with no significant change in phospho-Smad2/3 staining, when compared with PBS-and Ad-βGal –treated tumor controls (Fig. 6). Similarly, tumors treated with Ad-TβRII or Ad-TβRII + Ad-TβRIII (data not shown) showed significantly increased amounts of apoptosis; however, these effects were seen in conjunction with increased phospho-Smad2/3 staining and p38 MAPK staining. These in vivo findings suggest that induction of apoptosis associated with overexpression of TβRIII is driven by the cytoplasmic domain of this receptor, independent of the TβR/Smad pathway, and thus corroborate our in vitro findings.
Fig. 6.

Immunohistochemical assessment of human renal cell carcinoma xenografts. Tumors were removed 7 d after the last adenoviral injection, and representative portions embedded in paraffin blocks. For TUNEL assays, the tissue sections were evaluated with the Tumor TACS in situ apoptosis detection kit. Immunoperoxidase staining was done after antigen retrieval using the Vectastatin Elite ABC kit as described in Materials and Methods. Magnification, ×400.
Discussion
TGF-β signaling transduction controls a diverse number of biological processes in a dynamic and context-dependent manner (20, 21). Although it is well known that TGF-β signaling components are misregulated in several human epithelial malignancies, the role of TGF-β family members in the initiation and progression of renal cell carcinoma has not been well defined (4-9, 22). Recently, we have shown that the transformation of normal renal epithelium to cancer was associated with loss of TβRIII, a critical event in early-stage renal cell carcinogenesis (3). Similarly, Criswell and colleagues (23) have recently shown that silencing of TβRIII in normal breast epithelial cells leads to formation of poorly differentiated tumors in athymic nude mice, associated loss of E-cadherin, and activation of nuclear factor-κB signaling, further implicating loss of TβRIII as a general phenomena important to carcinogenesis.
TβRIII has a short cytoplasmic domain with no known intrinsic kinase activity, and has classically been thought to act as a TGF-β coreceptor, concentrating ligand on the cell surface and enhancing ligand binding to the signaling receptor TβRII (12). Alternatively, TβRIII has been proposed to function by undergoing ectodomain shedding, with a soluble domain of TβRIII antagonizing autocrine TGF-β signaling and reducing invasiveness and angiogenesis (4, 13). Nonetheless, a strict requirement for TβRIII during the epithelial-mesenchymal transition process that leads to heart valve formation supports a more direct role in TGF-β signaling for TβRIII (24). Consequently, recent studies have challenged the notion of TβRIII as a nonsignaling receptor by showing an additional role for TβRIII through activity present in its cytoplasmic domain, which has been shown to interact with proteins, such as Gα-interacting protein and β-arrestin, potentially scaffolding TβRIII to Smad-independent signaling pathways including p38 MAPK (14-17, 25).
In this investigation, we have shown that reexpression of TβRIII in in vitro and in vivo clear cell renal cell carcinoma models induces striking apoptosis in a manner independent of the TGF-β ligand or the presence of a functional TβRII/Smad signal transduction pathway. This ligand independence was corroborated by using antibodies that blocked TGF-β activity and by expression of a recombinant form of TβRIII lacking an extracellular TGF-β binding domain. Conversely, no induction of apoptosis was noted when a recombinant form of TβRIII lacking the cytoplasmic domain was used, showing that the cytoplasmic domain was sufficient for ligand-independent signaling. Santander et al. (17) have also shown that TGF-β–independent TβRIII signaling via p38 MAPK regulates TGF-β–responsive genes in myoblasts and fibroblasts; however, in their experiments, a functional TβRII was necessary for this TGF-β–independent signal transduction. We used three experimental designs to show that TβRIII-induced apoptosis was TβRII/ TβRI/Smad independent: (a) the UMRC3 cell line, which lacks detectable levels of TβRII and TβRIII mRNA or protein expression; (b) the UMRC6 cell line stably transected with a dominant-negative form of TβRII; and (c) the UMRC6 cell line pretreated with a small-molecule TβRI inhibitor. Lack of TβR/Smad signaling in these TβRII knockdown models was confirmed using a reporter plasmid containing Smad response elements and by assessment of Smad2/3 phosphorylation levels after overexpression of Ad-TβRIII or Ad-TβRIIICD. We found that full-length TβRIII and recombinant TβRIII lacking the extracellular TGF-β binding domain were both capable of inducing marked apoptosis under conditions absent of functional TβRII and TβR/Smad signal transduction. To our knowledge, this is the first demonstration linking apoptosis to the cytoplasmic domain of TBRIII.
As previously mentioned, an activated TβR complex can activate signaling through non-Smad pathways such as MAPK, ERK, and SAPK/JNK (26-28). Interestingly, we found a significant increase in phosphorylated p38 MAPK in Ad-TβRIII– and Ad-TβRIIICD–infected cells compared with uninfected cells and cells infected with adenoviral controls, whereas no difference in ERK and SAPK/JNK phosphorylation was seen. Furthermore, pretreatment of renal cell carcinoma cells with a specific p38 MAPK inhibitor before infection completely prevented apoptotic cell death. These results show that signal transduction through the p38 pathway is essential for TβR/Smad–independent induction of apoptosis by the cytoplasmic domain of TβRIII. While our article was in preparation, You et al. (25) published that TβRIII contributes to and enhances TGF-β–mediated growth inhibition in L6 rat myoblasts and primary human epithelial cells through both the TβRI/Smad3– dependent and p38 MAPK pathways. This growth inhibition seems to be mediated in part by the cytoplasmic domain of TβRIII. These data corroborate our findings but differ distinctly in requirements for canonical TGF-β/TβR/Smad–mediated signaling. These differences may be due to the use of normal primary cells versus that of cancer cells or due to tissue specificity.
There is ample evidence implicating the p38 MAPK pathway in the suppression of tumorigenesis and induction of apoptosis (29, 30). Increased p38 MAPK signaling has been shown to inhibit cell growth and potentiate apoptosis by decreasing the expression of cyclin D1, inhibition of Cdc25 phosphatase activity, activation of the p16/Rb and p19ARF/ p53 tumor suppressor pathways, and phosphorylation of members of the Bcl-2 family (31-34). The selectivity of p38 signaling in tumor suppression is unclear; however, Dolado et al. (35) and others have reported that p38 MAPK selectively functions as a sensor of oxidative stress during the initiation of tumorigenesis. This intriguing observation, combined with data presented herein, further strengthens our hypothesis that TβRIII loss is an early and critical event in renal cell carcinoma carcinogenesis. Renal proximal tubule epithelial cells, which give rise to the clear cell variant of renal cell carcinoma, are especially susceptible to oxidative stress; consequently, an intact TβRIII/p38 MAPK signaling axis may be an important cellular death mechanism under conditions of oxidative cell damage (36, 37). Conversely, loss of functional TβRIII signaling may contribute to apoptotic escape and tumorigenesis. Finally, these data provide a strong rationale for therapeutic modulation of the TβRIII/p38 MAPK axis in therapeutic strategies for clear cell renal cell carcinoma.
In summary, our work is the first to mechanistically link the cytoplasmic domain of TβRIII to activation of p38 MAPK to induce apoptosis. Furthermore, this mechanism seems to be independent of TGF-β and canonical TβRII/TβRI/Smad signaling. The mechanism by which the cytoplasmic domain of TβRIII mediates activation of p38 MAPK remains to be resolved, and ongoing experiments are addressing this question.
Acknowledgments
Grant support: American Urological Association Foundation (V. Margulis), National Cancer Institute grant R01CA104505 (J.A. Copland and C.G.Wood), and Dr. and Mrs. Ellis Brunton Rare Cancer Research Fund (J.A. Copland).
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Motzer RJ, Bacik J, Mazumdar M. Prognostic factors for survival of patients with stage IV renal cell carcinoma: Memorial Sloan-Kettering Cancer Center experience. Clin Cancer Res. 2004;10:6302–3S. doi: 10.1158/1078-0432.CCR-040031. [DOI] [PubMed] [Google Scholar]
- 3.Copland JA, Luxon BA, Ajani L, et al. Genomic profiling identifies alterations in TGFβ signaling through loss of TGFβ receptor expression in human renal cell carcinogenesis and progression. Oncogene. 2003;22:8053–62. doi: 10.1038/sj.onc.1206835. [DOI] [PubMed] [Google Scholar]
- 4.Dong M, How T, Kirkbride KC, et al. The type III TGF-β receptor suppresses breast cancer progression. J Clin Invest. 2007;117:206–17. doi: 10.1172/JCI29293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hempel N, How T, Dong M, Murphy SK, Fields TA, Blobe GC. Loss of betaglycan expression in ovarian cancer: role in motility and invasion. Cancer Res. 2007;67:5231–8. doi: 10.1158/0008-5472.CAN-07-0035. [DOI] [PubMed] [Google Scholar]
- 6.Turley RS, Finger EC, Hempel N, How T, Fields TA, Blobe GC. The type III transforming growth factor-β receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 2007;67:1090–8. doi: 10.1158/0008-5472.CAN-06-3117. [DOI] [PubMed] [Google Scholar]
- 7.Florio P, Ciarmela P, Reis FM, et al. Inhibin α-subunit and the inhibin coreceptor betaglycan are downregulated in endometrial carcinoma. Eur J Endocrinol. 2005;152:277–84. doi: 10.1530/eje.1.01849. [DOI] [PubMed] [Google Scholar]
- 8.Gordon KJ, Dong M, Chislock EM, Fields TA, Blobe GC. Loss of type III transforming growth factor β receptor expression increases motility and Invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis. 2007 doi: 10.1093/carcin/bgm249. [DOI] [PubMed] [Google Scholar]
- 9.Finger EC, Turley RS, Dong M, How T, Fields TA, Blobe GC. T RIII suppresses non-small cell lung cancer invasiveness and tumorigenicity. Carcinogenesis. 2008 doi: 10.1093/carcin/bgm289. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-β receptor system. Cell. 1991;67:785–95. doi: 10.1016/0092-8674(91)90073-8. [DOI] [PubMed] [Google Scholar]
- 11.Wang XF, Lin HY, Ng-Eaton E, Downward J, Lodish HF, Weinberg RA. Expression cloning and characterization of the TGF-β type III receptor. Cell. 1991;67:797–805. doi: 10.1016/0092-8674(91)90074-9. [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Casillas F, Wrana JL, Massague J. Betaglycan presents ligand to theTGF-β signaling receptor. Cell. 1993;73:1435–44. doi: 10.1016/0092-8674(93)90368-z. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Casillas F, Payne HM, Andres JL, Massague J. Betaglycan can act as a dual modulator of TGF-β access to signaling receptors: mapping of ligand binding and GAG attachment sites. J Cell Biol. 1994;124:557–68. doi: 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blobe GC, Liu X, Fang SJ, How T, Lodish HF. A novel mechanism for regulating transforming growth factor β (TGF-β) signaling. Functional modulation of type III TGF-β receptor expression through interaction with the PDZ domain protein, GIPC. J Biol Chem. 2001;276:39608–17. doi: 10.1074/jbc.M106831200. [DOI] [PubMed] [Google Scholar]
- 15.Blobe GC, Schiemann WP, Pepin MC, et al. Functional roles for the cytoplasmic domain of the type III transforming growth factor β receptor in regulating transforming growth factor β signaling. J Biol Chem. 2001;276:24627–37. doi: 10.1074/jbc.M100188200. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Kirkbride KC, How T, et al. Science. Vol. 301. New York, NY: 2003. β-Arrestin 2 mediates endocytosis of type III TGF-β receptor and down-regulation of its signaling; pp. 1394–7. [DOI] [PubMed] [Google Scholar]
- 17.Santander C, Brandan E. Betaglycan inducesTGF-β signaling in a ligand-independent manner, through activation of the p38 pathway. Cell Signal. 2006;18:1482–91. doi: 10.1016/j.cellsig.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Grossman HB, Wedemeyer G, Ren LQ. Human renal carcinoma: characterization of five new cell lines. J Surg Oncol. 1985;28:237–44. doi: 10.1002/jso.2930280320. [DOI] [PubMed] [Google Scholar]
- 19.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott RL, Blobe GC. Role of transforming growth factor β in human cancer. J Clin Oncol. 2005;23:2078–93. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 21.Zavadil J, Bitzer M, Liang D, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci U S A. 2001;98:6686–91. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 23.Criswell TL, Arteaga CL. Modulation of NFκB activity and E-cadherin by the type III transforming growth factor β receptor regulates cell growth and motility. J Biol Chem. 2007;282:32491–500. doi: 10.1074/jbc.M704434200. [DOI] [PubMed] [Google Scholar]
- 24.Brown CB, Boyer AS, Runyan RB, Barnett JV. Requirement of type III TGF-β receptor for endocardial cell transformation in the heart. Science (New York, NY) 1999;283:2080–2. doi: 10.1126/science.283.5410.2080. [DOI] [PubMed] [Google Scholar]
- 25.You HJ, Bruinsma MW, How T, Ostrander JH, Blobe GC. The type III TGF-β receptor signals through both Smad3 and the p38 MAP kinase pathways to contribute to inhibition of cell proliferation. Carcinogenesis. 2007;28:2491–500. doi: 10.1093/carcin/bgm195. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Hebert MC, Zhang YE. TGF-βreceptor-activated p38 MAP kinase mediates Smad-independentTGF-β responses. EMBOJ. 2002;21:3749–59. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moustakas A, Heldin CH. Non-SmadTGF-β signals. J Cell Sci. 2005;118:3573–84. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-β-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–73. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- 29.Bulavin DV, Fornace AJ., Jr p38 MAP kinase’s emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118. doi: 10.1016/S0065-230X(04)92005-2. [DOI] [PubMed] [Google Scholar]
- 30.Timofeev O, Lee TY, Bulavin DV. A subtle change in p38 MAPK activity is sufficient to suppress in vivo tumorigenesis. Cell Cycle. 2005;4:118–20. doi: 10.4161/cc.4.1.1342. [DOI] [PubMed] [Google Scholar]
- 31.Lavoie JN, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–16. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 32.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 33.Bulavin DV, Amundson SA, Fornace AJ. p38 and Chk1kinases:different conductors for the G2/M check-point symphony. Curr Opin Genet Dev. 2002;12:92–7. doi: 10.1016/s0959-437x(01)00270-2. [DOI] [PubMed] [Google Scholar]
- 34.Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 35.Dolado I, Swat A, Ajenjo N, DeVita G, Cuadrado A, Nebreda AR. p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 36.Dong J, Ramachandiran S, Tikoo K, Jia Z, Lau SS, Monks TJ. EGFR-independent activation of p38 MAPK and EGFR-dependent activation of ERK1/2 are required for ROS-induced renal cell death. Am J Physiol. 2004;287:F1049–58. doi: 10.1152/ajprenal.00132.2004. [DOI] [PubMed] [Google Scholar]
- 37.Weber TJ, Huang Q, Monks TJ, Lau SS. Differential regulation of redox responsive transcription factors by the nephrocarcinogen 2,3,5-tris(glutathion-S-yl) hydroquinone. Chem ResToxicol. 2001;14:814–21. doi: 10.1021/tx000190r. [DOI] [PubMed] [Google Scholar]